

ABSTRACT
The Epstein-Barr virus (EBV) gp350 glycoprotein interacts with the cellular receptor to mediate viral entry and is thought to be the major target for neutralizing antibodies. To better understand the role of EBV-specific antibodies in the control of viral replication and the evolution of sequence diversity, we measured EBV gp350-specific antibody responses and sequenced the gp350 gene in samples obtained from individuals experiencing primary EBV infection (acute infectious mononucleosis [AIM]) and again 6 months later (during convalescence [CONV]). EBV gp350-specific IgG was detected in the sera of 17 (71%) of 24 individuals at the time of AIM and all 24 (100%) individuals during CONV; binding antibody titers increased from AIM through CONV, reaching levels equivalent to those in age-matched, chronically infected individuals. Antibody-dependent cell-mediated phagocytosis (ADCP) was rarely detected during AIM (4 of 24 individuals; 17%) but was commonly detected during CONV (19 of 24 individuals; 79%). The majority (83%) of samples taken during AIM neutralized infection of primary B cells; all samples obtained at 6 months postdiagnosis neutralized EBV infection of cultured and primary target cells. Deep sequencing revealed interpatient gp350 sequence variation but conservation of the CR2-binding site. The levels of gp350-specific neutralizing activity directly correlated with higher peripheral blood EBV DNA levels during AIM and a greater evolution of diversity in gp350 nucleotide sequences from AIM to CONV. In summary, we conclude that the viral load and EBV gp350 diversity during early infection are associated with the development of neutralizing antibody responses following AIM.
IMPORTANCE Antibodies against viral surface proteins can blunt the spread of viral infection by coating viral particles, mediating uptake by immune cells, or blocking interaction with host cell receptors, making them a desirable component of a sterilizing vaccine. The EBV surface protein gp350 is a major target for antibodies. We report the detection of EBV gp350-specific antibodies capable of neutralizing EBV infection in vitro. The majority of gp350-directed vaccines focus on glycoproteins from lab-adapted strains, which may poorly reflect primary viral envelope diversity. We report some of the first primary gp350 sequences, noting that the gp350 host receptor binding site is remarkably stable across patients and time. However, changes in overall gene diversity were detectable during infection. Patients with higher peripheral blood viral loads in primary infection and greater changes in viral diversity generated more efficient antibodies. Our findings provide insight into the generation of functional antibodies, necessary for vaccine development.
KEYWORDS: EBV, gp350, antibodies, neutralization, ADCP, Epstein-Barr virus
Epstein-Barr virus (EBV) establishes a persistent infection in most adults by the fourth decade of life. While most EBV-infected individuals do not develop clinical sequelae (1), primary infection in older children and adults is often symptomatic (acute infectious mononucleosis [AIM]) and is linked to an increased risk of multiple sclerosis (2). EBV frequently causes lymphoproliferative disorders in immunosuppressed individuals and is associated with common cancers (3, 4).
The development of an EBV vaccine is thus a high priority and will likely require the generation of neutralizing antibodies (5). Cellular entry is initiated by EBV gp350 glycoprotein binding to the virus host cell receptors (CR2/CD21 [6] or CR1/CD35 [7]). gp350 is thought to be a major target of neutralizing antibodies (8–10), and vaccine development efforts are focused on generating gp350-specific neutralizing antibodies (11–15). Most immunogens are based on sequences from lab-adapted strains, as are assays used to measure neutralizing antibodies. These lab-adapted strains may differ significantly from circulating infectious strains.
Until recently, it was commonly accepted that the genomes of double-stranded DNA (dsDNA) viruses, such as herpesviruses, are fairly stable, particularly in comparison to those of RNA viruses. Recent studies, including our own, have demonstrated that dsDNA viruses can exhibit levels of variation similar to those observed in some RNA viruses, such as West Nile virus and HIV-1 (16, 17). Most published EBV gp350 sequences have originated from either transformed B-cell lines or cancerous tissue; at present, only a very limited number of primary EBV gp350 sequences are available (18). A better understanding of the diversity of circulating EBV gp350 sequences, as well as the potential role of EBV-specific antibodies in the evolution of EBV gp350 diversity over time in infected individuals, would be helpful to inform vaccine development.
Here we report the findings of studies investigating the generation of EBV gp350-specific antibodies, characterizing binding and function (antibody-dependent cell-mediated phagocytosis [ADCP]-stimulating neutralization) over time following primary EBV infection. Plasma IgG and saliva samples were obtained from individuals at presentation with AIM and 6 months later. EBV gp350-binding antibodies and neutralizing activity were detected during AIM in the majority of individuals; titers increased with time after infection. We also report some of the first sequences of gp350 amplified directly from circulating virus rather than from transformed cell lines or disease-associated tissue. While we observed interpatient sequence variability, the gp350-binding site was highly conserved and remarkably stable over time. The high viral load during AIM and the increased viral gene variation from AIM to convalescence (CONV) correlated positively with the strength of the neutralizing antibody response during CONV. Altogether, our data demonstrate the detection of neutralizing antibodies earlier than previously reported and identify key factors associated with the generation of neutralizing antibodies during primary viral infection.
RESULTS
EBV gp350-specific binding antibodies are readily detected in patient plasma at presentation with AIM.
EBV gp350-specific binding antibodies were detected in the purified IgG fraction of plasma from 17 (71%) individuals at presentation with AIM and from all of the same individuals 6 months later (during CONV) (Fig. 1). gp350-binding antibody levels increased significantly over time from AIM (median, 4,648 mean fluorescence intensity [MFI]; interquartile ratio [IQR], 3,858 MFI) to CONV (median, 16,817 MFI; IQR, 9,874 MFI) (Wilcoxon matched-pairs signed-rank test for AIM versus CONV, P < 0.0001). The gp350-specific IgG levels measured during CONV were similar to those measured in age-matched chronically infected (CHRON) individuals (Fig. 1A; median, 17,728 MFI; IQR, 11,825 MFI; Mann-Whitney test, P = 0.96). EBV gp350-binding antibodies were detected primarily in the IgG1 (13 patients; 54%) and IgG3 (16 patients; 67%) fractions of plasma during AIM; IgG2 gp350-binding antibodies were detected in only 6 (25%) patients during AIM. gp350-specific IgG1, IgG2, and IgG4 binding antibody titers increased significantly from AIM to CONV (P = 0.0001, P = 0.034, and P = 0.0001, respectively) to levels equal to those in CHRON individuals (Fig. 1B, C, and E). gp350-specific IgG3 binding antibody titers were significantly lower in CHRON individuals than during CONV (Fig. 1D) (P < 0.001).
FIG 1.

Levels of EBV gp350-binding immunoglobulin (IgG) during AIM, CONV, and chronic EBV infection. IgG was isolated from the serum of 24 patients during primary EBV infection (AIM) and 6 months later (during CONV). Binding of gp350 was compared to that in 12 age-matched chronically infected individuals (CHRON) and 12 age-matched EBV-seronegative individuals (Sero NEG). Binding of total patient IgG, as well as that of each IgG subclass, to EBV B95.8 gp350 (aa 4 to 440), was determined by detection of fluorescently labeled secondary antibodies. % Pos, percentage of individuals who were positive (indicated on the bottom line of each panel). P values were determined by the Mann-Whitney and Wilcoxon matched-pairs signed-rank tests. *, P < 0.05; **, P < 0.01; †, P < 0.001.
Patient IgG mediates ADCP during CONV.
Antibody-dependent cell-mediated phagocytosis (ADCP) activity was detected in the plasma-derived IgG of only 4 (17%) individuals with AIM but was detected in the plasma-derived IgG of 19 (79%) individuals during CONV (median phagocytosis score, 211,941; IQR of the phagocytosis score, 109,561; AIM versus CONV, P < 0.0001) (Fig. 2A) at levels similar to those in age-matched CHRON individuals (median phagocytosis score, 227,150; IQR of the phagocytosis score, 109,199; CONV versus CHRON individuals, P was not significant).
FIG 2.

IgG from EBV-infected donors stimulates ADCP in cultured phagocytes. (A) Patient IgG samples were used to stimulate phagocytosis of gp350-coated microbeads by THP-1 cells. Data for 24 matched patient samples obtained during AIM and CONV, 12 age-matched chronically infected individuals (CHRON), and 12 age-matched serologically EBV-negative donors (Sero NEG) are included. % Pos, percentage of individuals who were positive (indicated on the bottom line). †, P < 0.001; NS, not significant. (B to F) Correlations between phagocytosis scores and total anti-gp350 IgG, anti-gp350 IgG1, anti-gp350 IgG2, anti-gp350 IgG3, or anti-gp350 IgG4 relative titers (phagocytosis score versus relative MFI). Results only for samples obtained from 19 individuals during CONV and 11 CHRON individuals positive for ADCP activity are shown. P values were determined by the Mann-Whitney and Wilcoxon matched-pairs signed-rank tests.
A positive correlation between total anti-gp350 IgG levels and ADCP activity was observed in samples obtained during CONV and in CHRON individuals (19 samples obtained during CONV and 11 samples from CHRON individuals; Spearman r value = 0.480, P < 0.01) (Fig. 2B). ADCP activity was also associated with anti-gp350 IgG1 levels but did not reach significance (P = 0.06), suggesting that phagocytosis was largely mediated by IgG1 (Fig. 2C).
Neutralizing activity of AIM and CONV IgG in a B-cell line.
The EBV enhanced green fluorescent protein (eGFP)-tagged Raji cell neutralization assay performs comparably to a previously used transformation assay (19, 20). Using this assay, IgG neutralizing activity was not detected in any AIM patients (Fig. 3A) but was detected in 14 (58%) patients during CONV (median CONV neutralization score, 14.8%; IQR, 29%; AIM versus CONV, P < 0.0001) at titers similar to those detected in chronically infected individuals (median neutralization score in CHRON individuals, 14.8%; IQR, 21.2%). The murine monoclonal antibody 72A1, specific for the CR2-gp350-binding site, was used as a positive control and neutralized EBV infection by 90% (Fig. 3A). Neutralizing activity was positively correlated with total IgG levels (Spearman r value = 0.702, P < 0.0001), IgG1 levels (Spearman r value = 0.444, P = 0.006), and IgG4 levels (Spearman r value = 0.410, P = 0.013) but not with IgG2 or IgG3 levels (not shown).
FIG 3.

IgG recovered from patients during AIM and CONV neutralizes EBV infection of primary and cultured B cells. (A) IgG from AIM patient sera does not neutralize EBV infection of the Raji cell line; neutralizing activity was detected for serum IgG from the majority of individuals during CONV (58%) and the majority of CHRON individuals (72%). (B) In contrast, IgG recovered from most individuals during AIM (83%), all individuals during CONV, and all CHRON individuals neutralized EBV infection of primary cord blood B cells. (C and D) Correlation of individual patient blood viral load values during AIM with neutralization of EBV-infected Raji cells (C) or primary B cells (D). (E) Neutralization of EBV Akata infection by patient IgG is mediated by gp350-specific antibodies. Addition of 0.625 μg soluble gp350 to the neutralization reaction mixture reduced the effect of patient-specific neutralization of EBV infection in an IgG-dependent manner. P values were determined by the Mann-Whitney and Wilcoxon matched-pairs signed-rank tests *, P < 0.05; **, P < 0.01; †, P < 0.001; NS, not significant.
Neutralizing activity of AIM and convalescent-phase IgG in primary B cells.
We repeated our neutralization assays using EBV-negative, primary human cord blood B cells as targets and quantitative PCR (qPCR) to quantify the levels of infection (see Materials and Methods). As in the Raji cell assay, incubation of eGFP-tagged EBV Akata with monoclonal antibody 72A1 prior to the addition of target cells resulted in a 90% reduction in the level of infection, suggesting that virus detected in this assay required an interaction between gp350 and the host cell CD21/CD35 receptors (Fig. 3A and B). Neutralizing activity against Akata infection was detected in the plasma-derived IgG of 20 (83%) individuals with AIM (median neutralization score in individuals with AIM, 28.6%; IQR, 25.3%) (Fig. 3B) and in all individuals during CONV (median neutralization score in individuals during CONV, 63.2%; IQR, 18.6%) (AIM versus CONV, P < 0.0001) at levels similar to those in samples from CHRON individuals (median neutralization score for CHRON individuals, 65.5%; IQR, 19.7%) (Fig. 3B). The neutralization of Akata infection of primary B cells was positively correlated with total anti-gp350 IgG levels (r = 0.546, P = 0.001) and anti-gp350 IgG1 levels (r = 0.420, P = 0.029) but not with anti-gp350 IgG2, IgG3, or IgG4 levels (not shown). Finally, neutralization of Akata infection by samples from individuals during CONV in the primary B-cell assay (r = 0.366 and P = 0.039) and cultured Raji B-cell assay (r = 0.587 and P = 0.003) was positively correlated with ADCP activity.
Neutralizing antibody titers during CONV correlate with the peripheral blood EBV load in AIM.
Recent studies have reported that a high viral load during acute HIV-1 infection is associated with the development of broadly neutralizing antibodies (bnAbs) during the chronic phase of infection (21, 22). The EBV load in the blood of individuals with AIM correlated with the neutralization activity of plasma obtained from the same patients during CONV in Raji cell assays (Spearman r value = 0.89; P < 0.0001) (Fig. 3C) and in primary cell assays (Spearman r value = 0.84; P < 0.001) (Fig. 3D). Additionally, the peripheral blood EBV load in individuals with AIM was positively correlated with the total anti-gp350 IgG levels during CONV (Spearman r value = 0.512, P = 0.05) but not with anti-gp350 IgG1 levels during CONV (Spearman r value = 0.415; P = 0.11) (data not shown). No correlation between the EBV load during CONV and the neutralizing antibody activity or anti-gp350 IgG levels during CONV was observed.
Neutralization of EBV infection in Raji cells is inhibited by soluble gp350.
A blocking assay was used to further verify that the neutralizing activity measured in our assays was gp350 dependent. Patient IgG was incubated with 0.625 μg/reaction mixture of a soluble, truncated version of gp350 (amino acids [aa] 4 to 450, containing the CR2-binding site) prior to incubation of IgG with EBV and subsequent addition of Raji cells (23). As Fig. 3E illustrates, the addition of soluble gp350 reduced the neutralization mediated by IgG in a sample collected from a patient during CONV (patient E1477). Together with previously published reports (24, 25), our data suggest that the neutralization of EBV infection by IgG, as measured by our assays, is in large part, though not perhaps entirely, dependent on antibodies directed against the viral envelope gp350. The low residual levels of neutralization in the presence of IgG (at concentrations of 0.1 to 1 μg/ml) and soluble gp350 (Fig. 3E) might reflect the presence of antibodies directed against other viral envelope glycoproteins capable of mediating infection.
EBV gp350 gene sequences amplified from AIM patients display polymorphisms at both the nucleotide and protein levels.
We sequenced the gp350 genes from six patients to determine whether the observed interpatient differences in neutralizing activity could be explained by sequence differences between individual patient strains and lab-adapted strains (Fig. 4A). All patient sequences clustered with type I EBV reference strains (strains B95.8, Mutu, Akata, GD1, and GD2) and were distinctly separate from the sequence of the prototypical type II EBV strain AG876 (Fig. 4B). Furthermore, all AIM patient gp350 gene sequences clustered most closely with the B95.8 gp350 gene sequence, apart from viral sequences isolated from nasopharyngeal carcinoma-associated EBV strains (GD1, GD2) and the Burkitt's lymphoma-derived Akata sequence. Alignment of each AIM patient gp350 nucleotide sequence with the B95.8 and Akata gp350 nucleotide sequences demonstrated the conservation of bases encoding the binding site between gp350 and the CR2 cellular receptor; likewise, all variations from the consensus sequence observed in the amino acid sequence variants were located outside this binding region (Fig. 4C and D) (26–28).
FIG 4.
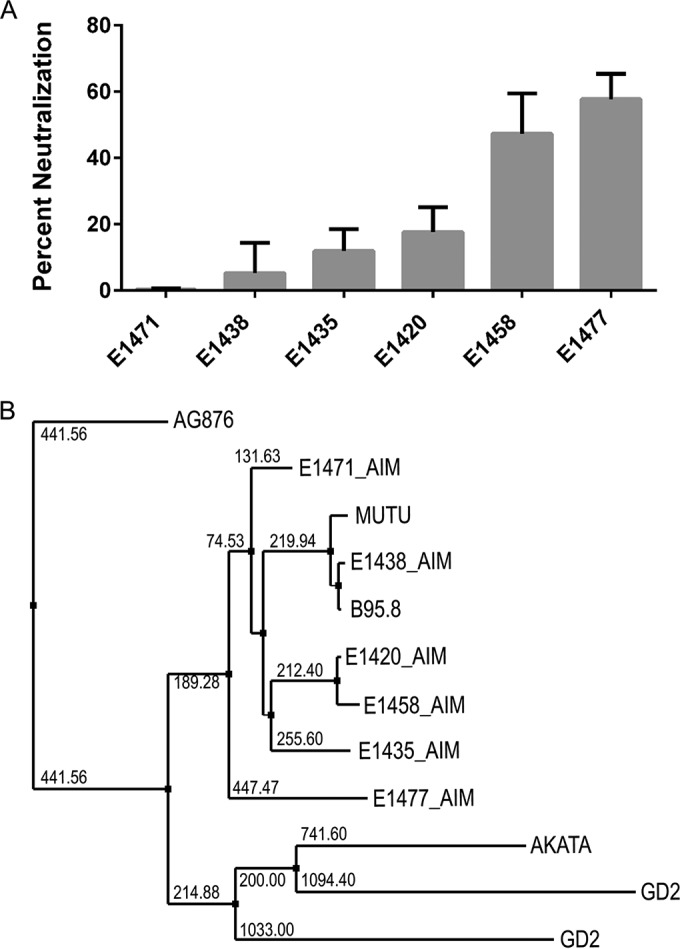


Neutralizing antibody responses and EBV gp350 genetic variability. (A) IgG from the plasma of 6 individuals chosen for EBV gp350 deep sequencing demonstrates variable activity for neutralization of EBV infection of Raji cells. The 6 numbers listed here and following denote individual patient study codes. (B) A nearest-neighbor tree for consensus EBV gp350 DNA sequences, along with gp350 sequences from GenBank. (C and D) Alignments of the AIM patient consensus gp350 nucleotide sequences (C) and protein sequences (D) to the strain B95.8 and Akata reference sequences; the alignments display both highly conserved and variable regions of the gene. Red boxes highlight the nucleotides (C) and amino acids (D) that encode and comprise the gp350-CR2-binding site.
Little variation in primary gp350 amino acid sequences from AIM to CONV.
To investigate whether the EBV gp350-specific neutralizing antibodies detected in our in vitro assays may have exerted selective pressures on the EBV gp350 glycoprotein over time following AIM, we also sequenced the EBV gp350 gene from saliva samples during CONV. Alignment of major variants revealed very few changes in the protein sequences of the EBV gp350 gene from five of six patients over the 6-month study period (Fig. 5). Three of four patients with low neutralizing activity levels (patients E1420, E1435, and E1438) showed no change in gp350 protein sequences over this time period; the gp350 protein sequence of patient E1471 displayed only a single amino acid change. Similarly, only a single amino acid in the gp350 protein sequence of a patient (patient E1458) whose plasma showed robust neutralizing activity was observed to change from AIM to CONV. The greatest amino acid sequence variability (14 aa) was detected during CONV in EBV gp350 sequences from a patient (patient E1477) with correspondingly high neutralizing antibody activity during CONV. Due to the large number of amino acid changes detected over time in patient E1477, gp350 DNA sequences were reamplified from original samples collected during AIM and CONV, and major variants were verified using Sanger sequencing. Despite the increased level of variation detected in patient E1477, all gp350 sequences present in the patient samples demonstrated high degrees of conservation in the CR2-binding region, and the few observed amino acid changes occurred outside the CR2-binding region (25).
FIG 5.

EBV gp350 sequences from all but one oral wash sample display little variation over time. The major variant amino acid sequence of each patient sample during AIM and CONV was determined on the basis of the results of next-generation sequencing. Paired patient sequences were aligned with each other, with the sequences from other patients in the cohort, and with the sequences of reference genomes (strains B95.8 and Akata). The consensus sequence for all samples is shown at the bottom of the alignment, and only nonconsensus amino acids are shown for each patient sample. Multiple-sequence alignment performed using the MUSCLE program (www.ebi.ac.uk/Tools/msa/muscle/), and the alignment was arranged using Jalview software (53).
Robust neutralizing antibody responses are associated with more variable patient viral pools.
Comparison of consensus gp350 protein sequences determined for the viral pool during AIM and CONV failed to indicate any significant effect of neutralizing antibody presence on gp350 gene evolution. Although limited changes in the gp350 protein sequence were observed (e.g., 1 amino acid change in patient E1458 and 14 amino acid changes in patient E1477), no variation in the CR2-binding site was detected for any of the patients (Fig. 4D, red boxes). However, a 9-amino-acid deletion was detected in gp350 from patient E1477 during AIM (relative to the strain B95.8 reference sequence), and this deletion was absent from the virus sequenced from the patient E1477 sample obtained during CONV (Fig. 5). Having already verified the fidelity of both samples, the absence of this deletion in the sample during CONV suggested either that recombination had occurred or that a minor variant became dominant through some imposed selection. This prompted us to consider and investigate the composition of the minor variant pool, i.e., nucleotides present in the mapped reads at percentages of less than 49%, for each patient at the time points indicated below. The nucleotide usage at each individual position of the gp350 gene was determined, and only nucleotides present at a frequency of greater than 2% were considered valid single nucleotide variants (SNV). The 2% change in frequency was selected as a conservative cutoff between sequencing error (determined by resequencing B95.8) and the establishment of new EBV variants that arose through ongoing replication in the oropharynx.
We observed measurable diversity in minor variant gp350 pools during both AIM and CONV in all patient samples. Patients E1438 and E1471, who had the lowest levels of EBV-neutralizing activity, showed the fewest number of SNV during both AIM and CONV. In contrast, patients E1420, E1435, and E1458, who generated stronger neutralizing antibody responses, demonstrated increased levels of overall viral diversity both in the total number of variants and as the relative proportion of variants in the viral pool. The notable exception to this trend was patient E1477, who displayed less overall diversity in gp350 viral pools during both AIM and CONV.
Additional analysis of the nucleotide usage of EBV from patient E1477 indicated that while the overall diversity of circulating EBV remained low, there were substantial fluctuations in nucleotide usage between the two time points. Several positions (n = 14) within the gp350 gene sequenced from patient E1477 displayed a complete change in nucleotide identity from AIM to CONV, suggesting the rapid emergence of a second variant during the study period. Fluctuations in the viral pools of the remaining patients were also investigated; the nucleotide frequency during AIM was subtracted from the frequency of that same nucleotide during CONV. Only two alternative nucleotides were detected at any position for all samples sequenced in this study. SNV frequency changes from AIM to CONV were highly correlated with the IgG neutralizing activity measured in the same patient (Spearman r value = 0.94, P = 0.016) (Fig. 6A). Interestingly, SNV frequency changes also correlated strongly with the peripheral blood EBV viral load during AIM (Spearman r value = 1.0, P = 0.003) (Fig. 6B).
FIG 6.

The convalescent-phase neutralizing antibody response correlates directly with changes in EBV gp350 gene diversity. The total change in the diversity of each patient's viral gp350 population, as measured by changes in single nucleotide variant (SNV) frequency (Freq), was quantified for each patient. SNVs, considered here to identify individual EBV strains, were identified from gp350 gene sequences in which alternative nucleotides were detected in at least 2% of mapped reads. An increase in the number of variants whose frequency changed >2% from AIM to CONV correlated positively with the strength of the neutralizing antibody response during CONV (Spearman r value = 0.94, P = 0.016) (A) and the peripheral blood viral load during AIM (Spearman r value = 1.0, P = 0.003) (B). The SNV frequency at each position of the gp350 gene was determined for the indicated patient samples during AIM and CONV, and the change was quantified by subtracting the nucleotide frequencies during AIM from the frequency during CONV (×100) to obtain percent total reads; all changes are reported as absolute values regardless of the direction of change.
DISCUSSION
This study is among the first to leverage longitudinal samples to quantify and characterize EBV-specific antibody responses and viral sequence evolution concurrently during primary infection. gp350-binding antibodies were detected in the majority of individuals with AIM at symptom onset (Fig. 1A) and increased in titer from AIM through CONV, with the levels during CONV being similar to those in healthy, age-matched EBV-seropositive donors; these findings are compatible with those of recently reported studies (20, 29).
Our data suggest that IgG1 antibodies mediate the most binding and functional antibody activity throughout infection (Fig. 1 to 3). These data are consistent with observations for patients infected with other viruses, such as HIV-1, in which early increases in anti-HIV IgG1 (e.g., to p55, gp120, or Nef) are sustained following acute HIV-1 infection, while the levels of anti-IgG3 decrease rapidly (30). The association of patient anti-gp350 IgG1 binding titers with phagocytosis scores and neutralizing activity suggests that IgG1 is the major mediator of ADCP and neutralization.
The lack of detectable EBV-specific neutralizing activity in Raji cell assays from samples collected during AIM is consistent with previously reported data from studies using either a similar assay (20) or a B-cell transformation assay (29). However, using a more sensitive PCR-based assay with primary B cells, we detected neutralizing antibody activity in the plasma of the majority (83%) of AIM patients. In assays with both primary B cells and Raji cells, the addition of the antibody 72A1 reduced EBV genome copy numbers by 90% (Fig. 3A and B), and neutralizing activity was not detected in seronegative controls. EBV gp350 blocking of patient samples confirmed the gp350 specificity of the neutralizing activity in the Raji cell infection assay (Fig. 3E). A likely reason for the discrepancy in the detection of neutralizing activity in primary B cells versus Raji cells is the difference in the readout used to measure infection; our qPCR-based viral load assay is highly sensitive and directly measures gp350-dependent attachment and, presumably, the entry of EBV virions into the host cell by directly measuring viral genome copy numbers. This readout is independent of transformation potential or viral gene expression, such as the amount of the viral genome-encoded green fluorescent protein (GFP) quantified by the Raji cell assay. It may thus provide a more sensitive and accurate measurement of viral envelope access to and interaction with the host cell receptor than B-cell transformation or a GFP readout. A recent study using in situ hybridization to quantify EBV genomes in AIM patient B cells demonstrated an average of 12 to 13 viral genomes per cell (31). Further, Delecluse and colleagues recently reported that only 10 to 15% of EBV genomes successfully transformed B cells, suggesting that B-cell transformation may underrepresent virus binding to and entry into host cells (32).
The recently determined crystal structure of the partial gp350 envelope (aa 4 to 443) has demonstrated it to be a heavily glycosylated, L-shaped structure, with the glycans shielding almost the entirety of the protein (33). The notable exception is the CR2-binding site, located in a glycan-free, negatively charged cleft formed from the two amino-terminal β-barrels and linker regions of the protein. Peptides that inhibit the neutralizing effects of gp350-binding antibody 72A1 on EBV infection map to the gp350 cleft; negatively charged amino acids required for the interaction between gp350 and CR2 also localize to this area (24, 25). Amino acid changes in the CR2-binding cleft were not observed in any of the gp350 sequences isolated from our patient cohort. We also failed to note any detectable changes in this binding site at the minor variant level. The data suggest that even in the presence of strongly neutralizing antibodies, the functional requirement for an interaction between gp350 and CR2 likely limits sequence evolution in this region.
Available data suggest that gp350 is the major viral envelope glycoprotein required for infection; gp350 CR2-specific antibodies are sufficient to block EBV infection (9, 19). We observed marked interpatient variability in neutralization titers and found a positive correlation between patient peripheral blood viral load during AIM and the patient neutralization titer in primary and cultured B cells during CONV (Fig. 3C and D). In several longitudinal studies of HIV-infected individuals that went on to generate broadly neutralizing antibodies (bnAbs) to the viral Env, a relationship between a high viral load during the acute phase of infection and the breadth of the antibody response was reported, although the authors were clear to state that a high viral load alone was likely not sufficient to cause bnAb generation (21, 22, 34). They postulated that the viral load and a higher frequency of T follicular helper cells led to the formation of germinal centers, which resulted in high levels of somatic hypermutation. Our data suggest that EBV infection may offer an additional model with which to study factors important in the generation of high-affinity and broadly neutralizing antibodies against persistent viral infections.
We report limited intrapatient differences in viral sequences at the time points studied and interpret our data to be consistent with a model in which a single founder virus rather than multiple, diverse viruses establishes infection. In contrast, it appears that one individual (patient E1477) may have been infected with at least two viruses. This is suggested by the presence in patient E1477 of a major strain with a 9-amino-acid deletion (relative to the B95.8 sequence) in gp350 during AIM and the almost complete absence of the deletion in the sequence from patient E1477 during CONV, suggestive of the presence of a second strain. Further evidence for this hypothesis can be seen in the rapid and complete change in nucleotide identity at no less than 16 different positions of gp350 and a corresponding change of 14 amino acids. Previous studies have reported cases in which multiple EBV genomes were present and detectable in both healthy and immunocompromised patients, although whether they were detecting subtle changes in major variant frequencies is unknown (35, 36). A more detailed longitudinal study would be required to distinguish whether the patient was infected by multiple variants or experienced EBV superinfection.
Several studies have highlighted the importance of diversity in the HIV-1 env gene in chronically infected patients that develop broadly neutralizing anti-HIV-1 antibodies (34, 37, 38). If patient E1477 was indeed infected with multiple EBV founder viruses or serially infected, this, combined with the high viral load observed during AIM, may help to explain the generation of strongly neutralizing antibodies in this individual.
Our data are consistent with a model of antibody generation in which an unlinked B-cell and T-cell response elicits higher-affinity and stronger neutralizing antibodies. In this model, CD4 T cells recognize any viral epitope expressed via the major histocompatibility complex on a B cell with a B-cell receptor specific for a viral surface protein. High levels of circulating EBV in the peripheral blood could serve to prime the generation of neutralizing antibodies by providing higher levels of antigen for gp350-specific B cells as well as ample structural proteins for CD4 T-cell recognition. Changes in viral populations over time may enable the constant recruitment of CD4 T cells recognizing the different nonlinked epitopes. This unlinked recognition can result in germinal center formation, hypersomatic mutation, and the generation of strongly neutralizing antibodies (39). Evidence for this model has been shown previously in studies describing the correlation of strongly neutralizing antibodies against the HIV-1 envelope with CD4 T-cell recognition of Gag epitopes (40) and the enhancement of antihemagglutinin-specific antibody responses by the adoptive transfer of CD4 T cells that recognized influenza virus matrix and nucleocapsid proteins (39); also, the expression of multiple, different vaccinia virus envelope proteins generated stronger antibody responses to individual envelopes (41). The EBV genome encodes fewer proteins than the vaccinia virus genome but many more than the genome of HIV or influenza virus, providing ample structural proteins to serve as CD4 T-cell epitopes. In agreement with our findings, previous analyses of available full-length EBV genomes, although mostly from cell lines and tumors, have shown the gp350 gene to be one of the least variable genes, suggesting that our measurements of gp350 gene variation may significantly underrepresent the total viral genome variation (18).
In summary, EBV gp350-binding antibodies were readily detected in patients with AIM and increased in titer through convalescence. Despite the ready detection of gp350-binding antibodies during AIM, ADCP and neutralizing antibodies were not readily detected using commonly used assays. In contrast, IgG gp350-specific neutralizing activity was reproducibly detected during AIM using a highly sensitive PCR readout in an assay using primary B cells as target cells. ADCP and neutralizing antibody activity were detected in all patient serum samples during CONV. A higher viral load and the increase in EBV gp350 gene diversity from AIM through CONV were positively correlated with the development of neutralizing antibody responses following primary EBV infection. Our findings improve our understanding of the development of functional gp350-specific antibodies and will be helpful in informing the design of future EBV vaccines.
MATERIALS AND METHODS
Study cohort.
Plasma and saliva samples were obtained during AIM and 6 months later (during convalescence [CONV]) from 24 young adults presenting with symptoms compatible with AIM (University of Massachusetts Amherst Health Services). Twelve age-matched individuals chronically infected (CHRON) with EBV and 12 age-matched individuals not infected with EBV (serologically negative) were studied as controls (42). The infection status of all participants was confirmed by serology.
Isolation and quantification of total plasma IgG.
IgG fractions were purified from patient plasma samples using Melon gel IgG purification kits (Thermo Scientific, Waltham, MA), quantified using a colorimetric assay (Bio-Rad, Carlsbad, CA), and diluted to 1 mg/ml.
Quantification of IgG and IgG subclass antibodies to EBV gp350.
The titers of EBV gp350-specific total and subclass IgG antibodies to recombinant gp350 (aa 4 to 450 of the B95.8 strain of EBV; Immune Technologies, San Mateo, CA) were measured using a Luminex-based assay and a Bio-Rad Bio-Plex 200 system (Bio-Rad, Carlsbad, CA) (43). All secondary antibodies, anti-human IgG-phycoerythrin (PE), anti-human IgG1-PE, anti-human IgG2-PE, anti-human IgG2-PE, and anti-human IgG4-PE, were purchased from Southern Biotech (Birmingham, AL).
Cell separation and cell line culture.
Cells of the THP-1 and Raji cell lines were maintained according to the supplier's protocols (American Type Culture Collection [ATCC], Manassas, VA). The EBV Akata-positive AGS gastric carcinoma cell line was cultured in Kaighn's modification of F-12 medium with 10% fetal bovine serum (FBS) and 500 U/ml G418 (44). Human primary cord blood lymphocytes (Umbilical Cord Blood Core, University of Massachusetts Medical School) were isolated by Ficoll separation.
Phagocytosis assay.
Phagocytosis assays were performed as previously described using recombinant gp350 (Immune Technologies); the phagocytosis score was calculated as the percent bead positive × bead-positive MFI and is given in percent (45).
Measurement of peripheral blood EBV load.
B cells were isolated from whole blood using a RosetteSep human B-cell kit (Stemcell Technologies, Vancouver, BC, Canada). DNA was extracted using a Qiagen DNeasy blood and tissue kit (Valencia, CA), and blood viral loads were measured using a Roche LightCycler EBV quantification kit (Roche, Indianapolis, IN) (17, 46).
Preparation of EBV stocks and virus titration.
eGFP-tagged EBV (EBV-eGFP) Akata isolates were recovered from EBV-eGFP-infected AGS cell supernatants as previously described (47). Virus was concentrated from filtered supernatant by high-speed centrifugation (17,000 × g, 90 min, 4°C) and stored frozen under liquid nitrogen prior to use.
Viral titration and neutralization assays in Raji cell lines and primary cord blood B cells.
Viruses, prepared as detailed above, were titrated on both Raji and primary cord blood B cells, and the amount of virus that was neutralized 90% by 10 μg of 72A1 antibody (ATCC) was used in each assay.
A flow cytometry-based assay was used to measure the neutralization of infection of Raji cells with the eGFP-tagged Akata isolate (Akata-eGFP); this method has been adopted as a suitable substitute for the traditional primary B-cell transformation assay (19, 20, 48). Briefly, plasma IgG (10 μl of a 1-mg/ml stock) was mixed with a known titer of virus and incubated for 1 h at 37°C. The Akata-eGFP–IgG mixture was added to 4.0 × 105 Raji cells resuspended in a minimal volume of RPMI supplemented with 10% heat-inactivated FBS; this mixture was incubated for 1 h at 37°C. Unbound virus was removed by washing with phosphate-buffered saline (PBS) (2 times), and the cells were plated and cultured. Three days later, cells were collected, washed with PBS (2 times), and resuspended in fluorescence-activated cell sorting buffer (0.5% bovine serum albumin and 2.0% paraformaldehyde in 1× PBS); GFP-positive cells, as determined in the fluorescein isothiocyanate channel of a FACSCalibur flow cytometer (BD Biosciences), were considered positive for EBV infection.
To measure the ability of EBV gp350-specific antibodies to neutralize EBV infection of primary B cells, EBV Akata-eGFP isolates were incubated with patient IgG for 1 h before they were mixed with primary cord blood B lymphocytes (1.0 ×106 total peripheral blood mononuclear cells [PBMCs] from cord blood) at 37°C for 2 h. After incubation of the IgG-virus mixture with primary B cells, unbound virus was removed by washing with PBS (2 times), and cells were resuspended in a minimal volume (200 μl) of RPMI supplemented with 10% heat-inactivated FBS and cultured for 3 days. Cells were removed from the culture and washed again with PBS (2 times) prior to DNA extraction using a DNeasy blood and tissue kit (Qiagen). Cell-associated DNA was measured using a multiplexed qPCR (CFX96 real-time system; Bio-Rad), as described below.
qPCR assay to measure EBV DNA in a primary cord blood B-cell neutralization assay.
A qPCR assay was used to simultaneously measure CCR5 and BALF5 (CFX96 real-time system; Bio-Rad). Cellular DNA was extracted from infected PBMCs from cord blood using a DNeasy blood and tissue kit (Qiagen). The EBV copy number was determined using primers and probes directed against the EBV DNA polymerase gene (BALF5): forward primer 5′-CGGAAGCCCTCTGGACTTC-3′, reverse primer 5′-CCCTGTTTATCCGATGAATG-3′, and probe 5′-FAM-TGTACACGCACGAGAAATGCGCCT-BHQ-1-3′, where FAM is 6-carboxyfluorescein and BHQ1 is black hole quencher 1. Human genome copy numbers were quantified using primers and probes directed against human C-C chemokine receptor type 5 (CCR5): forward primer 5′-GCTGTGTTTGCGTCTCTCCCAGGA-3′, reverse primer 5′-CTCACAGCCCTGTGCCTCTTCTTC-3′, and probe 5′-Cy5-AGCAGCGGCAGGACCAGCCCCAAG-BHQ-1-3′. Namalwa cells containing 2 EBV genomes per cell were used to generate standard curves for both EBV and CCR5 copy numbers in all assays (49). EBV copy numbers were reported as the BALF5 copy number per 1.0 × 106 PBMCs. Standards contained a range of from 100,000 copies to 1 copy of EBV per reaction mixture, with 10 copies being consistently detected and therefore being the assay minimum.
Percent neutralization of Raji and primary cells was determined by subtracting the number of EBV-infected cells in the presence of patient IgG from the total number of EBV-infected cells in an untreated sample divided by the untreated sample score (48).
Amplification of viral genomic DNA and library preparation.
EBV DNA was extracted from oral wash samples using a High Pure viral nucleic acid kit (Roche, Indianapolis, IN), and the gp350 gene was amplified using overlapping PCR. Custom-designed primers specific for gp350 generated four discrete products with three regions of overlapping DNA. These primers were also used to amplify the identical region of the EBV B95.8 bacterial artificial chromosome (BAC; provided by Fred Wang) (50).
PCR fragments were mixed in equimolar ratios, sheared via ultrasonication (Bioruptor; Diagenode), and end repaired using an EpiCenter end-repair kit (EpiCenter, Madison, WI). Bar-coded sequencing adaptors (New England BioLabs, Ipswich, MA) were ligated, prior to size selection and sequencing (Illumina HiSeq; UMASS Sequencing Core). Patient samples from both time points (during AIM and CONV) were sequenced in the same run, along with a gp350 control library generated using the B95.8 BAC as a template.
Sequence analysis.
Adaptors were trimmed using the Cutadapt (version 1.3) program, and reads were aligned to the B95.8 reference genome (GenBank accession no. NC_007605.1) using the BWA (version 0.7.5a) software package. PCR duplicates were removed using Picard's mark duplicates (http://picard.sourceforge.net), and the additional preprocessing steps recommended for the genome analysis tool kit (GATK) were performed. Variants were then called with GATK, setting the ploidy to 10 (51).
The frequency distribution of bases mapped to each position was calculated on the basis of the pileups of bases called with a minimum phred score of 17 and used to identify minor variants. Consensus sequences for gp350 were constructed for each sample by selecting from the maximum-frequency base at each nucleotide position. Multiple-sequence alignments and phylogenetic trees were generated using the MUSCLE (version 3.8.31) program, and publication-quality figures were made using Jalview (version 2.8.1) software (52, 53).
Statistical analysis.
Statistical analyses were performed using GraphPad Prism (version 6.05) software for Windows (GraphPad Software, San Diego, CA, USA). Correlations for nonnormally distributed data were calculated using Spearman's correlation. Comparison of data for AIM patients during AIM and CONV were calculated using the Wilcoxon matched-pairs signed-rank test; comparisons between groups were calculated using the Mann-Whitney test. All statistical tests were two-sided, and a P value of <0.05 was considered statistically significant.
Ethics statement.
All study participants provided written informed consent, and the University of Massachusetts Medical School Institutional Review Board approved these studies.
Accession number(s).
Consensus sequence data from this study are available from the NCBI database and are listed under GenBank accession numbers KX271782 through KX271793.
ACKNOWLEDGMENTS
We thank the study participants for their commitment to the research. We thank Alan Calhoun, Jessica Conrad, George Corey, and Gail Gnatek at the University of Massachusetts Amherst Student Health Service for providing clinical care and research samples.
REFERENCES
- 1.Luzuriaga K, Sullivan JL. 2010. Infectious mononucleosis. N Engl J Med 362:1993–2000. doi: 10.1056/NEJMcp1001116. [DOI] [PubMed] [Google Scholar]
- 2.Nielsen TR, Rostgaard K, Nielsen NM, Koch-Henriksen N, Haahr S, Sorensen PS, Hjalgrim H. 2007. Multiple sclerosis after infectious mononucleosis. Arch Neurol 64:72–75. doi: 10.1001/archneur.64.1.72. [DOI] [PubMed] [Google Scholar]
- 3.Hjalgrim H, Engels EA. 2008. Infectious aetiology of Hodgkin and non-Hodgkin lymphomas: a review of the epidemiological evidence. J Intern Med 264:537–548. doi: 10.1111/j.1365-2796.2008.02031.x. [DOI] [PubMed] [Google Scholar]
- 4.Thacker EL, Mirzaei F, Ascherio A. 2006. Infectious mononucleosis and risk for multiple sclerosis: a meta-analysis. Ann Neurol 59:499–503. doi: 10.1002/ana.20820. [DOI] [PubMed] [Google Scholar]
- 5.Cohen JI, Fauci AS, Varmus H, Nabel GJ. 2011. Epstein-Barr virus: an important vaccine target for cancer prevention. Sci Transl Med 3:107fs7. doi: 10.1126/scitranslmed.3002878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fingeroth JD, Weis JJ, Tedder TF, Strominger JL, Biro PA, Fearon DT. 1984. Epstein-Barr virus receptor of human B lymphocytes is the C3d receptor CR2. Proc Natl Acad Sci U S A 81:4510–4514. doi: 10.1073/pnas.81.14.4510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ogembo JG, Kannan L, Ghiran I, Nicholson-Weller A, Finberg RW, Tsokos GC, Fingeroth JD. 2013. Human complement receptor type 1/CD35 is an Epstein-Barr virus receptor. Cell Rep 3:371–385. doi: 10.1016/j.celrep.2013.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hoffman GJ, Lazarowitz SG, Hayward SD. 1980. Monoclonal antibody against a 250,000-dalton glycoprotein of Epstein-Barr virus identifies a membrane antigen and a neutralizing antigen. Proc Natl Acad Sci U S A 77:2979–2983. doi: 10.1073/pnas.77.5.2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thorley-Lawson DA, Geilinger K. 1980. Monoclonal antibodies against the major glycoprotein (gp350/220) of Epstein-Barr virus neutralize infectivity. Proc Natl Acad Sci U S A 77:5307–5311. doi: 10.1073/pnas.77.9.5307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thorley-Lawson DA, Poodry CA. 1982. Identification and isolation of the main component (gp350-gp220) of Epstein-Barr virus responsible for generating neutralizing antibodies in vivo. J Virol 43:730–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moutschen M, Leonard P, Sokal EM, Smets F, Haumont M, Mazzu P, Bollen A, Denamur F, Peeters P, Dubin G, Denis M. 2007. Phase I/II studies to evaluate safety and immunogenicity of a recombinant gp350 Epstein-Barr virus vaccine in healthy adults. Vaccine 25:4697–4705. doi: 10.1016/j.vaccine.2007.04.008. [DOI] [PubMed] [Google Scholar]
- 12.Sokal EM, Hoppenbrouwers K, Vandermeulen C, Moutschen M, Leonard P, Moreels A, Haumont M, Bollen A, Smets F, Denis M. 2007. Recombinant gp350 vaccine for infectious mononucleosis: a phase 2, randomized, double-blind, placebo-controlled trial to evaluate the safety, immunogenicity, and efficacy of an Epstein-Barr virus vaccine in healthy young adults. J Infect Dis 196:1749–1753. doi: 10.1086/523813. [DOI] [PubMed] [Google Scholar]
- 13.Rees L, Tizard EJ, Morgan AJ, Cubitt WD, Finerty S, Oyewole-Eletu TA, Owen K, Royed C, Stevens SJ, Shroff RC, Tanday MK, Wilson AD, Middeldorp JM, Amlot PL, Steven NM. 2009. A phase I trial of Epstein-Barr virus gp350 vaccine for children with chronic kidney disease awaiting transplantation. Transplantation 88:1025–1029. doi: 10.1097/TP.0b013e3181b9d918. [DOI] [PubMed] [Google Scholar]
- 14.Cui X, Cao Z, Sen G, Chattopadhyay G, Fuller DH, Fuller JT, Snapper DM, Snow AL, Mond JJ, Snapper CM. 2013. A novel tetrameric gp350 1-470 as a potential Epstein-Barr virus vaccine. Vaccine 31:3039–3045. doi: 10.1016/j.vaccine.2013.04.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Coghill AE, Bu W, Nguyen H, Hsu WL, Yu KJ, Lou PJ, Wang CP, Chen CJ, Hildesheim A, Cohen JI. 2016. High levels of antibody that neutralize B-cell infection of Epstein-Barr virus and that bind EBV gp350 are associated with a lower risk of nasopharyngeal carcinoma. Clin Cancer Res 22:3451–3457. doi: 10.1158/1078-0432.CCR-15-2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Renzette N, Bhattacharjee B, Jensen JD, Gibson L, Kowalik TF. 2011. Extensive genome-wide variability of human cytomegalovirus in congenitally infected infants. PLoS Pathog 7:e1001344. doi: 10.1371/journal.ppat.1001344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Renzette N, Somasundaran M, Brewster F, Coderre J, Weiss ER, McManus M, Greenough T, Tabak B, Garber M, Kowalik TF, Luzuriaga K. 2014. Epstein-Barr virus latent membrane protein 1 genetic variability in peripheral blood B cells and oropharyngeal fluids. J Virol 88:3744–3755. doi: 10.1128/JVI.03378-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Palser AL, Grayson NE, White RE, Corton C, Correia S, Ba Abdullah MM, Watson SJ, Cotten M, Arrand JR, Murray PG, Allday MJ, Rickinson AB, Young LS, Farrell PJ, Kellam P. 2015. Genome diversity of Epstein-Barr virus from multiple tumor types and normal infection. J Virol 89:5222–5237. doi: 10.1128/JVI.03614-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sashihara J, Burbelo PD, Savoldo B, Pierson TC, Cohen JI. 2009. Human antibody titers to Epstein-Barr virus (EBV) gp350 correlate with neutralization of infectivity better than antibody titers to EBV gp42 using a rapid flow cytometry-based EBV neutralization assay. Virology 391:249–256. doi: 10.1016/j.virol.2009.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bu W, Hayes GM, Liu H, Gemmell L, Schmeling DO, Radecki P, Aguilar F, Burbelo PD, Woo J, Balfour HH Jr, Cohen JI. 2016. Kinetics of Epstein-Barr virus (EBV) neutralizing and virus-specific antibodies after primary infection with EBV. Clin Vaccine Immunol 23:363–369. doi: 10.1128/CVI.00674-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Piantadosi A, Panteleeff D, Blish CA, Baeten JM, Jaoko W, McClelland RS, Overbaugh J. 2009. Breadth of neutralizing antibody response to human immunodeficiency virus type 1 is affected by factors early in infection but does not influence disease progression. J Virol 83:10269–10274. doi: 10.1128/JVI.01149-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gray ES, Madiga MC, Hermanus T, Moore PL, Wibmer CK, Tumba NL, Werner L, Mlisana K, Sibeko S, Williamson C, Abdool Karim SS, Morris L, CAPRISA002 Study Team . 2011. The neutralization breadth of HIV-1 develops incrementally over four years and is associated with CD4+ T cell decline and high viral load during acute infection. J Virol 85:4828–4840. doi: 10.1128/JVI.00198-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tanner J, Whang Y, Sample J, Sears A, Kieff E. 1988. Soluble gp350/220 and deletion mutant glycoproteins block Epstein-Barr virus adsorption to lymphocytes. J Virol 62:4452–4464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Urquiza M, Lopez R, Patino H, Rosas JE, Patarroyo ME. 2005. Identification of three gp350/220 regions involved in Epstein-Barr virus invasion of host cells. J Biol Chem 280:35598–35605. doi: 10.1074/jbc.M504544200. [DOI] [PubMed] [Google Scholar]
- 25.Young KA, Herbert AP, Barlow PN, Holers VM, Hannan JP. 2008. Molecular basis of the interaction between complement receptor type 2 (CR2/CD21) and Epstein-Barr virus glycoprotein gp350. J Virol 82:11217–11227. doi: 10.1128/JVI.01673-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nemerow GR, Houghten RA, Moore MD, Cooper NR. 1989. Identification of an epitope in the major envelope protein of Epstein-Barr virus that mediates viral binding to the B lymphocyte EBV receptor (CR2). Cell 56:369–377. doi: 10.1016/0092-8674(89)90240-7. [DOI] [PubMed] [Google Scholar]
- 27.Zhang PF, Klutch M, Armstrong G, Qualtiere L, Pearson G, Marcus-Sekura CJ. 1991. Mapping of the epitopes of Epstein-Barr virus gp350 using monoclonal antibodies and recombinant proteins expressed in Escherichia coli defines three antigenic determinants. J Gen Virol 72(Pt 11):2747–2755. [DOI] [PubMed] [Google Scholar]
- 28.Tanner JE, Coincon M, Leblond V, Hu J, Fang JM, Sygusch J, Alfieri C. 2015. Peptides designed to spatially depict the Epstein-Barr virus major virion glycoprotein gp350 neutralization epitope elicit antibodies that block virus-neutralizing antibody 72A1 interaction with the native gp350 molecule. J Virol 89:4932–4941. doi: 10.1128/JVI.03269-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Panikkar A, Smith C, Hislop A, Tellam N, Dasari V, Hogquist KA, Wykes M, Moss DJ, Rickinson A, Balfour HH Jr, Khanna R. 2015. Impaired Epstein-Barr virus-specific neutralizing antibody response during acute infectious mononucleosis is coincident with global B-cell dysfunction. J Virol 89:9137–9141. doi: 10.1128/JVI.01293-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yates NL, Lucas JT, Nolen TL, Vandergrift NA, Soderberg KA, Seaton KE, Denny TN, Haynes BF, Cohen MS, Tomaras GD. 2011. Multiple HIV-1-specific IgG3 responses decline during acute HIV-1: implications for detection of incident HIV infection. AIDS 25:2089–2097. doi: 10.1097/QAD.0b013e32834b348e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Calattini S, Sereti I, Scheinberg P, Kimura H, Childs RW, Cohen JI. 2010. Detection of EBV genomes in plasmablasts/plasma cells and non-B cells in the blood of most patients with EBV lymphoproliferative disorders by using Immuno-FISH. Blood 116:4546–4559. doi: 10.1182/blood-2010-05-285452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shannon-Lowe C, Baldwin G, Feederle R, Bell A, Rickinson A, Delecluse HJ. 2005. Epstein-Barr virus-induced B-cell transformation: quantitating events from virus binding to cell outgrowth. J Gen Virol 86:3009–3019. doi: 10.1099/vir.0.81153-0. [DOI] [PubMed] [Google Scholar]
- 33.Szakonyi G, Klein MG, Hannan JP, Young KA, Ma RZ, Asokan R, Holers VM, Chen XS. 2006. Structure of the Epstein-Barr virus major envelope glycoprotein. Nat Struct Mol Biol 13:996–1001. doi: 10.1038/nsmb1161. [DOI] [PubMed] [Google Scholar]
- 34.Moore PL, Williamson C, Morris L. 2015. Virological features associated with the development of broadly neutralizing antibodies to HIV-1. Trends Microbiol 23:204–211. doi: 10.1016/j.tim.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Srivastava G, Wong KY, Chiang AK, Lam KY, Tao Q. 2000. Coinfection of multiple strains of Epstein-Barr virus in immunocompetent normal individuals: reassessment of the viral carrier state. Blood 95:2443–2445. [PubMed] [Google Scholar]
- 36.Walling DM, Brown AL, Etienne W, Keitel WA, Ling PD. 2003. Multiple Epstein-Barr virus infections in healthy individuals. J Virol 77:6546–6550. doi: 10.1128/JVI.77.11.6546-6550.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carter CC, Wagner GA, Hightower GK, Caballero G, Phung P, Richman DD, Pond SL, Smith DM. 2015. HIV-1 neutralizing antibody response and viral genetic diversity characterized with next generation sequencing. Virology 474:34–40. doi: 10.1016/j.virol.2014.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cortez V, Wang B, Dingens A, Chen MM, Ronen K, Georgiev IS, McClelland RS, Overbaugh J. 2015. The broad neutralizing antibody responses after HIV-1 superinfection are not dominated by antibodies directed to epitopes common in single infection. PLoS Pathog 11:e1004973. doi: 10.1371/journal.ppat.1004973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Scherle PA, Gerhard W. 1988. Differential ability of B cells specific for external vs. internal influenza virus proteins to respond to help from influenza virus-specific T-cell clones in vivo. Proc Natl Acad Sci U S A 85:4446–4450. doi: 10.1073/pnas.85.12.4446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ranasinghe S, Soghoian DZ, Lindqvist M, Ghebremichael M, Donaghey F, Carrington M, Seaman MS, Kaufmann DE, Walker BD, Porichis F. 2016. HIV-1 antibody neutralization breadth is associated with enhanced HIV-specific CD4+ T cell responses. J Virol 90:2208–2220. doi: 10.1128/JVI.02278-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yin L, Calvo-Calle JM, Cruz J, Newman FK, Frey SE, Ennis FA, Stern LJ. 2013. CD4+ T cells provide intermolecular help to generate robust antibody responses in vaccinia virus-vaccinated humans. J Immunol 190:6023–6033. doi: 10.4049/jimmunol.1202523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hess RD. 2004. Routine Epstein-Barr virus diagnostics from the laboratory perspective: still challenging after 35 years. J Clin Microbiol 42:3381–3387. doi: 10.1128/JCM.42.8.3381-3387.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brown EP, Licht AF, Dugast AS, Choi I, Bailey-Kellogg C, Alter G, Ackerman ME. 2012. High-throughput, multiplexed IgG subclassing of antigen-specific antibodies from clinical samples. J Immunol Methods 386:117–123. doi: 10.1016/j.jim.2012.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Marquitz AR, Mathur A, Chugh PE, Dittmer DP, Raab-Traub N. 2014. Expression profile of microRNAs in Epstein-Barr virus-infected AGS gastric carcinoma cells. J Virol 88:1389–1393. doi: 10.1128/JVI.02662-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ackerman ME, Moldt B, Wyatt RT, Dugast AS, McAndrew E, Tsoukas S, Jost S, Berger CT, Sciaranghella G, Liu Q, Irvine DJ, Burton DR, Alter G. 2011. A robust, high-throughput assay to determine the phagocytic activity of clinical antibody samples. J Immunol Methods 366:8–19. doi: 10.1016/j.jim.2010.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Greenough TC, Straubhaar JR, Kamga L, Weiss ER, Brody RM, McManus MM, Lambrecht LK, Somasundaran M, Luzuriaga KF. 2015. A gene expression signature that correlates with CD8+ T cell expansion in acute EBV infection. J Immunol 195:4185–4197. doi: 10.4049/jimmunol.1401513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Speck P, Longnecker R. 1999. Epstein-Barr virus (EBV) infection visualized by EGFP expression demonstrates dependence on known mediators of EBV entry. Arch Virol 144:1123–1137. doi: 10.1007/s007050050574. [DOI] [PubMed] [Google Scholar]
- 48.Chesnokova LS, Hutt-Fletcher LM. 2011. Fusion of Epstein-Barr virus with epithelial cells can be triggered by alphavbeta5 in addition to alphavbeta6 and alphavbeta8, and integrin binding triggers a conformational change in glycoproteins gHgL. J Virol 85:13214–13223. doi: 10.1128/JVI.05580-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lawrence JB, Villnave CA, Singer RH. 1988. Sensitive, high-resolution chromatin and chromosome mapping in situ: presence and orientation of two closely integrated copies of EBV in a lymphoma line. Cell 52:51–61. doi: 10.1016/0092-8674(88)90530-2. [DOI] [PubMed] [Google Scholar]
- 50.Chen A, Divisconte M, Jiang X, Quink C, Wang F. 2005. Epstein-Barr virus with the latent infection nuclear antigen 3B completely deleted is still competent for B-cell growth transformation in vitro. J Virol 79:4506–4509. doi: 10.1128/JVI.79.7.4506-4509.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, Banks E, Garimella KV, Altshuler D, Gabriel S, DePristo MA. 2013. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics 43:11.10.1–11.10.33. doi: 10.1002/0471250953.bi1110s43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ. 2009. Jalview version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics 25:1189–1191. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]