

ABSTRACT
The highly conserved herpesvirus glycoprotein complex gB/gH-gL mediates membrane fusion during virion entry and cell-cell fusion. Varicella-zoster virus (VZV) characteristically forms multinucleated cells, or syncytia, during the infection of human tissues, but little is known about this process. The cytoplasmic domain of VZV gB (gBcyt) has been implicated in cell-cell fusion regulation because a gB[Y881F] substitution causes hyperfusion. gBcyt regulation is necessary for VZV pathogenesis, as the hyperfusogenic mutant gB[Y881F] is severely attenuated in human skin xenografts. In this study, gBcyt-regulated fusion was investigated by comparing melanoma cells infected with wild-type-like VZV or hyperfusogenic mutants. The gB[Y881F] mutant exhibited dramatically accelerated syncytium formation in melanoma cells caused by fusion of infected cells with many uninfected cells, increased cytoskeleton reorganization, and rapid displacement of nuclei to dense central structures compared to pOka using live-cell confocal microscopy. VZV and human transcriptomes were concurrently investigated using whole transcriptome sequencing (RNA-seq) to identify viral and cellular responses induced when gBcyt regulation was disrupted by the gB[Y881F] substitution. The expression of four vital VZV genes, ORF61 and the genes for glycoproteins gC, gE, and gI, was significantly reduced at 36 h postinfection for the hyperfusogenic mutants. Importantly, hierarchical clustering demonstrated an association of differential gene expression with dysregulated gBcyt-mediated fusion. A subset of Ras GTPase genes linked to membrane remodeling were upregulated in cells infected with the hyperfusogenic mutants. These data implicate gBcyt in the regulation of gB fusion function that, if unmodulated, triggers cellular processes leading to hyperfusion that attenuates VZV infection.
IMPORTANCE The highly infectious, human-restricted pathogen varicella-zoster virus (VZV) causes chickenpox and shingles. Postherpetic neuralgia (PHN) is a common complication of shingles that manifests as prolonged excruciating pain, which has proven difficult to treat. The formation of fused multinucleated cells in ganglia might be associated with this condition. An effective vaccine against VZV is available but not recommended for immunocompromised individuals, highlighting the need for new therapies. This study investigated the viral and cellular responses to hyperfusion, a condition where the usual constraints of cell membranes are overcome and cells form multinucleated cells. This process hinders VZV and is regulated by a viral glycoprotein, gB. A combination of live-cell imaging and next-generation genomics revealed an alteration in viral and cellular responses during hyperfusion that was caused by the loss of gB regulation. These studies reveal mechanisms central to VZV pathogenesis, potentially leading to improved therapies.
KEYWORDS: varicella-zoster virus, fusion, glycoprotein B, herpesvirus, RNA-seq, pathogenesis
INTRODUCTION
Varicella-zoster virus (VZV) is a medically important human alphaherpesvirus that causes varicella (chickenpox) and, upon reactivation from latently infected sensory ganglia, leads to zoster (shingles) (1). Varicella can be serious and is life-threatening in immunocompromised patients (2–5). Zoster in the elderly often triggers severe postherpetic neuralgia (PHN), and VZV can cause severe morbidity and mortality in individuals with impaired immune function (6–8). The varicella and zoster vaccines currently available are effective, but these live viral vaccines are not safe for those with immunodeficiencies (9–11). Acyclovir and related drugs are useful for acute infections but not for effective treatment of PHN. Importantly, current vaccines and drugs do not prevent VZV latency (9–15). A better understanding of the regulation of critical events in VZV pathogenesis, such as cell-cell fusion, has the potential to guide the design for new therapeutics and next-generation vaccines.
VZV infection is restricted to the human host, and its pathogenesis is mediated via tropism for T cells, skin, and neurons (16–18). Cell-cell fusion of epidermal cells generates polykaryocytes, also known as syncytia, typical of varicella and zoster skin lesions (19). Strikingly, replication in ganglia also results in neuron-satellite cell fusion (20). Thus, a hallmark of VZV infection is its capacity to overcome the usual constraint against fusion between fully differentiated host cells in vivo. VZV is a valuable model pathogen for investigating this phenomenon because VZV triggers syncytium formation in vitro, as well as in skin and sensory ganglia xenografts infected in vivo in the SCID mouse model of VZV pathogenesis (18, 21–25).
Enveloped viruses, including herpesviruses, enter host cells via fusion of the virion membrane with cellular membranes. Herpesviruses accomplish this by using a minimum of three essential, highly conserved, virally encoded glycoproteins, gB, gH, and gL (26). Currently, gB is proposed to be the fusion protagonist because X-ray crystal structures of this molecule from several herpesviruses show trimer formation reminiscent of that of viral fusion proteins (27–30). The role of the herpesvirus gH-gL heterodimer is uncertain, but it is required to trigger gB-induced fusion (26). Importantly, monoclonal antibodies derived from natural infection that target VZV gH neutralize the virus and inhibit fusion, potentially through preventing binding to gB (31–33). In contrast to fusion of the virion envelope with cell membranes during entry, little is known about virus-induced cell-cell fusion, which is a prominent feature of VZV pathogenesis (19, 20). The VZV gB and gH/gL are necessary and sufficient for this process (34, 35). In addition, VZV gB was demonstrated to be central to the regulation of VZV-induced cell-cell fusion and dependent on an immunoreceptor tyrosine-based inhibition motif (ITIM) ([SIVL]xYxx[IVL]) in the gB cytoplasmic domain (gBcyt) (36). Canonical ITIMs in the cytoplasmic domains of cellular receptors act to inhibit receptor-induced signaling, require tyrosine phosphorylation to be functional, and play an important role in maintaining homeostasis (37–39). A tyrosine-to-phenylalanine (Y881F) substitution in the VZV gBcyt ITIM, which prevents phosphorylation, resulted in hyperfusion when coexpressed with gH/gL in a cell-cell fusion assay (36). The hyperfusion phenotype was reproduced in the context of infection when the gB[Y881F] substitution was incorporated into the VZV genome, leading to extensive cell-cell fusion in cultured melanoma cells and human skin xenografts in vivo. Moreover, the gB[Y881F] substitution significantly reduced skin pathogenesis, indicating that dysregulation of canonical VZV-induced cell-cell fusion is detrimental to VZV propagation.
The purpose of this study was to define the process of VZV-induced cell fusion and its regulation by gBcyt. The propagation of infection in melanoma cells, which recapitulates syncytium formation observed in human tissues, was examined after inoculation with wild-type (WT) pOka or the hyperfusogenic gB[Y881F] mutant using live-cell confocal microscopy. This comparison revealed both the characteristics of the cell fusion process regulated by the VZV gBcyt and the consequences for cell-cell interactions when this regulatory control was disrupted. The influence of dysregulated cell fusion related to VZV particle assembly and trafficking was established using electron microscopy of cells infected with pOka or the gB[Y881F] mutant. To investigate the effect of dysregulated fusion on viral and human transcriptomes, VZV and host cell gene expression profiles were quantified by whole transcriptome sequencing (RNA-seq) as syncytium formation progressed in melanoma cells infected with pOka and the gB[Y881F] hyperfusogenic mutants. Accelerated cell fusion had significant effects on the kinetics and levels of viral gene expression, especially of ORF14 which encodes VZV gC, as well as ORF61, gE and gI. As expected, the host cell transcriptome was altered in virus-infected cells but substantial differences in cell gene expression were triggered by the hyperfusogenic gBcyt mutants compared with pOka. These data demonstrate that the dysregulation of VZV-induced cell-cell fusion has multifactorial effects and support the hypothesis that the fusogenic potential of gB, by which VZV overcomes the barriers to host cell fusion, must be regulated by its own cytoplasmic domain in order to support VZV propagation.
RESULTS
gBcyt regulates syncytium formation in VZV-infected cells.
To track VZV-induced syncytium formation and to determine how it was altered by hyperfusogenic mutants, melanoma cells expressing green fluorescent protein (GFP) (LifeAct-tGFP melanoma cells) were infected with pOka or the gB[Y881F] mutant. Each virus expressed a thymidine kinase (TK)-red fluorescent protein (RFP) chimeric protein (inoculum, 20 PFU/cm2) and was used for live-cell confocal microscopy of syncytia starting at 24 h postinfection (hpi) (T0), with images captured for an additional 20 hpi (T20). LifeAct-tGFP enabled visualization of actin filaments, and VZV TK expression, detected as RFP, was a marker for early-late infection. Small syncytia were observed in pOka-TK-RFP-infected cells at 24 hpi (T0), consisting of several nuclei visualized with Hoechst 33342 and surrounded by an RFP signal within LifeAct-tGFP-expressing cells (Fig. 1; see Movie S1 at https://purl.stanford.edu/wc992yg2549). The spread of VZV in the cell monolayer was successfully tracked, as cells that were RFP negative at T0 became RFP positive by T5, signifying infection, and were followed through to T20 (Fig. 1A, box 1). Infected cells and cells that were uninfected came into close proximity repeatedly in this region of the monolayer where syncytia had not yet formed by T0. While an immediate early (IE) stage of infection was not excluded, these cells remained RFP negative over the following 9 h, when early/late proteins are made in VZV-infected cells (40). Uninfected cells adjacent to infected cells frequently made direct contact via filopodium-like structures containing actin filaments, visualized by the LifeAct-tGFP (see seconds 0 to 5 of Movie S1 at https://purl.stanford.edu/wc992yg2549, box 1 region). Occasionally, RFP-negative cells fused with the infected RFP-positive cells. Between T15 and T20, cellular protrusions from uninfected cells became elongated and attached to infected cells, but these contacts often did not initiate cell-cell fusion, suggesting a constraint on the fusion process (see seconds 28 to 32 of Movie S1, box 1 region).
FIG 1.


Regulation of syncytium formation by the gBcyt in VZV-infected cells. Live-cell confocal microscopy images of LifeAct-tGFP melanoma cells infected with pOka-TK-RFP (A) or gB[Y881F]-TK-RFP (B) captured from Movies S1 and S2 at https://purl.stanford.edu/wc992yg2549 at five time points, beginning at 24 hpi (T0) and at 5 (T5), 10 (T10), 15 (T15) and 20 (T20) hpi, are shown. Infected cells are detected by the RFP signal (red), actin filaments express LifeAct-tGFP (green), and nuclei are stained with Hoechst 33342 (blue). White boxes in the panels in the first column (1 to 3) indicate three areas within the monolayer that are shown at higher magnification in the columns at the right, labeled 1 to 3, for each of the five time points, (T0, T5, T10, T15, and T20). Arrowheads identify representative cells that have not undergone fusion within the syncytia (open arrowheads) or nuclei not expressing RFP (closed arrowheads). These images highlight events in cell-cell fusion that are visualized in Movies S1 and S2, available at https://purl.stanford.edu/wc992yg2549.
Small syncytia that were detectable in pOka-TK-RFP-infected monolayers at T0 evolved with two different outcomes (Fig. 1A, boxes 2 and 3; see Movie S1 at https://purl.stanford.edu/wc992yg2549). The syncytium central to the field of view at T0 contained 11 nuclei, and infected cells were seen in direct contact with the periphery of the syncytium but had not fused (Fig. 1A, box 2). Overall, the syncytium exhibited little lateral movement during the live imaging period, but the periphery of the syncytium was dynamic, undergoing waves of expansion and contraction at the plasma membrane. Numerous cells were incorporated into this syncytium, while others remained adjacent without undergoing fusion (Fig. 1, box 2; see Movie S1 at https://purl.stanford.edu/wc992yg2549). For example, five cells that moved into the area of the syncytium did not fuse for several hours (see seconds 8 to 24 s of Movie S1, box 2 region). Some of these cells appeared to be inside the syncytium, but this was an artifact created from the static image derived from a single confocal plane. By T20, substantial surface blebbing was apparent, typical of cells undergoing programmed cell death, and most cells forming the syncytium had detached from the coverslip.
A second small syncytium in pOka-TK-RFP-infected cells had four nuclei at T0 and progressed to encompass 17 nuclei visible in the confocal plane at T20 (Fig. 1A, box 3). This developing syncytium made substantial and prolonged contact with the syncytium highlighted in Fig. 1A, box 2, for 13 h (seconds 8 to 29 of Movie S1 at https://purl.stanford.edu/wc992yg2549) and prior to its disassociation from the coverslip. Similar to the case for the syncytium in Fig. 1A, box 2, a number of infected and RFP-negative cells interacted considerably with the plasma membrane of the syncytium but failed to fuse. Another small syncytium fused with the original within the last 90 min (see seconds 30 to 32 of Movie S1, box 3 region) and contributed three additional nuclei (see Movie S1). Overall, the live imaging demonstrated that pOka-induced syncytium formation was stochastic and that fusion was not triggered simply by contact between infected and uninfected cells or between infected cells.
In contrast to the case with pOka-TK-RFP, the expansion of syncytia was dramatically accelerated due to frequent cell fusion events when LifeAct-tGFP cells were inoculated with the hyperfusogenic gB[Y881F]-TK-RFP mutant (Fig. 1B, boxes 1 to 3; see Movie S2 at https://purl.stanford.edu/wc992yg2549). Several characteristics differentiated the events that were observed during gB[Y881F]-TK-RFP infection compared to that with pOka-TK-RFP. The threshold for cell fusion was lower, with both uninfected and RFP-positive infected cells being incorporated rapidly into each syncytium. The LifeAct-tGFP-labeled actin filaments were more abundant and dynamic at the periphery of the gB[Y881F]-TK-RFP syncytium during its initial formation (Fig. 1B, box 2). These actin structures became increasingly prominent during the expansion of the syncytia from T0 to T20, forming dense extensions along the periphery of the syncytium (Fig. 1B, boxes 1 to 3; see Movie S2 at https://purl.stanford.edu/wc992yg2549). Both uninfected and RFP-positive infected cells were rapidly incorporated into each syncytium. Strikingly, once cell fusion had occurred, the nucleus of the fused cell became rapidly amalgamated with the cluster of nuclei that was central to the developing syncytium. Based on the pattern of their movement, nuclei entering the syncytium appeared to became tethered, presumably to the cytoskeletal network, and were rapidly sequestered to the spheroid of nuclei at the center (see Movie S2). When displacement of cell nuclei was quantified using TrackMate to measure the distance that nuclei traversed in the monolayer from T0 to T20, the mean was 323 μm ± 7.7 for gB[Y881F]-TK-RFP, which was 3.4-fold greater than nuclear displacement in pOka-TK-RFP-infected monolayers (Fig. 2A). In addition, both the mean and maximum velocities of nuclear movement were significantly higher with gB[Y881F]-TK-RFP infection (Fig. 2B and C). While the mean difference was 0.2 μm/s faster for gB[Y881F]-TK-RFP than for pOka-TK-RFP, the maximum velocity (8.1 μm/s) for gB[Y881F]-TK-RFP nuclei was more than twice that for pOka nuclei (4.0 μm/s).
FIG 2.

Displacement of cell nuclei during dysregulated syncytium formation in VZV-infected cells. (A to C) The displacement (A), mean velocity (B), and maximum velocity (C) of nuclei (n = 200) were calculated using the TrackMate plugin of FiJi. Error bars show standard error of the mean. (D and E) Quantification of plaque sizes (n = 50) and immunohistochemistry of representative plaques at 3 days postinfection (dpi) (size is given in the bottom right hand corners) in LifeAct-tGFP melanoma cells infected with pOka or gB[Y881F]. A two-tailed t test was used to calculate significant differences between pOka and gB[Y881F] (****, P < 0.001.
In association with the more rapid fusion and accumulation of cells into gB[Y881F]-TK-RFP syncytia, many nuclei were TK-RFP negative, indicating absent or very early infection at the time of fusion (see Movie S2 at https://purl.stanford.edu/wc992yg2549). Once incorporated in the syncytium, some nuclei became RFP positive as a result of infection prior to fusion or accumulation of TK-RFP from the syncytium. Notably, the dense actin filaments at the periphery of the gB[Y881]-TK-RFP syncytia extended well into the intercellular spaces and contacted many uninfected cells, compared to those for pOka-TK-RFP (see Movies S1 and S2 at https://purl.stanford.edu/wc992yg2549). Initially, the syncytia were adherent to the glass coverslip, but highly multinucleated giant cells formed by gB[Y881]-TK-RFP infection began to detach as infection progressed and the syncytium size increased. Consequently, gB[Y881F]-TK-RFP plaques were smaller and exhibited an absence of infected cells at their centers compared to pOka-infected cells, demonstrating the poor propagation characteristics of hyperfusogenic VZV (Fig. 2D to F).
gBcyt-regulated fusion occurs predominantly between VZV-infected cells at an early/late stage of infection.
To clarify whether syncytium formation progressed primarily by fusion between individual infected cells or by fusion between infected cells and cells that were uninfected or had not progressed to early/late infection and whether this process was altered by dysregulation of gBcyt control, melanoma cells were labeled with the plasma membrane marker FM4-64FX. Live-cell imaging was performed with the labeled melanoma cells infected with the enhanced GFP (eGFP)-expressing viruses pOka-TK-eGFP and gB[Y881F]-TK-eGFP (inocula, 20 PFU/cm2). The FM4-64FX fluorescent marker provided contrast between the plasma membrane and the eGFP signal of infected cells demonstrating an intact cell membrane prior to fusion (Fig. 3A and B; see Movies S3 and S4 at https://purl.stanford.edu/wc992yg2549).
FIG 3.


Regulation of fusion between uninfected and VZV-infected cells by gBcyt. Live-cell confocal microscopy images of FM4-64FX melanoma cells infected with pOka-TK-eGFP (A) or gB[Y881F]-TK-eGFP (B) captured from Movies S3 and S4 at https://purl.stanford.edu/wc992yg2549 at five time points, beginning at 24 hpi (T0) and at 4 (T4), 9 (T9), 13 (T13), and 17 (T17) hpi, are shown. Infected cells are detected by the eGFP signal (green), plasma membranes are labeled with FM4-64FX (red), and nuclei are stained with Hoechst 33342 (blue). White boxes in the panels in the first column (1 to 3) indicate three areas within the monolayer that are shown at higher magnification in the columns at the right, labeled 1 to 3, for each of the five time points, (T0, T4, T9, T13, and T17). Arrowheads identify representative cells that have not undergone fusion within the syncytia. These images highlight events in cell-cell fusion that are visualized in Movies S3 and S4, available at https://purl.stanford.edu/wc992yg2549.
Initially, pOka-TK-eGFP-infected monolayers showed both single eGFP-expressing cells and small syncytia containing 3 to 4 nuclei at 24 hpi (T0), similar to the case for pOka-TK-RFP infection of LifeAct-tGFP-labeled melanoma cells (Fig. 3A, boxes 1 to 3; see Movie S3 at https://purl.stanford.edu/wc992yg2549). FM4-64FX staining was detected both on the plasma membranes and internally, presumably from endocytosed or pinocytosed membranes. By T4, the frequency of infected cells had increased and the small syncytia began to enlarge by cell-cell fusion, sequestering more nuclei (Fig. 3A, boxes 1 and 2; see Movie S3). Motile cells that were TK-eGFP negative, indicating either absent or very early infection, did not readily fuse with these syncytia, as shown by tracking the individual eGFP-negative cells (see Movie S3). In addition, single infected cells were in contact with single uninfected cells frequently without triggering cell fusion over the 17-hour imaging period (Fig. 3A, box 3). As the syncytia evolved, it was apparent that most fusion events were between eGFP-expressing cells, which typified early/late time points of infection (Fig. 3A, boxes 1 and 2). At T9 and later times, the FM4-64FX membrane label localized to the centers of the rings of nuclei in larger syncytia, appearing as orange in combination with eGFP, consistent with the sequestration of endosomes at the center of syncytia during VZV infection (36). Blebbing, indicating programmed cell death highlighted by the FM4-64FX membrane label, was seen for numerous cells infected with pOka-TK-eGFP, which detached from the coverslip without infecting adjacent cells. This was similar to the case for pOka-TK-RFP-infected LifeAct-tGFP cells, suggesting that cell fusion between infected cells and those that are uninfected or in the IE phase of infection is restricted when the gBcyt regulatory function is intact.
As observed with gB[Y881F]-TK-RFP infection at T0 (24 hpi), the syncytia in FM4-64FX melanoma cells infected with gB[Y881F]-TK-eGFP were considerably larger than pOka-TK-eGFP syncytia at T0 (Fig. 3B; see Movie S4 at https://purl.stanford.edu/wc992yg2549). Fusion of infected cells with neighboring infected cells was observed during gB[Y881F]-TK-eGFP infection, similar to the case for the gB[Y881F]-TK-RFP-infected LifeAct-tGFP cells (see Movies S2 and S4 at https://purl.stanford.edu/wc992yg2549). However, labeling cell membranes with FM4-64FX made it possible to demonstrate a notable increase in fusion between uninfected and gB[Y881F]-TK-eGFP-infected cells. The membranes of eGFP-negative cells disappeared rapidly as their nuclei were sequestered into the evolving syncytium (Fig. 3B, boxes 2 and 3; see Movie S4). The engulfment of cells at the periphery of the syncytium was evident as the live imaging progressed (see seconds 11 to 45 of Movie S4, box 2 and 3 region). Infrequently, eGFP-negative cells were observed to traverse the syncytium without fusing, suggesting that these cells remained uninfected. As seen with gB[Y881F]-TK-RFP infection, the periphery of the gB[Y881F]-TK-eGFP syncytium was highly dynamic. Thus, dysregulated gBcyt fusion caused the plasma membranes of infected cells to acquire a heightened capacity to fuse with those of uninfected cells, whereas fusion associated with an intact gBcyt was ordered and occurred predominantly between infected cells. This effect on cell membrane function, along with aberrant cell protrusions due to an altered actin cytoskeleton, resulted in syncytia sequestering a greater quantity of cells during the live imaging period.
Dysregulated gBcyt-mediated cell fusion affects infected-cell ultrastructure.
It was postulated that the abundance of uninfected nuclei and cytoskeletal reorganization caused by gB[Y881F]-mediated dysregulated fusion could interfere with the production of nascent virions. As expected, numerous virus particles were apparent on the surfaces of pOka-TK-RFP-infected cells when examined by electron microscopy (Fig. 4). In contrast, the number of intact cell surface virions in gB[Y881F]-TK-RFP-infected cells was markedly reduced. Importantly, because gB is also involved in nuclear egress, accumulation of capsids in the perinuclear space was not observed for pOka-TK-RFP, as expected, or for the gB[Y881F]-TK-RFP mutant. Notably, cellular protrusions at the periphery of infected cells were more extensive for the hyperfusogenic gB[Y881F]-TK-RFP than for pOka-TK-RFP. These differences suggest that the alterations of the cytoskeletal network by gB[Y881F]-TK-RFP infection interfered with the trafficking of virus particles through the cytoplasm after exiting the nucleus and exocytosis to the cell surface.
FIG 4.

Dysregulated gBcyt-mediated cell fusion affects infected-cell ultrastructure. Electron microscopy of melanoma cells infected with pOka-TK-RFP or the hyperfusogenic mutant gB[Y881F]-TK-RFP at 48 hpi is shown. The cytosol (C) and extracellular space (E) of infected cells are indicated on each micrograph.
Dysregulated gBcyt-mediated cell fusion affects the VZV transcriptome.
Transcription from VZV open reading frames (ORFs) was quantified at 12, 24, and 36 hpi using RNA-seq in melanoma cell monolayers infected with pOka, gB[Y920F], a control virus that has a pOka phenotype, and gB[Y881] and gB[Y881/920F] which are both hyperfusogenic. The inoculum titers used for RNA-seq were 1.5 × 105 PFU/cm2, leading to the majority of the monolayer being infected at 36 hpi, as determined by the localization of IE62 in cell nuclei by confocal microscopy (Fig. 5). The frequency of infected cells ranged from 30 to 45% at 12 hpi and from 90 to 95% by 36 hpi. As expected, the frequency of VZV transcripts increased during the course of pOka infection in melanoma cells (Table 1; Fig. 6). By 36 hpi, transcripts from ORF9 were the most abundant, as shown by the high frequency of transcripts per kilobase million (TPM) values associated with this ORF (Table 1). ORF68, which encodes gE, expressed the next most abundant transcripts at 36 hpi, followed by ORF57 and ORF58. ORF67, which encodes gI, and ORF61 were also abundantly expressed by 36 hpi. ORF56, -6, -55, -28, -2, -65, listed in the order of increasing TPM values, were the least abundantly transcribed. As expected, the transcript abundances of VZV ORFs did not correlate with their transcriptional class as defined by herpes simplex virus 1 (HSV-1) homologs in these experiments because infection was not synchronous. ORF61 protein is expressed at the earliest times after VZV infection and is considered to be an IE gene by putative α transcriptional class (40). ORF2, -6, and -28 are putative early genes (the β class). gE protein is detected beginning at early times in VZV-infected cells (40, 41). ORF9, -33, -56, -58, -65, and -67 are all considered to be in the γ class (late gene expression).
FIG 5.

Enumeration of infected nuclei in melanoma cells inoculated with pOka and the gBcyt mutants. (A) Confocal microscopy of melanoma cells infected with pOka, gB[Y818F], gB[Y920F], and gB[Y881/920F] that were fixed at 12, 24, and 36 hpi and stained for VZV IE62 (red) and nuclei (Hoechst 33342; blue). (B) Frequencies of nuclei infected (IE62 positive) with pOka, gB[Y818F], gB[Y920F], and gB[Y881/920F] at 12, 24, and 36 hpi. The percentage of infected nuclei for each virus was determined from three fields of view.
TABLE 1.
VZV ORF TPM values of pOka and the gB mutants gB[Y881F], gB[Y920F], and gB[Y881/920F] during infection of melanoma cells from 12 to 36 h postinfection
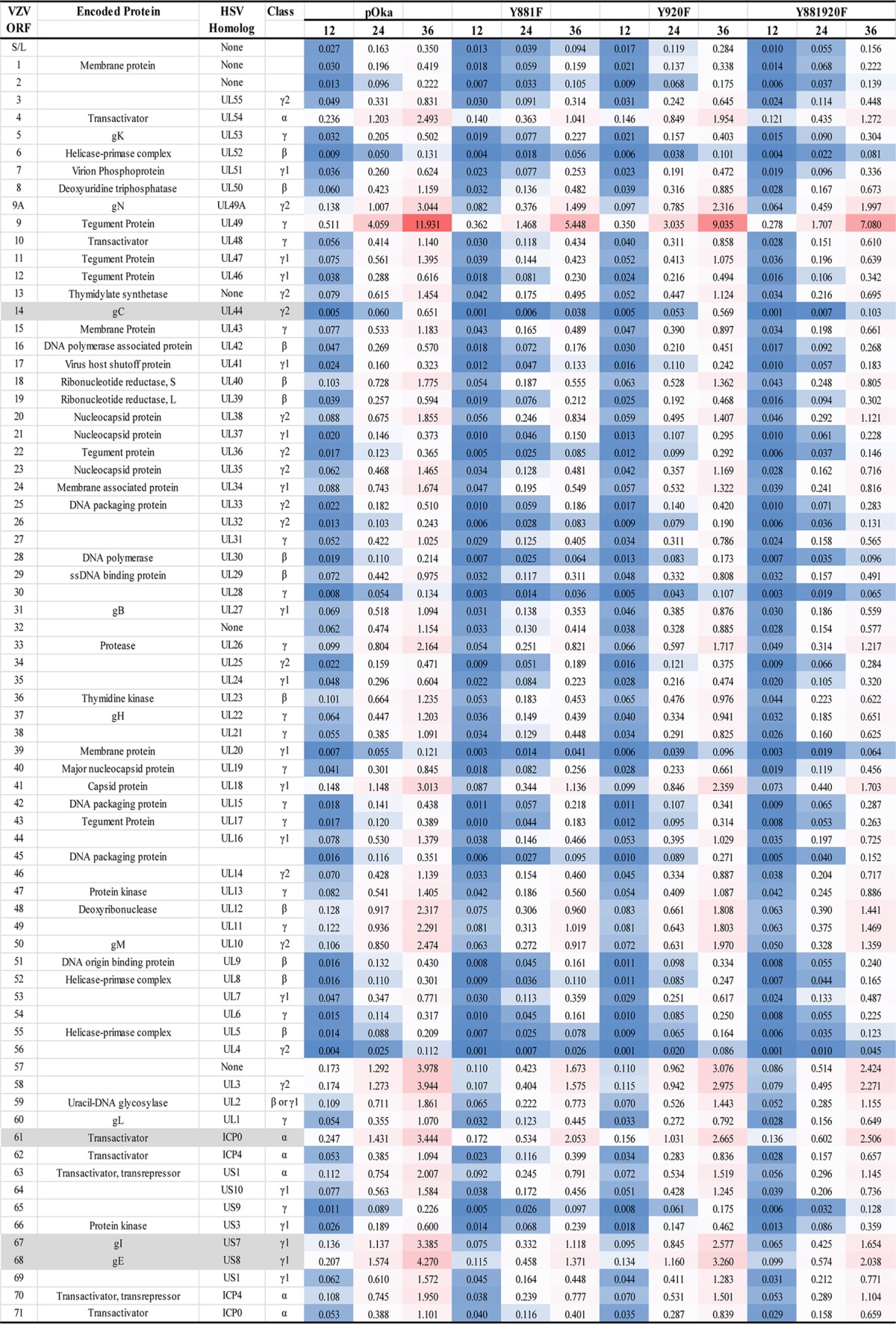
FIG 6.

Dysregulated gBcyt-mediated cell fusion affects VZV gene expression during infection. Integrated genomics viewer (IGV) of RNA-seq reads from pOka-, gB[Y818F]-, gB[Y920F]-, and gB[Y881/920F]-infected melanoma cells at 12, 24 and 36 hpi that mapped to the VZV pOka genome is shown. The histograms represent the frequency of RNA-seq reads that mapped to the pOka genome. The red bars in ORF31 highlight the locations of nucleotide substitutions leading to the Y881F and Y920F mutations. The blue bars at the foot of the IGV represent pOka ORFs. The arrowheads highlight the location of ORF14, which encodes gC.
There was a predictable relationship between the phenotype observed in VZV-infected cells and VZV transcriptome profiles. The transcription from the gB[Y920F] ORFs was comparable to that from the pOka ORFs at 12, 24, and 36 hpi (Table 1). ORF9 was the most abundant transcript, followed by ORF68, -57, -58, -61, and -67. The smallest amount of transcription was from ORF56, again similar to the case for pOka. Transcription profiles for both of the hyperfusogenic mutants, gB[Y881F] and gB[Y881/920F], had overall similarities to that for pOka, consistent with the production of infectious virus in melanoma cells. However, transcript abundance was reduced compared to that for pOka at 12, 24, and 36 hpi, with several ORFs showing altered transcription kinetics in association with the hyperfusion phenotype (Table 1). An important difference was the proportional reduction of ORF14[gC] in the hyperfusogenic mutants (Fig. 6 and 7). Compared to ORF9, which had the most abundant transcripts and was used as an internal transcriptional control (100%), the proportions of RNA-seq TPM values for ORF14 from the hyperfusogenic mutants were 0.7% ± 0.01% (Y881F) and 1.4% ± 0.22% (Y881/920F). These values were 3.8- to 7.8-fold lower than that for pOka and 4.4- to 9-fold lower than that for the wild-type-like gB[Y920F] mutant. The ORF14 transcript is considered to be produced very late during VZV infection (41, 42). This decrease is consistent with the poor propagation of the hyperfusogenic mutants. Three additional ORFs, ORF61, -67, and -68, had significantly different proportions of transcripts relative to ORF9 in cells infected with the hyperfusogenic mutants than pOka and the wild-type-like mutant gB[Y920F] (Fig. 7), which is particularly notable because these transcripts are among the most abundant in association with the pOka phenotype. Proteins from each of these ORFs are essential to VZV propagation, and reduced transcription would be a barrier to replication for the hyperfusogenic mutants.
FIG 7.

Dysregulated gBcyt-mediated cell fusion affects the expression kinetics of vital ORFs during infection. The proportions of RNA-seq TPM values, ORF9/ORF, for ORF14 (A), ORF61 (B), ORF67 (C), and ORF68 (D) that were significantly different between wild-type-like VZV (pOka and gB[Y920F]) and the hyperfusogenic mutants (gB[Y881F] and gB[Y881/920F]) are shown.
Dysregulated gBcyt-mediated cell fusion affects the host cell transcriptome.
To determine the effects of VZV replication on cell gene transcription, transcriptomes of uninfected and pOka-infected melanoma cells at 12, 24, and 36 hpi were compared using RNA-seq. Significant differential gene expression, calculated using a ≥2-fold change in reads per kilobase per million (RPKM) values as a cutoff, was absent at 12 hpi, but 20 genes were differentially expressed by 24 hpi, which increased significantly to 449 genes by 36 hpi (Table 2). At 24 hpi, 95% (19) of these genes were upregulated rather than downregulated, whereas the proportion of upregulated genes was 48% (215) at 36 hpi. The frequency of differentially expressed cell genes also increased at 24 and 36 hpi in gB[Y920F]-, gB[Y881F]-, and gB[Y881/920F]-infected cells. In contrast to pOka and gB[Y920F], fewer cell genes were differentially expressed in both gB[Y881F]- and gB[Y881/920F]-infected cells, and the majority of differentially expressed genes were upregulated.
TABLE 2.
Frequencies of differentially expressed genes in melanoma cells at 12 to 36 h postinfection with pOka or the gB[Y881F], gB[Y920F], and gB[Y881/920F] mutants
| Virus | hpi | Total no. of genes | No. (%) of genes: |
|
|---|---|---|---|---|
| Upregulated | Downregulated | |||
| pOka (WT) | 12 | 0 | NAa | NA |
| 24 | 20 | 19 (95) | 1 (5) | |
| 36 | 449 | 215 (48) | 234 (52) | |
| gB[Y881F] | 12 | 0 | NA | NA |
| 24 | 3 | 3 (100) | NA | |
| 36 | 220 | 186 (85) | 34 (15) | |
| gB[Y920F] | 12 | 0 | NA | NA |
| 24 | 41 | 40 (98) | 1 (2) | |
| 36 | 444 | 250 (56) | 194 (44) | |
| gB[Y881/920F] | 12 | 0 | NA | NA |
| 24 | 2 | 2 (100) | NA | |
| 36 | 180 | 150 (83) | 30 (17) | |
NA, not applicable.
A correlation between gene expression and the gB[Y881F]-induced hyperfusion phenotype was identified when hierarchical clustering was performed based on the 449 differentially expressed genes identified in pOka at 36 hpi (Fig. 8A). In contrast, an association between phenotype and gene expression was not apparent at 12 hpi. As expected, the correlation between gene expression profiles and the hyperfusion phenotype was also identified when hierarchical clustering was performed based on the 220 differentially expressed genes identified in gB[Y881F]-infected cells at 36 hpi (Fig. 8B). These data suggested that a subset of differentially expressed genes associated with the hyperfusion phenotype was independently regulated compared to those in pOka and the wild-type-like mutant gB[Y920F].
FIG 8.

Dysregulated gBcyt-mediated cell fusion affects the host transcriptional response to VZV infection. (A, B, F, and G) Hierarchical clustering of the RPKM gene expression values for the entire RNA-seq data set by using either the 449 genes for pOka (A), the 220 genes for gB[Y881F] (B), the 537 genes associated with wild-type-like (F), or the 267 genes associated with hyperfusion (G) that were differentially expressed in melanoma cells at 36 hpi. Gene expression data have been normalized by reallocating gene RPKM values to a mean of zero, scaled to a standard deviation of one, and expressed as a heat map with a range from −4.5 (blue) to +4.5 (red). The dendrograms above the heat maps show the grouping of RNA-seq samples. The colored boxes highlight the groupings of uninfected cells (orange) at 12 to 36 hpi and the wild-type phenotype (blue) and the hyperfusion phenotype (red) at 24 and 36 hpi. (C to E and H to J) Venn diagrams show the frequencies of differentially expressed genes (C and H), upregulated genes (D and I), and downregulated genes (E and J) associated with pOka and the three gBcyt mutant viruses (C to E) or VZV phenotype (H to J) in melanoma cells at 36 hpi.
Gene associations with the hyperfusion phenotype were established by generating Venn diagrams based on the genes that were differentially expressed at 36 hpi. Common to all four viruses, 84 cell genes were differentially expressed, of which 66 were upregulated and 18 were downregulated (Fig. 8C to E). There were 43 genes exclusively associated with the gB[Y881F] mutants that were all upregulated. In addition, there were 48 and 20 differentially expressed genes that were associated with gB[Y881F] or gB[Y881/920F], respectively.
To ascertain the relationship between phenotype and cell gene expression, RPKM values were combined for cells infected with wild-type (pOka and gB[Y920F]) or the hyperfusogenic (gB[Y881F] and gB[Y881/920F]) viruses and compared to those for uninfected cells. As observed in the analyses using individual viruses, differential gene expression was not detected at 12 hpi (Table 3). By 24 hpi, 13 genes were differentially regulated in association with the wild-type phenotype, and three were differentially regulated with the hyperfusion phenotype. By 36 hpi, 537 genes were differentially expressed in association with the wild-type phenotype compared to 267 genes for the hyperfusogenic phenotype, with a majority being upregulated.
TABLE 3.
Frequencies of differentially expressed genes in melanoma cells at 12 to 36 h postinfection with viruses with a wild-type-like or hyperfusion phenotype
| Phenotype | hpi | Total no. of genes | No. (%) of genes: |
|
|---|---|---|---|---|
| Upregulated | Downregulated | |||
| Wild type-like | 12 | 0 | NAa | NA |
| 24 | 13 | 12 (92) | 1 (8) | |
| 36 | 537 | 288 (54) | 249 (46) | |
| Hyperfusion | 12 | 0 | NA | NA |
| 24 | 3 | 2 (67) | 1 (33) | |
| 36 | 267 | 216 (81) | 51 (19) | |
NA, not applicable.
The wild-type-like or the hyperfusion mutants grouped independently by phenotype when hierarchical clustering was performed on the differentially expressed genes identified at 36 hpi (Fig. 8G and H). The constellations for pOka and gB[Y920F] and for gB[Y881F] and gB[Y881/920F], were retained at 24 hpi and 36 hpi, which was consistent with hierarchical clustering performed with differentially expressed genes identified for pOka or gB[Y881F] at 36 hpi (Fig. 8A and B). At 36 hpi, 162 genes were associated with both the wild-type-like and hyperfusion phenotypes, as shown in Venn diagrams of the differentially expressed genes (Fig. 8H to J). Of the differentially expressed genes associated with the Y881F mutants, 105 were specific to the hyperfusion phenotype. More differentially expressed genes were associated with the wild-type phenotype, which was attributed to the overall 2-fold increase in the number of genes differentially expressed compared to the hyperfusion phenotype.
Dysregulated gBcyt-mediated cell fusion affects the categorization of differentially expressed cell genes in VZV-infected melanoma cells.
Substantial similarities and some differences in biogroups associated with canonical pathways, protein families, regulatory motifs, and gene ontology were identified when differentially expressed genes from pOka, gB[Y920F], gB[Y881F], or gB[Y881/920F] were analyzed using the BaseSpace Correlation Engine (Table 4; see the supplemental material). To identify biogroups associated with the two phenotypes, Correlation Engine analyses were performed using the differentially expressed genes identified for these phenotypes at 36 hpi; the top 10 biogroups were listed for each of the four categories (Table 4). In the canonical pathways category, all differentially expressed genes were upregulated; the wild-type phenotype shared five of nine biogroups with the hyperfusion phenotype. One other biogroup in the top 10 for the wild-type category, genes involved in class A/1 (rhodopsin-like receptors), was also associated with the hyperfusogenic mutants but was not in the top 10 (see the supplemental material). In addition, four biogroups identified in the hyperfusion phenotype were associated with the wild-type phenotype but, again, not found in the top 10. Only one in the canonical pathways biogroup category, basal cell carcinoma, was associated with the hyperfusion but not the wild-type phenotype (Table 4; see the supplemental material).
TABLE 4.
Top 10 biogroup categories of cell genes differentially expressed in melanoma cells at 36 h after infection with wild-type-like or hyperfusogenic VZVa
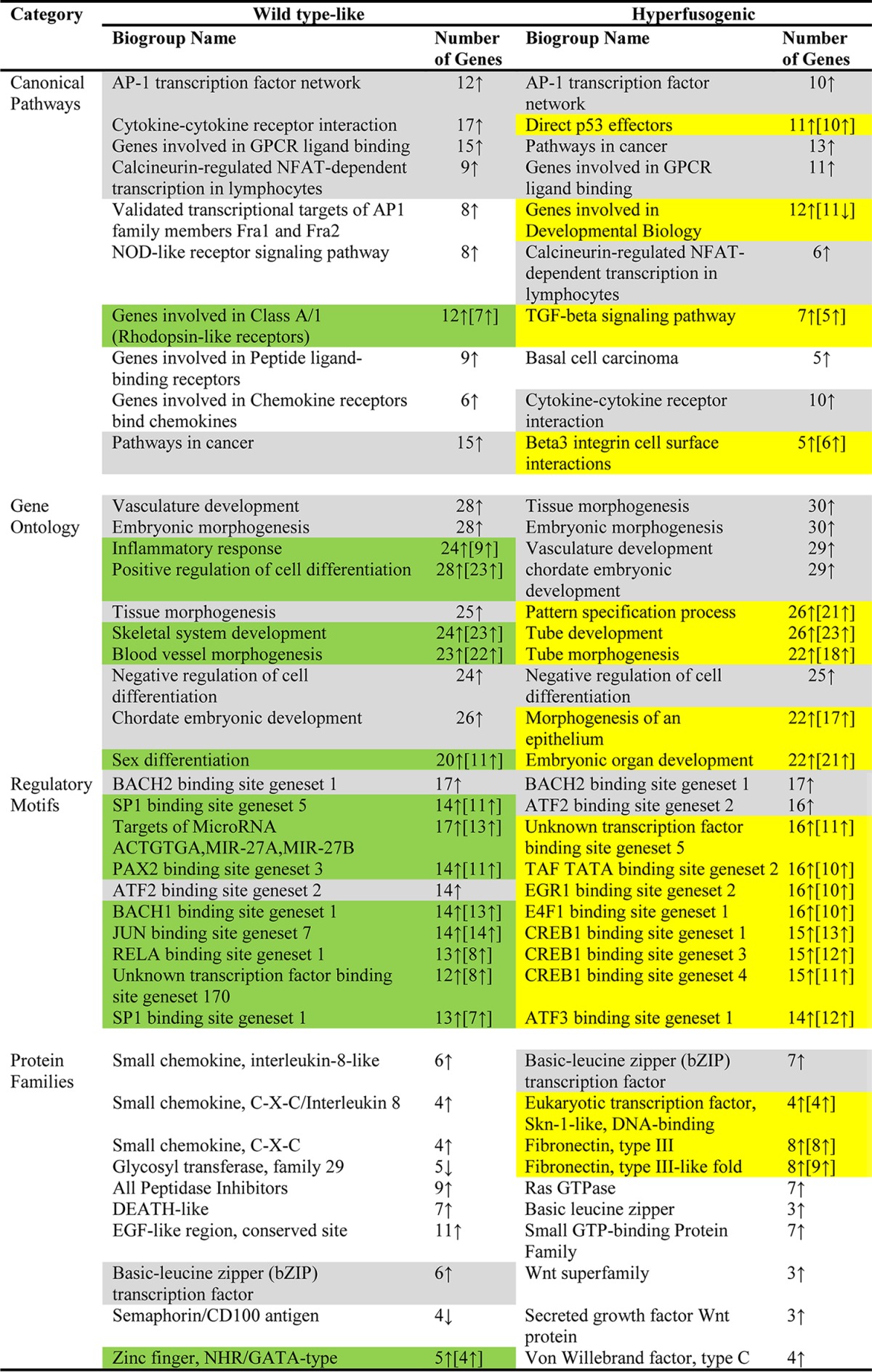
Shading: gray, biogroups in the top 10 for both phenotypes; green, biogroups in the top 10 for the wild-type phenotype and also within the hyperfusogenic category with a P value of <0.0001 but outside the top ten; yellow, biogroups in the top 10 for the hyperfusogenic phenotype and also within the wild-type-like category with a P value of <0.0001 but outside the top ten (see the supplemental material).
Biogroups associated with the wild-type and hyperfusion phenotypes in the gene ontology category were all similar and upregulated (Table 4). Although only five biogroups were shared in the top 10 for each phenotype, the remaining biogroups were also associated with each phenotype outside the top 10 (see the supplemental material). All genes were predominantly upregulated for all of the top 10 associated biogroups for each phenotype. In contrast to the gene ontology category, the regulatory motifs category had few biogroups that were represented in the top 10 for both of the wild-type and hyperfusion phenotypes. Two biogroups, BACH2 binding site gene set 1 and ATF2 binding site gene set 3, were associated with differentially expressed genes from both wild-type and hyperfusion phenotypes (Table 4). The remaining eight biogroups within the regulatory motifs category for each phenotype were also represented, although these fell outside the top 10 (see the supplemental material). All of the genes associated with these biogroups were predominantly upregulated. This suggested that influences on cell transcriptional regulation were similar for viruses that produced wild-type or the hyperfusion phenotypes.
In contrast, within the protein families category, biogroups associated with phenotype shared little commonality. The only mutual biogroup within the top 10 was the basic leucine zipper (bZIP) transcription factor (Table 4). Interestingly, two biogroups associated with the wild-type phenotype, glycosyl transferase family 29 and semaphorin/CD100 antigen, had genes that were predominantly downregulated, whereas as all other biogroups in the protein families category were predominantly upregulated. The zinc finger, NHR/GATA type was the only other wild-type-like associated biogroup that was also present in the hyperfusion phenotype, albeit not in the top 10 (see the supplemental material). Some biogroup duplication was associated with the hyperfusion phenotype, with the basic leucine zipper, fibronectin, and Wnt represented in at least two categories in the top 10.
Interestingly, the Ras GTPases and small GTP binding proteins, which are involved in cytoskeletal reorganization, were strongly represented in the hyperfusion phenotype based on RNA-seq. This association of genes in the Ras GTPase biogroup with hyperfusion, including ARL4D, GEM, RASD1, RASL11B, RHOV, RND1, and RRAD, was verified using quantitative reverse transcription-PCR (qRT-PCR). In each case, mRNA transcription was increased in gB[Y881F]-infected melanoma cells compared to both uninfected and pOka-infected melanoma cells at 36 hpi (Fig. 9). These data indicate the involvement of Ras GTPases in the hyperfusion phenotype of the gB[Y881F] mutant, suggesting a link between these genes and the extensive cytoskeletal reorganization observed during live-cell imaging.
FIG 9.

Validation of differentially transcribed Ras GTPase genes associated with the gB[Y881F] hyperfusion phenotype. (A) RPKM values for transcripts from melanoma cells at 12, 24, and 36 hpi with pOka and the three gBcyt mutants gB[Y881F], gB[Y920], and gB[Y881/920F] for cellular Ras GTPase genes ARL4D, GEM, RASD1, RASL11B, RHOV, RND1, and RRAD associated with the hyperfusion phenotype. (B) Transcript abundance calculated by qRT-PCR (ΔCq) for the cellular Ras GTPase genes associated with the hyperfusion phenotype in cells infected with pOka and gB[Y881F] at 48 hpi.
DISCUSSION
VZV characteristically overcomes the intrinsic barriers to cell-cell fusion in skin and sensory ganglion pathogenesis. This study demonstrates that this fusogenic function of gB, in the context of infection, requires continuous modulation by its own cytoplasmic domain. Loss of gBcyt regulation has consequences that are highly detrimental for VZV propagation. A novel finding, illuminated by the live-cell imaging comparing the gB[Y881F] mutant with pOka, was that fusion must be modulated early in VZV infection; otherwise, infected cells fuse rapidly with large numbers of uninfected cells. This process proceeds unchecked and is accompanied by marked changes in actin filament organization. In contrast, canonical syncytium formation evolved slowly, preceded by cell-cell fusion primarily between infected cells at early/late time points in replication. Significantly reduced transcription of four major VZV genes, encoding the ORF61 regulatory protein and three crucial glycoproteins, gC (ORF14), gE (ORF68), and gI (ORF67), was associated with gBcyt dysregulation, leading to reduced viral propagation likely compounded by poor virion egress, as demonstrated by electron microscopy. As expected, pOka infection induced differential regulation of numerous cell genes of interest for further investigation. However, transcriptome profiling did not identify manipulation of specific cell signaling pathways in association with gBcyt-regulated cell-cell fusion. Unsurprisingly, both wild-type (pOka and gB[920F]) and hyperfusogenic (gB[Y881F] and gB[Y881/920F]) viruses had substantial overlap in transcriptome profiles, since these viruses replicate in melanoma cells and VZV characteristically induces cell-cell fusion. However, a subset of Ras-GTPase family genes was associated with the hyperfusion phenotype, supporting the notion that gBcyt regulates molecular mechanisms involved in actin cytoskeleton and membrane remodeling.
The live-cell imaging of pOka- and gB[Y881F]-infected melanoma cells suggests two possible mechanisms underlying diminished VZV propagation caused by hyperfusion. First, accelerated fusion caused a rapid uptake of uninfected cell nuclei into evolving syncytia. In contrast to those of pOka-infected cells, the nuclei accumulating at the centers of the exaggerated gB[Y881F] syncytia showed limited evidence of viral protein expression, indicating that these intracellular conditions interfered with docking of incoming virions at the nuclear membranes and transfer of viral DNA. Therefore, many nuclei were effectively unavailable to support VZV replication. Second, the substantial cytoskeletal reorganization and constant dynamic changes of actin filament protrusions on the outer membranes of the gB[Y881F] syncytia were associated with sparse VZV particles on cell surfaces. Thus, altered cell properties caused by gBcyt dysregulation interfered with virion trafficking and egress to extracellular spaces. The phenomenon of reduced virus particles on cell surfaces was reported for VZV gB-36, another VZV hyperfusogenic gB mutant (43). In contrast, pOka-infected melanoma cells had an abundance of cell surface virions, as previously observed in VZV-infected fibroblasts (40). This earlier study showed that VZV particles trafficked to cell surfaces by 9 h and spread to adjacent cells without requiring cell-cell fusion to initiate infection, with fusion events increasing as infection progressed beyond 12 h, when many progeny virions had been produced. Premature fusion appears to impair VZV propagation by the dual effects of sequestering many nuclei within syncytia and actin cytoskeleton disruption.
The aberrant plasma membranes associated with gBcyt dysregulation might also affect virion assembly in the cytoplasm. Although F-actin is not required for transport of nucleocapsids during infection with the related alphaherpesvirus pseudorabies virus, the trans-Golgi network (TGN) and other membrane-associated cytoplasmic structures are vital for herpesvirus virion assembly (44, 45). Herpesvirus glycoproteins must traffic to plasma membranes and back to the TGN, where secondary envelopment occurs. Dynamin-dependent endocytosis is required for HSV-1 glycoprotein trafficking to cytoplasmic assembly compartments (46). Plasma membrane distortions caused by gB[Y881F] mutants could affect these processes, limiting the availability of glycoproteins for virion incorporation and generating virus particles that are poorly infectious, as was observed with reduced gI in VZV particles (47).
The dysregulation of gBcyt fusion control also negatively affected the kinetics of VZV gene expression. The pOka transcriptome profile in melanoma cells was similar to that found in previous RNA-seq-based studies in human fetal lung fibroblasts, neurons derived from human-induced pluripotent stem cells, and neonatal primary human epidermal keratinocytes (HEKn), which also identified ORF9 and -57 as the most abundantly transcribed VZV genes (48, 49). However, regulated cell fusion was important for the canonical expression of vital VZV genes, because the gB[Y881F] substitution significantly reduced transcription of ORF61, a major regulatory protein, gC, and the heterodimer-forming glycoproteins gE and gI. The ORF61 protein is critical for VZV replication and skin pathogenesis, as shown when ORF61 expression is reduced by disrupting its transactivation by the cellular transcription factor SP1 (50). Importantly, ORF61 has several functions in innate antiviral responses, as it has SUMO binding activity important for the degradation of PML bodies in skin cells, degrades activated IRF3 to abrogate IRF3-depedent innate immunity, and influences JNK/SAPK and p38/MAPK phosphorylation (51–55). Consistent with previous observations in melanoma cells, fibroblasts, and undifferentiated HEKn cells, VZV gC was expressed during pOka infection (41, 42, 48, 49). However, ORF14 transcription was markedly delayed in gB[Y881F]-infected cells. Although gC is dispensable for virus replication in cell culture, it is required for effective VZV propagation in human skin xenografts and ex vivo skin cultures (56, 57). In contrast to that of other alphaherpesviruses, VZV gE, encoded by ORF68, is essential for viral replication, and reducing ORF68 transcription by promoter mutations substantially impairs cell-cell spread and skin pathogenesis (58, 59). In addition, deletion of gI, encoded by ORF67, also leads to a small-plaque phenotype and severe attenuation in skin (57, 58, 60). Consequently, the gE/gI heterodimer is important for cell-cell spread in vitro and in vivo (23, 61, 62). Thus, failure to control gB-induced fusion has detrimental consequences at the basic level of VZV gene expression.
The kinetic analysis of cellular gene expression provided an insight into the complex cellular transcriptional response to VZV replication as infection progressed. As expected, the increased sensitivity of RNA-seq detected more differentially expressed genes than were found by hybridization-based microarrays in pOka-infected human T cells (189 genes) or human foreskin fibroblasts (51 genes) at 48 hpi or in human skin xenografts (104 genes) at 21 to 28 dpi (63). RNA-seq studies of HEKn cells, differentiated HEKn cells, Hutchinson-Gilford progeria syndrome (HGPS) fibroblasts, and senescent normal human dermal fibroblasts (NHDF) also identified more VZV-induced cell gene changes (49, 64). As expected and consistent with transcriptome studies of HFF and HEKn cells, few genes associated with interferon responses were differentially expressed in pOka-infected cells at 36 hpi (49, 63). Upregulation of integrin signaling was a common response identified in the present and previous studies. These data support the recent finding that VZV requires the αV integrin subunit for effective propagation, suggesting that virion binding of integrins triggers cell signaling pathways during entry (65).
Dysregulation of gB-dependent cell-cell fusion was associated with similarities as well as significant differences in cell gene transcriptional responses to pOka and gB[Y881F]. The Ras GTPases were identified to be significantly upregulated in association with the gB[Y881F] hyperfusion phenotype, which included the seven genes ARL4D, GEM, RASD1, RASL11B, RHOV, RND1, and RRAD. Although ARL4D and GEM were upregulated to similar levels in pOka-infected cells the remaining five differentially expressed Ras GTPases were all expressed in greater quantities during gB[Y881F] infection. These genes were also increased, but to a lesser extent, in pOka-infected cells. Given the natural propensity of VZV to induce cell-cell fusion, these data fit well with the syncytial phenotype of wild-type infection and higher levels of Ras GTPase gene expression in cells infected with the hyperfusogenic mutants.
While information about the functions of the Ras GTPases identified in the present study is limited, their known associations with membrane remodeling, extracellular matrix interactions, modulation of cell shape, and actin reorganization are consistent with pOka-induced changes in cell structure properties and the correspondingly exaggerated effects of the gB[Y881F] hyperfusion mutants. Briefly, ARL4D, a member of the ADP-ribosylation factor ARF-like protein of Ras-related GTPases family, localizes to the plasma membrane, where it recruits cytohesin-2 (Arf nucleotide binding site opener [ARNO]) and other cytohesin proteins that regulate integrin adhesiveness (66). ARF6, which is linked with ARL4D actin remodeling, is a GTPase guanine nucleotide exchange factor (GEF) that regulates actin rearrangements involved in the formation of plasma membrane protrusions, cell adhesion, cell migration, and endosomal trafficking (66–69). Importantly, ARF6 activation requires ARNO recruitment from the cytosol to the plasma membrane. Therefore, ARL4D has been proposed to be part of the signaling pathway that regulates the cytohesins and ARF6. The interaction of ARF6 with ARL4D might explain the increase in gene expression of ARL4D as a modulating response to the extensive membrane reorganization during melanoma cell infection with hyperfusogenic gBcyt mutants.
Both GTP binding protein overexpressed in skeletal muscle (GEM) (kir/gem) and ras-related associated with diabetes (RADD) belong to the RGK family of small GTP binding proteins, which are characterized by 14-3-3 protein and calmodulin binding domains and negatively regulate the RHO-RHO kinase pathway (70). The binding of 14-3-3 and calmodulin to GEM modulates the subcellular distribution of GEM between the cytoplasm and the nucleus, downregulating calcium channel activity and cell shape remodeling by reducing dendrite formation (71–73). RRAD has also been associated with cell shape remodeling (74, 75). Although GEM and RRAD were associated with the hyperfusion phenotype, they were also upregulated, to a lesser extent, in pOka-infected cells, supporting the notion that these genes are linked with VZV gB-induced membrane fusion.
Little is known about the canonical functions of RASD1, RASL11B, and RHOV. RASD1 (Ras-related dexamethasone induced 1) is a member of the Ras superfamily of small GTPases normally induced by dexamethasone with a likely role in dexamethasone-induced alterations in cell morphology, growth, and cell-extracellular matrix interactions and inhibition of G protein-coupled receptor (GPR) signaling (76–78). RASL11B is another small GTPase, but information about its function is limited (79). RHOV is an atypical RAS homolog family member, which is transiently induced by the Wnt family of proteins and affects the actin cytoskeleton via activation of the JNK pathway (80). Notably, Wnts were also associated with the hyperfusion phenotype, suggesting a mechanism for RHOV induction in VZV-infected cells.
The Rho GTPase RND1 has been implicated in the regulation of the actin cytoskeleton and microtubule dynamics in response to extracellular growth factors through actin reorganization in response to extracellular interactions with plexin/semaphorin (81–84). Plexin-B1 directly binds RND1. Intriguingly, genes associated with the semaphorin/CD100 biogroup were significantly downregulated in melanoma cells in response to wild-type infection, perhaps limiting cell-cell fusion. In addition, overexpression of RND1 causes disassembly of actin stress fibers and branching cellular processes (81). Similar structures were seen in the gB[Y881F]-infected cells, suggesting a potential link with this protein and the filopodium-like structures produced during hyperfusion. Overall, the differential expression of a constellation of Ras GTPases in VZV-infected cells and their association with the hyperfusion phenotype warrant further study into their influence on VZV-induced cell-cell fusion and syncytium formation.
In summary, syncytium formation is a natural process during VZV pathogenesis. The data derived from studies of the hyperfusogenic gB mutants provide new insights into how the process affects VZV replication and virion production. Moreover, defining the viral and cellular molecular mechanisms of cell-cell fusion in sensory ganglia that occurs during VZV reactivation could be an avenue to develop therapeutics that block these effects and offer new options to prevent postherpetic neuralgia.
MATERIALS AND METHODS
Viruses.
The viruses pOka, gB[Y881F], gB[Y920F], and gB[Y881/920F] were derived from the self-excisable bacterial artificial chromosome (BAC), pPOKA-BAC-DX, as described previously (36). The gB[Y920F] mutant has a pOka phenotype, whereas the gB[Y881F] and gB[Y881/920F] mutants are hyperfusogenic. The pOka-TK-eGFP and gB[Y881F]-TK-eGFP mutants, which express eGFP conjugated to the thymidine kinase (TK) encoded by open reading frame 36 (ORF36), were also previously generated with the pPOKA-BAC-DX (85). Two new viruses, pOka-TK-RFP and gB[Y881F]-TK-RFP, which express monomeric RFP conjugated to TK, were also generated from pPOKA-BAC-DX. Briefly, a TK-RFP shuttle vector was generated by amplifying monomer RFP (mRFP) from the EP mRFP vector (86) with Accuprime Pfx (Invitrogen) using oligonucleotides 5′TKmRFP (AATGCGCATGGAATATATTGAAAGAGCTTGTTAATGCCGTTCAGGACAACACTTCCGCCTCCTCCGAGGACGTCATCAAG)and 3′TKmRFP (AAAGATATCATCTTTTTACTGGTACATACGTAAATACTAGGTATATTTAGCCTCCTCCGAGGACGTCATC) and TOPO TA cloning the product into pCR4.0 (Invitrogen). To confirm that spurious mutations from the PCR had not been incorporated, the insert was sequenced. The EcoRV/ApaL1 fragment from the TK-RFP shuttle vector was used for RED recombination with the pPOKA-ΔORF31 BAC (36) to generate pPOKA-ΔORF31-TK-RFP BAC. A gB-Kan cassette containing either wild-type gB or gB[Y881F] was used to reconstitute ORF31 into the pOka-ΔORF31-TK-RFP BAC to generate pPOKA-TK-RFP and pPOKA-gB[Y881F]-TK-RFP as previously described (36). Melanoma cells were transfected with these new pPOKA BACs using Lipofectamine 2000 (Invitrogen) following the manufacturer's instructions to generate the pOka-TK-RFP and gB[Y881F]-TK-RFP viruses.
Cells.
Melanoma cells expressing GFP (LifeAct-tGFP melanoma cells) were generated from melanoma cells (MeWo) with a pGIPZ-LifeAct-tGFP vector using replication-incompetent lentiviruses in accordance with protocols from Open Biosystems (GE Dharmacon). The LifeAct sequence, a short, 17-amino-acid peptide from Saccharomyces cerevisiae that binds F-actin, was incorporated at the N terminus of the tGFP sequence in the commercially available pGIPZ vector (Thermo Scientific). Oligonucleotides 5HindIII_LA-tGFP (AATAAGCTTGCCACCATGGGTGTCGCAGATTTGATCAAGAAATTCGAAAGCATCTCAAAGGAAGAAGAGAGCGACGAGAGCGGCCTGC) and 3XhoI_tGFP (AATCTCGAGTCATTATTCTTCACCGGCATCTGCATCCG) were used to amplify the tGFP gene from the pGIPZ vector using Accuprime Pfx (Invitrogen), and the product was TOPO TA cloned to generate pCR4.0-LifeAct-tGFP for sequence verification. The HindIII/XhoI fragment of pCR4.0-LifeAct-tGFP was subcloned to generate the pCDNA3.1-LifeAct-tGFP mammalian expression vector. Actin filament-associated GFP expression was confirmed by transient transfection of pCDNA3.1-LifeAct-tGFP into melanoma cells using Lipofectamine 2000 (Invitrogen) following the manufacturer's instructions. Oligonucleotides pGIPZ_XbaI (TGCTGCAGGTCCGAGGTTCTAGACG)/pGIPZ_5CMV ([PHOS]GGTGGCAGATCCTCTAGTAGAGTCG) and 5LifeAct ([PHOS]ATGGGTGTCGCAGATTTGATCAAG)/5tGFP[BsrGI] (TACTTGTACATTATTCTTCACCGGCATCTGCATCC) were used to generate PCR products that were then used for a triple ligation into the XbaI/BsrGI site of the pGIPZ vector to generate the pGIPZ-LifeAct-tGFP vector. Melanoma cells and transduced LifeAct-tGFP melanoma cells were propagated in culture medium (minimal essential medium [MEM] supplemented with 10% fetal bovine serum [Life Technologies], nonessential amino acids [100 μM; Cellgro], penicillin G-streptomycin [100 units/ml; Life Technologies], and amphotericin [0.5 mg/ml; Cellgro]). The LifeAct-tGFP melanoma cells were kept under puromycin selection (5 μg/ml; Invitrogen).
Live-cell confocal microscopy.
Melanoma cells or LifeAct-tGFP melanoma cells were seeded in 2-well Lab-Tek II chambered cover glass slides (Lab-Tek) at 2 × 105/cm2 24 h prior to infection with pOka or the gB[Y881F] mutant carrying ORF36[TK]-eGFP or -mRFP. At 24 h postinfection, the medium was changed to contain either 2 μg/ml Hoechst 33342 for LifeAct-tGFP melanoma cells infected with the mRFP-tagged viruses or 2 μg/ml Hoechst 33342 and 5 μg/ml FM4-64FX (Invitrogen), which labels plasma membranes, for melanoma cells infected with eGFP-tagged viruses. Live imaging was performed at the Stanford Cell Sciences and Imaging Facility (SCSIF) using a spinning-disc confocal microscope (Nikon) controlled with the NIS Elements software (Nikon). A 4-by-4 array was captured every 6 min for 17 to 20 h. Images were exported using NIS Elements, and files were converted using Photoshop (Adobe) and processed into an (AVI) format using Premiere Pro (Adobe). The tracking of nuclei was performed using the TrackMate plugin in for FiJi (ImageJ 1.50g). All movies associated with the live-cell confocal microscopy can be downloaded from the Stanford Digital Repository at https://purl.stanford.edu/wc992yg2549.
Electron microscopy.
Transmission electron microscopy was performed as previously described (36). Briefly, melanoma cells were seeded at 2 × 105 per well of 24-well plates containing 10-mm glass coverslips and then infected with 3.0 log10 PFU of either pOka-TK-RFP or gB[Y881F]-TK-RFP. Cells were fixed in 2% glutaraldehyde and 4% paraformaldehyde at 48 h postinfection (hpi). Fixed cells were dehydrated with increasing concentrations of ethanol followed by propylene oxide and then flat-embedded in epoxy resin. Ultrathin sections of 70 nm thickness were prepared with a diamond knife (Diatome, Hatfield, PA) using an ultramicrotome (Leica Microsystems Inc.). Sections were stained with 3.5% aqueous uranyl acetate (Electron Microscopy Sciences) and then 0.2% lead citrate (Electron Microscopy Sciences) for 1 min and air dried. Sections were visualized at the SCSIF using a JEOL 1400 transmission electron microscope (TEM) (Jeol USA Inc.) at 80 kV, and digital photographs were captured with a GATAN Multiscan 701 digital camera.
Confocal microscopy.
Melanoma cells infected with pOka or the gBcyt mutants were fixed with 4% formaldehyde for 10 min. Cells were stained with an anti-IE62 monoclonal antibody (MAB8616; EMD Millipore) because IE62 is expressed within 30 min after infection (40). The primary antibody was detected using donkey anti-mouse IgG, and nuclei were stained with Hoechst 33342 (Life Technologies). Images were captured at the SCSIF using a Zeiss LSM780 multiphoton laser scanning confocal microscope. Image processing was performed using Zen 2.1 (Black Software; Zeiss).
RNA-seq.
Melanoma cells were seeded at a density of 1.5 × 105/cm2 in 60-cm2 dishes (Corning) 16 h prior to inoculation with log10 4.8 PFU of pOka or the gBcyt mutants gB[Y881F], gB[Y920F] or gB[Y881/920F]. The infected cells were harvested at 12, 24, and 36 h postinfection with buffer RLT Plus following the manufacturer's instructions (Qiagen). RNA was purified from lysates using an RNeasy Plus minikit (Qiagen). RNA-seq libraries were prepared from 1,250 ng of total RNA using a TruSeq RNA preparation kit (Illumina). The size range of the RNA-seq libraries and their quality were checked using a 2100 Bioanalyzer (Agilent). Libraries were sequenced at the Stanford Stem Cell Institute Genome Center using the HiSeq 2000 platform (Illumina), which generated reads of 50 bp. The quality of the FASTQ files generated from the libraries was checked with FastQC (Babraham Bioinformatics) and aligned to the human genome (hg19) using STAR (87). The complete data set is available from the NCBI GEO repository (Reviewer access link, http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=uvwtskminpezzmz&acc=GSE85493). The RNA-seq pipeline in Partek Genomics Suite v6.6 (Partek Inc.) was used to analyze the BAM files generated by STAR and to generate RPKM values to determine differential gene expression. Statistically significant gene expression was calculated if there was a ≥2-fold change with a P value of <0.05 and a false discovery rate (FDR) of <0.001. RNA-seq reads were aligned to the VZV pOka genome (accession number AB097933) using Bowtie (88).
The BaseSpace Correlation Engine is part of the BaseSpace Informatics Suite (Illumina), which is a cloud-based solution to compare experimental omics molecular profiles with results from a large curated repository of open-access and controlled-access public data sets. This was used to generate biogroups for gene ontology, canonical pathways, regulatory motifs, and protein families. As defined by Illumina, a biogroup is a collection of genes that are associated with a specific biological function, pathway, or similar criteria. Numerical information is not directly associated with a biogroup. Gene lists represented as biogroups in the Correlation Engine were derived from gene ontology (biological processes, cellular components, and molecular functions), MSigDB (canonical pathways, positional gene sets, and regulatory motif gene sets), InterPro (protein families), and TargetScan (predicted miRNA targets). Subcategories of genes were ranked based on the number of genes that were differentially regulated within the subcategory, as determined at 36 hpi. Each biogroup with a P value of <0.0001 was identified (see the supplemental material), and the top 10 biogroups for each category were tabulated.
qRT-PCR for mRNA quantification.
The synthesis of cDNA from 1 μg of RNA was performed using oligo(dT) and Superscript III following the manufacturer's instructions (Invitrogen). qRT-PCR primers were designed using Primer-BLAST (http://www.ncbi.nlm.nih.gov/tools/primer-blast/) from the National Center for Biotechnology information (Table 5). The SSoAdvanced universal SYBR green supermix (Bio-Rad) was used for qRT-PCR with the CFX384 real-time PCR system (Bio-Rad). The CFX Manager Software (Bio-Rad) was used to calculate ΔCq for each gene.
TABLE 5.
Primers for quantification of Ras GTPase transcripts by qRT-PCR
| Gene | Primer name | Primer sequence | Product length (bp) |
|---|---|---|---|
| ARL4D | ARL4D-01F | TGTTTCACCGGGAAAGGCTC | 142 |
| ARL4D-01R | CGCCATCTCAGTCAAGTGGT | ||
| GEM | GEM-01F | AGAAAAGAACCCCTGGAACGTG | 173 |
| GEM-01R | GTACTGGTGGGGCTCTTTCTG | ||
| RASL11B | RASL11B-03F | TGGCTCTTCAGGTTCAAGACA | 113 |
| RASL11B-03R | GAAAACGATCACCACAGCATCT | ||
| RASD1 | RASD1-01F | ACTTCCACCGCAAGTTCTACT | 137 |
| RASD1-01R | ACTGAACACCAGGATGAAAACG | ||
| RHOV | RHOV-qPCR01 | GTCAGCTACACCTGCAATGG | 150 |
| RHOV-qPCR02 | AAGGGAACGAAGTCGGTCAA | ||
| RND1 | RND1-qPCR01 | AGACAGACCTGCGAACAGAC | 118 |
| RND1-qPCR02 | AGGTAGATTTCTGCACCCAGC | ||
| RRAD | RRAD-01F | AGCGTTTACAAGGTGCTGCT | 151 |
| RRAD-01R | GTGATGCCTCTTCTCCGTCC |
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge John Mulholland, Kitty Lee, and Cedric Espenel of the SCSIF and Somalee Datta, Ramesh Nair, and Yue Zhang of the Stanford Center for Genomics and Personalized Medicine. We thank Philip Lavori and Rob Tibshirani of the Department of Biomedicine Data Science for valuable discussions about data analysis. We also acknowledge Klaus Osterrieder (Freie Universität) for providing reagents.
This work was funded by NIH grants AI20459 and AI102546 and Stanford Cell Sciences and Imaging Facility electron microscopy equipment grant NIH SIG 1S10RR02678001.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JVI.01613-16.
REFERENCES
- 1.Arvin AM, Gilden D. 2013. Varicella Zoster Virus, p 2015–2184. In Fields BN, Knipe DM, Howley PM (ed), Fields virology, 6th ed, vol 2 Wolters Kluwer Health/Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Banovic T, Yanilla M, Simmons R, Robertson I, Schroder WA, Raffelt NC, Wilson YA, Hill GR, Hogan P, Nourse CB. 2011. Disseminated varicella infection caused by varicella vaccine strain in a child with low invariant natural killer T cells and diminished CD1d expression. J Infect Dis 204:1893–1901. doi: 10.1093/infdis/jir660. [DOI] [PubMed] [Google Scholar]
- 3.Gilden D, Cohrs RJ, Mahalingam R, Nagel MA. 2009. Varicella zoster virus vasculopathies: diverse clinical manifestations, laboratory features, pathogenesis, and treatment. Lancet Neurol 8:731–740. doi: 10.1016/S1474-4422(09)70134-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jean-Philippe P, Freedman A, Chang MW, Steinberg SP, Gershon AA, LaRussa PS, Borkowsky W. 2007. Severe varicella caused by varicella-vaccine strain in a child with significant T-cell dysfunction. Pediatrics 120:e1345–1349. doi: 10.1542/peds.2004-1681. [DOI] [PubMed] [Google Scholar]
- 5.Pahud BA, Glaser CA, Dekker CL, Arvin AM, Schmid DS. 2011. Varicella zoster disease of the central nervous system: epidemiological, clinical, and laboratory features 10 years after the introduction of the varicella vaccine. J Infect Dis 203:316–323. doi: 10.1093/infdis/jiq066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gilden DH, Mahalingham R, Deitch S, Randall CJ. 2006. Varicella zoster virus neuropathogenesis and latency, p 402 In Sandri-Goldin RM. (ed), Alpha herpesviruses: molecular and cellular biology. Caister Academic, Wymondham, United Kingdom. [Google Scholar]
- 7.Johnson RW. 2007. Zoster-associated pain: what is known, who is at risk and how can it be managed? Herpes 14(Suppl 2):S30–S34. [PubMed] [Google Scholar]
- 8.Schmader K. 2007. Herpes zoster and postherpetic neuralgia in older adults. Clin Geriatr Med 23:615–632, vii–viii. doi: 10.1016/j.cger.2007.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brunell PA, Geiser CF, Novelli V, Lipton S, Narkewicz S. 1987. Varicella-like illness caused by live varicella vaccine in children with acute lymphocytic leukemia. Pediatrics 79:922–927. [PubMed] [Google Scholar]
- 10.Marin M, Meissner HC, Seward JF. 2008. Varicella prevention in the United States: a review of successes and challenges. Pediatrics 122:e744–751. doi: 10.1542/peds.2008-0567. [DOI] [PubMed] [Google Scholar]
- 11.Pickering LK, Baker CJ, Freed GL, Gall SA, Grogg SE, Poland GA, Rodewald LE, Schaffner W, Stinchfield P, Tan L, Zimmerman RK, Orenstein WA, Infectious Diseases Society of America . 2009. Immunization programs for infants, children, adolescents, and adults: clinical practice guidelines by the Infectious Diseases Society of America. Clin Infect Dis 49:817–840. doi: 10.1086/605430. [DOI] [PubMed] [Google Scholar]
- 12.Han JY, Hanson DC, Way SS. 2011. Herpes zoster and meningitis due to reactivation of varicella vaccine virus in an immunocompetent child. Pediatr Infect Dis J 30:266–268. doi: 10.1097/INF.0b013e3181f63cf9. [DOI] [PubMed] [Google Scholar]
- 13.Krause PR, Klinman DM. 2000. Varicella vaccination: evidence for frequent reactivation of the vaccine strain in healthy children. Nat Med 6:451–454. doi: 10.1038/74715. [DOI] [PubMed] [Google Scholar]
- 14.Whitley RJ. 2008. Therapy of herpes virus infections in children. Adv Exp Med Biol 609:216–232. doi: 10.1007/978-0-387-73960-1_16. [DOI] [PubMed] [Google Scholar]
- 15.Whitley RJ, Volpi A, McKendrick M, Wijck A, Oaklander AL. 2010. Management of herpes zoster and post-herpetic neuralgia now and in the future. J Clin Virol 4(Suppl 1):S20–S28. [DOI] [PubMed] [Google Scholar]
- 16.Arvin AM, Moffat JF, Sommer M, Oliver S, Che X, Vleck S, Zerboni L, Ku CC. 2010. Varicella-zoster virus T cell tropism and the pathogenesis of skin infection. Curr Top Microbiol Immunol 342:189–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moffat J, Ku CC, Zerboni L, Sommer M, Arvin A. 2007. VZV: pathogenesis and the disease consequences of primary infection. In Arvin A, Campadelli-Fiume G, Mocarski E, Moore PS, Roizman B, Whitley R, Yamanishi K (ed), Human herpesviruses: biology, therapy, and immunoprophylaxis. Cambridge University Press, Cambridge, United Kingdom. [PubMed] [Google Scholar]
- 18.Zerboni L, Reichelt M, Arvin A. 2010. Varicella-zoster virus neurotropism in SCID mouse-human dorsal root ganglia xenografts. Curr Top Microbiol Immunol 342:255–276. [DOI] [PubMed] [Google Scholar]
- 19.Weigle KA, Grose C. 1983. Common expression of varicella-zoster viral glycoprotein antigens in vitro and in chickenpox and zoster vesicles. J Infect Dis 148:630–638. doi: 10.1093/infdis/148.4.630. [DOI] [PubMed] [Google Scholar]
- 20.Zerboni L, Reichelt M, Jones CD, Zehnder JL, Ito H, Arvin AM. 2007. Aberrant infection and persistence of varicella-zoster virus in human dorsal root ganglia in vivo in the absence of glycoprotein I. Proc Natl Acad Sci U S A 104:14086–14091. doi: 10.1073/pnas.0706023104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Besser J, Ikoma M, Fabel K, Sommer MH, Zerboni L, Grose C, Arvin AM. 2004. Differential requirement for cell fusion and virion formation in the pathogenesis of varicella-zoster virus infection in skin and T cells. J Virol 78:13293–13305. doi: 10.1128/JVI.78.23.13293-13305.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moffat JF, Stein MD, Kaneshima H, Arvin AM. 1995. Tropism of varicella-zoster virus for human CD4+ and CD8+ T lymphocytes and epidermal cells in SCID-hu mice. J Virol 69:5236–5242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oliver SL, Sommer MH, Reichelt M, Rajamani J, Vlaycheva-Beisheim L, Stamatis S, Cheng J, Jones C, Zehnder J, Arvin AM. 2011. Mutagenesis of varicella-zoster virus glycoprotein I (gI) identifies a cysteine residue critical for gE/gI heterodimer formation, gI structure, and virulence in skin cells. J Virol 85:4095–4110. doi: 10.1128/JVI.02596-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weller TH, Witton HM, Bell EJ. 1958. The etiologic agents of varicella and herpes zoster; isolation, propagation, and cultural characteristics in vitro. J Exp Med 108:843–868. doi: 10.1084/jem.108.6.843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zerboni L, Ku CC, Jones CD, Zehnder JL, Arvin AM. 2005. Varicella-zoster virus infection of human dorsal root ganglia in vivo. Proc Natl Acad Sci U S A 102:6490–6495. doi: 10.1073/pnas.0501045102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Connolly SA, Jackson JO, Jardetzky TS, Longnecker R. 2011. Fusing structure and function: a structural view of the herpesvirus entry machinery. Nat Rev Microbiol 9:369–381. doi: 10.1038/nrmicro2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Backovic M, Longnecker R, Jardetzky TS. 2009. Structure of a trimeric variant of the Epstein-Barr virus glycoprotein B. Proc Natl Acad Sci U S A 106:2880–2885. doi: 10.1073/pnas.0810530106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Burke HG, Heldwein EE. 2015. Crystal structure of the human cytomegalovirus glycoprotein B. PLoS Pathog 11:e1005227. doi: 10.1371/journal.ppat.1005227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chandramouli S, Ciferri C, Nikitin PA, Calo S, Gerrein R, Balabanis K, Monroe J, Hebner C, Lilja AE, Settembre EC, Carfi A. 2015. Structure of HCMV glycoprotein B in the postfusion conformation bound to a neutralizing human antibody. Nat Commun 6:8176. doi: 10.1038/ncomms9176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heldwein EE, Lou H, Bender FC, Cohen GH, Eisenberg RJ, Harrison SC. 2006. Crystal structure of glycoprotein B from herpes simplex virus 1. Science 313:217–220. doi: 10.1126/science.1126548. [DOI] [PubMed] [Google Scholar]
- 31.Birlea M, Owens GP, Eshleman EM, Ritchie A, Traktinskiy I, Bos N, Seitz S, Azarkh Y, Mahalingam R, Gilden D, Cohrs RJ. 2013. Human anti-varicella-zoster virus (VZV) recombinant monoclonal antibody produced after Zostavax immunization recognizes the gH/gL complex and neutralizes VZV infection. J Virol 87:415–421. doi: 10.1128/JVI.02561-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Suzuki K, Akahori Y, Asano Y, Kurosawa Y, Shiraki K. 2007. Isolation of therapeutic human monoclonal antibodies for varicella-zoster virus and the effect of light chains on the neutralizing activity. J Med Virol 79:852–862. doi: 10.1002/jmv.20838. [DOI] [PubMed] [Google Scholar]
- 33.Xing Y, Oliver SL, Nguyen T, Ciferri C, Nandi A, Hickman J, Giovani C, Yang E, Palladino G, Grose C, Uematsu Y, Lilja AE, Arvin AM, Carfi A. 2015. A site of varicella-zoster virus vulnerability identified by structural studies of neutralizing antibodies bound to the glycoprotein complex gHgL. Proc Natl Acad Sci U S A 112:6056–6061. doi: 10.1073/pnas.1501176112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Suenaga T, Satoh T, Somboonthum P, Kawaguchi Y, Mori Y, Arase H. 2010. Myelin-associated glycoprotein mediates membrane fusion and entry of neurotropic herpesviruses. Proc Natl Acad Sci U S A 107:866–871. doi: 10.1073/pnas.0913351107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vleck SE, Oliver SL, Brady JJ, Blau HM, Rajamani J, Sommer MH, Arvin AM. 2011. Structure-function analysis of varicella-zoster virus glycoprotein H identifies domain-specific roles for fusion and skin tropism. Proc Natl Acad Sci U S A 108:18412–18417. doi: 10.1073/pnas.1111333108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oliver SL, Brady JJ, Sommer MH, Reichelt M, Sung P, Blau HM, Arvin AM. 2013. An immunoreceptor tyrosine-based inhibition motif in varicella-zoster virus glycoprotein B regulates cell fusion and skin pathogenesis. Proc Natl Acad Sci U S A 110:1911–1916. doi: 10.1073/pnas.1216985110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barrow AD, Trowsdale J. 2006. You say ITAM and I say ITIM, let's call the whole thing off: the ambiguity of immunoreceptor signalling. Eur J Immunol 36:1646–1653. doi: 10.1002/eji.200636195. [DOI] [PubMed] [Google Scholar]
- 38.Daeron M, Jaeger S, Du Pasquier L, Vivier E. 2008. Immunoreceptor tyrosine-based inhibition motifs: a quest in the past and future. Immunol Rev 224:11–43. doi: 10.1111/j.1600-065X.2008.00666.x. [DOI] [PubMed] [Google Scholar]
- 39.O'Brien CD, Cao G, Makrigiannakis A, DeLisser HM. 2004. Role of immunoreceptor tyrosine-based inhibitory motifs of PECAM-1 in PECAM-1-dependent cell migration. Am J Physiol Cell Physiol 287:C1103-1113. doi: 10.1152/ajpcell.00573.2003. [DOI] [PubMed] [Google Scholar]
- 40.Reichelt M, Brady J, Arvin AM. 2009. The replication cycle of varicella-zoster virus: analysis of the kinetics of viral protein expression, genome synthesis, and virion assembly at the single-cell level. J Virol 83:3904–3918. doi: 10.1128/JVI.02137-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Storlie J, Jackson W, Hutchinson J, Grose C. 2006. Delayed biosynthesis of varicella-zoster virus glycoprotein C: upregulation by hexamethylene bisacetamide and retinoic acid treatment of infected cells. J Virol 80:9544–9556. doi: 10.1128/JVI.00668-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Storlie J, Carpenter JE, Jackson W, Grose C. 2008. Discordant varicella-zoster virus glycoprotein C expression and localization between cultured cells and human skin vesicles. Virology 382:171–181. doi: 10.1016/j.virol.2008.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Heineman TC, Hall SL. 2002. Role of the varicella-zoster virus gB cytoplasmic domain in gB transport and viral egress. J Virol 76:591–599. doi: 10.1128/JVI.76.2.591-599.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bosse JB, Virding S, Thiberge SY, Scherer J, Wodrich H, Ruzsics Z, Koszinowski UH, Enquist LW. 2014. Nuclear herpesvirus capsid motility is not dependent on F-actin. mBio 5:e01909-14. doi: 10.1128/mBio.01909-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Johnson DC, Baines JD. 2011. Herpesviruses remodel host membranes for virus egress. Nat Rev Microbiol 9:382–394. doi: 10.1038/nrmicro2559. [DOI] [PubMed] [Google Scholar]
- 46.Albecka A, Laine RF, Janssen AF, Kaminski CF, Crump CM. 2016. HSV-1 glycoproteins are delivered to virus assembly sites through dynamin-dependent endocytosis. Traffic 17:21–39. doi: 10.1111/tra.12340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Arvin AM, Oliver S, Reichelt M, Moffat JF, Sommer M, Zerboni L, Berarducci B. 2010. Analysis of the functions of glycoproteins E and I and their promoters during VZV replication in vitro and in skin and T-cell xenografts in the SCID mouse model of VZV pathogenesis. Curr Top Microbiol Immunol 342:129–146. [DOI] [PubMed] [Google Scholar]
- 48.Baird NL, Bowlin JL, Cohrs RJ, Gilden D, Jones KL. 2014. Comparison of varicella-zoster virus RNA sequences in human neurons and fibroblasts. J Virol 88:5877–5880. doi: 10.1128/JVI.00476-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jones M, Dry IR, Frampton D, Singh M, Kanda RK, Yee MB, Kellam P, Hollinshead M, Kinchington PR, O'Toole EA, Breuer J. 2014. RNA-seq analysis of host and viral gene expression highlights interaction between varicella zoster virus and keratinocyte differentiation. PLoS Pathog 10:e1003896. doi: 10.1371/journal.ppat.1003896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang L, Sommer M, Rajamani J, Arvin AM. 2009. Regulation of the ORF61 promoter and ORF61 functions in varicella-zoster virus replication and pathogenesis. J Virol 83:7560–7572. doi: 10.1128/JVI.00118-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rahaus M, Desloges N, Wolff MH. 2005. ORF61 protein of varicella-zoster virus influences JNK/SAPK and p38/MAPK phosphorylation. J Med Virol 76:424–433. doi: 10.1002/jmv.20373. [DOI] [PubMed] [Google Scholar]
- 52.Walters MS, Kyratsous CA, Silverstein SJ. 2010. The RING finger domain of varicella-zoster virus ORF61p has E3 ubiquitin ligase activity that is essential for efficient autoubiquitination and dispersion of Sp100-containing nuclear bodies. J Virol 84:6861–6865. doi: 10.1128/JVI.00335-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Walters MS, Kyratsous CA, Wan S, Silverstein S. 2008. Nuclear import of the varicella-zoster virus latency-associated protein ORF63 in primary neurons requires expression of the lytic protein ORF61 and occurs in a proteasome-dependent manner. J Virol 82:8673–8686. doi: 10.1128/JVI.00685-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhu H, Zheng C, Xing J, Wang S, Li S, Lin R, Mossman KL. 2011. Varicella-zoster virus immediate-early protein ORF61 abrogates the IRF3-mediated innate immune response through degradation of activated IRF3. J Virol 85:11079–11089. doi: 10.1128/JVI.05098-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang L, Oliver SL, Sommer M, Rajamani J, Reichelt M, Arvin AM. 2011. Disruption of PML nuclear bodies is mediated by ORF61 SUMO-interacting motifs and required for varicella-zoster virus pathogenesis in skin. PLoS Pathog 7:e1002157. doi: 10.1371/journal.ppat.1002157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Moffat JF, Zerboni L, Kinchington PR, Grose C, Kaneshima H, Arvin AM. 1998. Attenuation of the vaccine Oka strain of varicella-zoster virus and role of glycoprotein C in alphaherpesvirus virulence demonstrated in the SCID-hu mouse. J Virol 72:965–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang Z, Selariu A, Warden C, Huang G, Huang Y, Zaccheus O, Cheng T, Xia N, Zhu H. 2010. Genome-wide mutagenesis reveals that ORF7 is a novel VZV skin-tropic factor. PLoS Pathog 6:e1000971. doi: 10.1371/journal.ppat.1000971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mallory S, Sommer M, Arvin AM. 1997. Mutational analysis of the role of glycoprotein I in varicella-zoster virus replication and its effects on glycoprotein E conformation and trafficking. J Virol 71:8279–8288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Berarducci B, Sommer M, Zerboni L, Rajamani J, Arvin AM. 2007. Cellular and viral factors regulate the varicella-zoster virus gE promoter during viral replication. J Virol 81:10258–10267. doi: 10.1128/JVI.00553-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Moffat J, Ito H, Sommer M, Taylor S, Arvin AM. 2002. Glycoprotein I of varicella-zoster virus is required for viral replication in skin and T cells. J Virol 76:8468–8471. doi: 10.1128/JVI.76.16.8468-8471.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Berarducci B, Rajamani J, Reichelt M, Sommer M, Zerboni L, Arvin AM. 2009. Deletion of the first cysteine-rich region of the varicella-zoster virus glycoprotein E ectodomain abolishes the gE and gI interaction and differentially affects cell-cell spread and viral entry. J Virol 83:228–240. doi: 10.1128/JVI.00913-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Berarducci B, Rajamani J, Zerboni L, Che X, Sommer M, Arvin AM. 2010. Functions of the unique N-terminal region of glycoprotein E in the pathogenesis of varicella-zoster virus infection. Proc Natl Acad Sci U S A 107:282–287. doi: 10.1073/pnas.0912373107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jones JO, Arvin AM. 2003. Microarray analysis of host cell gene transcription in response to varicella-zoster virus infection of human T cells and fibroblasts in vitro and SCIDhu skin xenografts in vivo. J Virol 77:1268–1280. doi: 10.1128/JVI.77.2.1268-1280.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kim JA, Park SK, Kumar M, Lee CH, Shin OS. 2015. Insights into the role of immunosenescence during varicella zoster virus infection (shingles) in the aging cell model. Oncotarget 6:35324–35343. doi: 10.18632/oncotarget.6117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yang E, Arvin AM, Oliver SL. 2016. Role for the alphaV integrin subunit in varicella-zoster virus-mediated fusion and infection. J Virol 90:7567–7578. doi: 10.1128/JVI.00792-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hofmann I, Thompson A, Sanderson CM, Munro S. 2007. The Arl4 family of small G proteins can recruit the cytohesin Arf6 exchange factors to the plasma membrane. Curr Biol 17:711–716. doi: 10.1016/j.cub.2007.03.007. [DOI] [PubMed] [Google Scholar]
- 67.D'Souza-Schorey C, Li G, Colombo MI, Stahl PD. 1995. A regulatory role for ARF6 in receptor-mediated endocytosis. Science 267:1175–1178. doi: 10.1126/science.7855600. [DOI] [PubMed] [Google Scholar]
- 68.Palacios F, Price L, Schweitzer J, Collard JG, D'Souza-Schorey C. 2001. An essential role for ARF6-regulated membrane traffic in adherens junction turnover and epithelial cell migration. EMBO J 20:4973–4986. doi: 10.1093/emboj/20.17.4973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Radhakrishna H, Klausner RD, Donaldson JG. 1996. Aluminum fluoride stimulates surface protrusions in cells overexpressing the ARF6 GTPase. J Cell Biol 134:935–947. doi: 10.1083/jcb.134.4.935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ward Y, Yap SF, Ravichandran V, Matsumura F, Ito M, Spinelli B, Kelly K. 2002. The GTP binding proteins Gem and Rad are negative regulators of the Rho-Rho kinase pathway. J Cell Biol 157:291–302. doi: 10.1083/jcb.200111026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Beguin P, Mahalakshmi RN, Nagashima K, Cher DH, Ikeda H, Yamada Y, Seino Y, Hunziker W. 2006. Nuclear sequestration of beta-subunits by Rad and Rem is controlled by 14-3-3 and calmodulin and reveals a novel mechanism for Ca2+ channel regulation. J Mol Biol 355:34–46. doi: 10.1016/j.jmb.2005.10.013. [DOI] [PubMed] [Google Scholar]
- 72.Pan JY, Fieles WE, White AM, Egerton MM, Silberstein DS. 2000. Ges, A human GTPase of the Rad/Gem/Kir family, promotes endothelial cell sprouting and cytoskeleton reorganization. J Cell Biol 149:1107–1116. doi: 10.1083/jcb.149.5.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Piddini E, Schmid JA, de Martin R, Dotti CG. 2001. The Ras-like GTPase Gem is involved in cell shape remodelling and interacts with the novel kinesin-like protein KIF9. EMBO J 20:4076–4087. doi: 10.1093/emboj/20.15.4076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Beguin P, Mahalakshmi RN, Nagashima K, Cher DH, Takahashi A, Yamada Y, Seino Y, Hunziker W. 2005. 14-3-3 and calmodulin control subcellular distribution of Kir/Gem and its regulation of cell shape and calcium channel activity. J Cell Sci 118:1923–1934. doi: 10.1242/jcs.02321. [DOI] [PubMed] [Google Scholar]
- 75.Mahalakshmi RN, Ng MY, Guo K, Qi Z, Hunziker W, Beguin P. 2007. Nuclear localization of endogenous RGK proteins and modulation of cell shape remodeling by regulated nuclear transport. Traffic 8:1164–1178. doi: 10.1111/j.1600-0854.2007.00599.x. [DOI] [PubMed] [Google Scholar]
- 76.Graham TE, Prossnitz ER, Dorin RI. 2002. Dexras1/AGS-1 inhibits signal transduction from the Gi-coupled formyl peptide receptor to Erk-1/2 MAP kinases. J Biol Chem 277:10876–10882. doi: 10.1074/jbc.M110397200. [DOI] [PubMed] [Google Scholar]
- 77.Kemppainen RJ, Behrend EN. 1998. Dexamethasone rapidly induces a novel ras superfamily member-related gene in AtT-20 cells. J Biol Chem 273:3129–3131. doi: 10.1074/jbc.273.6.3129. [DOI] [PubMed] [Google Scholar]
- 78.Takesono A, Nowak MW, Cismowski M, Duzic E, Lanier SM. 2002. Activator of G-protein signaling 1 blocks GIRK channel activation by a G-protein-coupled receptor: apparent disruption of receptor signaling complexes. J Biol Chem 277:13827–13830. doi: 10.1074/jbc.M201064200. [DOI] [PubMed] [Google Scholar]
- 79.Stolle K, Schnoor M, Fuellen G, Spitzer M, Cullen P, Lorkowski S. 2007. Cloning, genomic organization, and tissue-specific expression of the RASL11B gene. Biochim Biophys Acta 1769:514–524. doi: 10.1016/j.bbaexp.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 80.Faure S, Fort P. 2011. Atypical RhoV and RhoU GTPases control development of the neural crest. Small GTPases 2:310–313. doi: 10.4161/sgtp.18086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nobes CD, Lauritzen I, Mattei MG, Paris S, Hall A, Chardin P. 1998. A new member of the Rho family, Rnd1, promotes disassembly of actin filament structures and loss of cell adhesion. J Cell Biol 141:187–197. doi: 10.1083/jcb.141.1.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Riou P, Villalonga P, Ridley AJ. 2010. Rnd proteins: multifunctional regulators of the cytoskeleton and cell cycle progression. Bioessays 32:986–992. doi: 10.1002/bies.201000060. [DOI] [PubMed] [Google Scholar]
- 83.Wang H, Hota PK, Tong Y, Li B, Shen L, Nedyalkova L, Borthakur S, Kim S, Tempel W, Buck M, Park HW. 2011. Structural basis of Rnd1 binding to plexin Rho GTPase binding domains (RBDs). J Biol Chem 286:26093–26106. doi: 10.1074/jbc.M110.197053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wang Y, He H, Srivastava N, Vikarunnessa S, Chen YB, Jiang J, Cowan CW, Zhang X. 2012. Plexins are GTPase-activating proteins for Rap and are activated by induced dimerization. Sci Signal 5:ra6. doi: 10.1126/scisignal.2002636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yang E, Arvin AM, Oliver SL. 2014. The cytoplasmic domain of varicella-zoster virus glycoprotein H regulates syncytium formation and skin pathogenesis. PLoS Pathog 10:e1004173. doi: 10.1371/journal.ppat.1004173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tischer BK, Kaufer BB, Sommer M, Wussow F, Arvin AM, Osterrieder N. 2007. A self-excisable infectious bacterial artificial chromosome clone of varicella-zoster virus allows analysis of the essential tegument protein encoded by ORF9. J Virol 81:13200–13208. doi: 10.1128/JVI.01148-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.