

ABSTRACT
20 (enfuvirtide) and other peptides derived from the human immunodeficiency virus type 1 (HIV-1) gp41 C-terminal heptad repeat (CHR) region inhibit HIV fusion by binding to the hydrophobic grooves on the N-terminal heptad repeat (NHR) trimer and blocking six-helix-bundle (6-HB) formation. Several strategies focusing on the binding grooves of the NHR trimer have been adopted to increase the antiviral activity of the CHR peptides. Here, we developed a novel and simple strategy to greatly enhance the potency of the existing peptide-based HIV fusion inhibitors. First, we identified a shallow pocket adjacent to the groove in the N-terminal region of NHR trimer as a new drug target, and then we designed several short artificial peptides to fit this target. After the addition of IDL (Ile-Asp-Leu) to the C terminus of CHR peptide WQ or MT-WQ, the conjugated peptides, WQ-IDL and MT-WQ-IDL, showed much more potent activities than WQ and T20, respectively, in inhibiting HIV-1 IIIB infection. WQ-IDL and MT-WQ-IDL were also more effective than WQ in blocking HIV-1 Env-mediated membrane fusion and had higher levels of binding affinity with NHR peptide N46. We solved the crystal structure of the 6-HB formed by MT-WQ-IDL and N46 and found that, besides the N-terminal MT hook tail, the IDL tail anchor of MT-WQ-IDL also binds with the shallow hydrophobic pocket outside the groove of the NHR trimer, resulting in enhanced inhibition of HIV-1 fusion with the target cell. It is expected that this novel approach can be widely used to improve the potency of peptidic fusion inhibitors against other enveloped viruses with class I fusion proteins.
IMPORTANCE The hydrophobic groove of the human immunodeficiency virus type 1 (HIV-1) gp41 NHR trimer has been known as the classic drug target to develop fusion inhibitors derived from the gp41 CHR. Here, we developed a novel and simple strategy to improve the existing peptide-based HIV fusion inhibitors. We identified a shallow pocket adjacent to the groove in the NHR trimer and added a short artificial peptide consisting of three amino acids (IDL) to the C terminus of a fusion inhibitor to fit this new target. The inhibition activity of this new conjugated peptide was significantly enhanced, by 77-fold, making it much more potent than T20 (enfuvirtide) and suggesting that the IDL tail can be adopted for optimizing existing HIV-1 CHR peptide fusion inhibitors. This new approach of identifying a potential binding pocket outside the traditional target and creating an artificial tail anchor can be widely applied to design novel fusion inhibitors against other class I enveloped viruses, such as Middle East respiratory syndrome coronavirus (MERS-CoV).
KEYWORDS: HIV, gp41, fusion inhibitor, six-helix bundle, peptide
INTRODUCTION
Human immunodeficiency virus (HIV) entry into the target cell is initiated by binding of the viral envelope glycoprotein (Env) surface subunit gp120 to the receptor CD4 and a coreceptor (CCR5 or CXCR4) on the target cell, triggering a cascade of conformational changes of the transmembrane subunit gp41. The C-terminal heptad repeat (CHR) and N-terminal heptad repeat (NHR) of gp41 interact with each other (Fig. 1A and B) to form a stable six-helix bundle (6-HB), in which three CHR helices pack into the hydrophobic grooves on the NHR trimer core in an antiparallel way to pull viral and target cell membranes into close proximity for fusion (1, 2).
FIG 1.

Schematic illustration of HIV-1 gp41 and 6-HB. (A) Domain distribution of HIV-1 gp41. CP, cytoplasm region; TM, transmembrane region; TR, tryptophan-rich region; CHR, C-terminal heptad repeat; PBD, pocket binding domain; NHR, N-terminal heptad repeat; PFD, pocket-forming domain; FP, fusion peptide region. (B) NHR and CHR bind through residues at the a and d sites and the e and g sites. (C) Crystal structure of gp41 NHR trimer (PDB 2X7R) is shown as an electrostatic surface. Red circle indicates N-terminal hydrophobic pocket formed by aa 544 to 549 of NHR, a potential new binding target on gp41 NHR for a fusion/entry inhibitor. Purple circle indicates the NHR C-terminal hydrophobic pocket. Green box indicates the hydrophobic groove for CHR binding.
In the early 1990s, Jiang et al. (3) reported that peptides derived from the gp41 CHR sequence exhibit potent anti-HIV type 1 (HIV-1) activity. One of the CHR peptides, T20 (enfuvirtide [Fuzeon]; Genentech), was approved by the U.S. FDA as the first HIV fusion/entry inhibitor for treatment of HIV-infected patients who fail to respond to the current antiretroviral drugs.
It has been reported that HIV-1 can spread directly from productively infected cells to neighboring cells via virological synapses, a process that is 100- to 1,000-fold more efficient than infection with cell-free virions (4). Recent studies have shown that HIV cell-cell transmission is responsible for the death of most (∼95%) CD4 T cells (5) and that the deletion of T cells is the main reason for most symptoms of AIDS (6). Therefore, entry/fusion inhibitors like T20 that can effectively block the cell-cell fusion induced by HIV Env have significant advantages in treating HIV infection and AIDS. However, the clinical application of T20 is limited by its low potency, high dosage, and susceptibility to drug resistance (7, 8). Therefore, developing new peptide fusion inhibitors with improved anti-HIV-1 activities and drug resistance profiles remains urgent.
Several strategies have been developed to optimize CHR-derived inhibitors, such as the introduction of EE-KK double mutations and salt bridges to enhance peptides' α-helicity and solubility (9–11), conversion of α-amino acids to β-amino acids to strengthen their antiprotease ability (12), and stabilization of CHR peptides by constructing trimers (13). Point mutations have been introduced to enhance the binding of CHR peptides to the hydrophobic grooves on the NHR trimer (14). A pocket-binding domain (PBD) was added to the N terminus of a CHR peptide (e.g., T20) to strengthen the interaction between the CHR peptide and the deep hydrophobic pocket in the C-terminal groove region on the NHR trimer (15). Adding Met and Thr, which form a hooklike structure, to the N terminus of the PBD in a CHR peptide could further increase the binding affinity of an inhibitor to the C terminus of the NHR trimer (16). Based on these results, we speculated that increasing the binding affinity of an inhibitor to the N terminus of the NHR trimer could also significantly improve the inhibitory activity. However, this strategy has never been reported yet, implying a significant challenge in enhancing interaction between an inhibitor and the N-terminal region of NHR, unless we could find more target binding site(s) besides the traditional hydrophobic groove.
In this study, we identified a new hydrophobic pocket outside the hydrophobic groove in the N-terminal region of the NHR trimer as an ideal new target to improve the activity of an inhibitor. Then, we intentionally added 2 or 3 artificial amino acid residues to the C terminus of a CHR peptide to act as a tail anchor to fit the new pocket-binding target. We found that this tail anchor could significantly enhance the inhibitory activity, by up to 77-fold. These results confirmed our assumptions and proved the great potential of this strategy in inhibitor optimization.
RESULTS AND DISCUSSION
Addition of an artificial peptide tail anchor, IDL (Ile-Asp-Leu), to the C terminus of the WQ peptide resulted in enhanced anti-HIV-1 activity.
To identify a new binding pocket outside the hydrophobic grooves of the NHR trimer as a novel target, we used computer modeling (17) to analyze the crystal structure of the NHR trimer (18) and observed a shallow pocket in the N-terminal region (L544 to V549) of the NHR domain, designated the N-terminal pocket (Fig. 1C). Since it is located far from the binding groove, CHR residues, such as Q653, cannot reach and bind to it. The distance between Q653 and the N-terminal pocket could accommodate 2 to 4 amino acid residues. Certain hydrophobic residues with long side chains, such as Met, Trp, Tyr, Ile, and Leu, are needed to bind to this hydrophobic pocket. Considering the possibility that this artificial tail might adopt a β-strand-like conformation, hydrophilic residues should also be included between adjacent hydrophobic ones.
Accordingly, we designed 5 artificial peptide tails, each containing 2 to 4 amino acid residues, including SM, SW, SY, SDLD, and LDL, and linked these peptide tails individually to the C terminus of WQ, a 26-mer CHR-derived peptide (Fig. 1A). These conjugated peptides were tested for their levels of inhibitory activity against infection by HIV-1 IIIB, a tier 1 X4 strain. As shown by the results in Fig. 2, the anti-HIV-1 IIIB activities of peptides WQ-SM, WQ-SW, WQ-SY, and WQ-SDLD were slightly (2- to 3-fold) better than that of WQ, while the WQ-LDL peptide was about 33-fold more potent than WQ.
FIG 2.

Inhibitory activities of the conjugated peptides against HIV-1 infection. (A) Inhibitory activities of WQ-SM, WQ-SW, WQ-SY, WQ-SDLD, WQ-LDL, and WQ against HIV-1 IIIB (X4 virus) infection. (B) Inhibitory activities of WQ, WQ-LDL, WQ-IDI, WQ-LDI, WQ-IDL, and T20 against HIV-1 Bal (R5 virus) infection. (C) Inhibitory activities of WQ, WQ-IDL, MT-WQ-IDL, MT-WQ, and MT-WQ-IDL scrambled peptide against HIV-1 Bal (R5 virus) infection. (D) Inhibitory activities of peptides against cell-cell fusion between H9/HIV-1IIIB cells and MT-2 cells. Error bars show standard deviations.
Based on the success of WQ-LDL, we further designed WQ-IDI, WQ-LDI, and WQ-IDL peptides (Fig. 1A) and compared their antiviral activities against HIV-1 IIIB. As shown by the results in Table 1, peptides WQ-IDI and WQ-LDI showed antiviral activities similar to that of WQ-LDL, while WQ-IDL was about 2.3-, 77.1-, and 44.9-fold more effective than WQ-LDL, WQ, and T20, respectively. This result suggests that IDL is the most effective peptide tail for binding the CHR peptide WQ to the newly identified N-terminal pocket on the NHR trimer.
TABLE 1.
Inhibitory activities and helicity of conjugated peptides and control peptides
| Peptide | Mean IC50 ± SD (nM)a |
Secondary structure (with N46) |
|||
|---|---|---|---|---|---|
| HIV-1 IIIB (tier 1) | HIV-1 Bal (tier 1) | Cell-cell fusion | Helicity (%) | Tm (°C) | |
| WQ | 123.4 ± 6.5 | 126.2 ± 9.2 | 159.5 ± 9.5 | 83.8 | 65.1 |
| WQ-LDL | 3.7 ± 1.1 | 3.6 ± 0.2 | 6.4 ± 2.7 | 86.0 | 80.9 |
| WQ-IDI | 3.7 ± 0.3 | 3.2 ± 0.2 | 6.4 ± 1.8 | 85.2 | 81.9 |
| WQ-LDI | 3.6 ± 0.2 | 3.1 ± 0.1 | 7.2 ± 1.0 | 84.2 | 80.5 |
| WQ-IDL | 1.6 ± 0.1 | 2.5 ± 0.1 | 5.6 ± 1.2 | 85.1 | 81.8 |
| MT-WQ-IDL | 0.6 ± 0.1 | 0.6 ± 0.1 | 1.2 ± 0.2 | 90.8 | 90.7 |
| MT-WQ | 8.8 ± 0.4 | 6.7 ± 0.5 | 14.8 ± 2.8 | 84.4 | 74.8 |
| T20 | 71.8 ± 2.2 | 70.5 ± 0.9 | 91.0 ± 1.2 | NAb | NA |
| C34 | 7.2 ± 0.2 | 7.6 ± 0.2 | 18.8 ± 1.6 | 85.0 | 67.9 |
IC50 data were derived from the results of three independent experiments.
NA, not applicable.
We then compared the antiviral activities of the conjugated peptides WQ-LDL, WQ-IDI, WQ-LDI, and WQ-IDL with those of WQ and T20 against infection by HIV-1 Bal, a tier 1 R5 strain. As shown by the results in Fig. 2B and Table 1, all four conjugated peptides were much more effective in inhibiting HIV-1 Bal infection than WQ or T20. WQ-IDL was the most effective among those four peptides, with a 50% inhibitory concentration (IC50) of 2.5 nM, about 50- and 28-fold more potent than WQ and T20, respectively, further confirming that the addition of the IDL peptide tail to the C terminus of a CHR peptide can enhance its anti-HIV-1 activity.
A previous study revealed that the addition of Met and Thr residues to the N terminus of SC22EK, a CHR peptide, could strengthen its activity because these two residues can form a hooklike structure that allows the N-terminal region of the CHR peptide to anchor tightly into the deep hydrophobic pocket in the groove of the C-terminal region of the NHR trimer (19). In this study, we also added Met and Thr to the N terminus of WQ-IDL and tested the antiviral activity of MT-WQ-IDL against HIV-1 IIIB infection. As expected, the IC50 of MT-WQ-IDL reached as low as 0.6 nM for inhibiting HIV-1 IIIB infection, which is about 2.7-, 206-, 14.7-, 12.0-, and 120-fold more potent than WQ-IDL, WQ, MT-WQ, C34, and T20, respectively (Table 1). Meanwhile, the scrambled version of peptide MT-WQ-IDL (Fig. 1A) had no anti-HIV-1 activity. This result indicated that the arrangement of the amino acid residues is highly relevant to its inhibition activity. Similarly, MT-WQ-IDL was about 4.2-, 210-, 11.2-, 12.7-, and 118-fold more potent than WQ-IDL, WQ, MT-WQ, C34, and T20, respectively, for inhibiting HIV-1 Bal infection (Fig. 2C), while the scrambled MT-WQ-IDL peptide showed no inhibition activity. These results suggest that the simultaneous addition of both MT and IDL tails to the N and C termini, respectively, of the WQ peptide can further increase the potency of WQ, possibly because these two tails allow the N- and C-terminal regions of the peptide to bind more strongly and stably with the C- and N-terminal pockets, respectively, on the NHR trimer.
The IDL-conjugated peptides were more effective than the CHR peptides without an IDL tail in inhibiting HIV-1 Env-mediated cell-cell fusion.
We then compared the WQ-IDL and MT-WQ-IDL peptides to the CHR peptides without tails, including WQ, MT-WQ, C34, and T20, for their inhibitory activities against HIV-1 Env-mediated cell-cell fusion, using MT-2 cells that express CD4 and coreceptor CXCR4 and H9/HIV-1IIIB cells that stably express HIV-1 Env protein as the target and effector cells, respectively (20). The result showed that H9/HIV-1IIIB cells fused with MT-2 cells in the absence of peptide fusion inhibitors and that this fusion process was blocked by WQ-IDL in a dose-dependent way. As shown by the results in Table 1 and Fig. 2D, the IC50 of the WQ-IDL peptide was 5.6 nM, which is about 28.5-, 2.6-, 3.4-, and 16.2-fold more potent than WQ, MT-WQ, C34, and T20, respectively, while MT-WQ-IDL was about 132.9-, 4.7-, 12.3-, 15.7-, and 75.8-fold more potent than WQ, WQ-IDL, MT-WQ, C34, and T20, respectively, for inhibiting HIV-1 Env-mediated cell-cell fusion. This result suggests that the addition of the IDL tail to the C terminus of a CHR peptide can indeed increase its fusion inhibitory activity.
MT-WQ-IDL inhibits HIV-1 infection by targeting the early stage of viral replication.
To understand which stage of HIV-1 replication is targeted by MT-WQ-IDL, we conducted a time-of-addition assay as previously described (21, 22). As shown by the results in Fig. 3, MT-WQ-IDL exhibited significantly decreased inhibitory activity against infection by either the X4 or R5 HIV-1 strain when it was added 1 or 2 h postinfection, suggesting that MT-WQ-IDL may be an HIV-1 entry inhibitor. In contrast, zidovudine (AZT), an HIV reverse transcriptase inhibitor, maintained its inhibitory activity even when it was added to cells 8 h postinfection. This result indicates that MT-WQ-IDL inhibits early-stage events of HIV-1 replication, such as viral entry.
FIG 3.
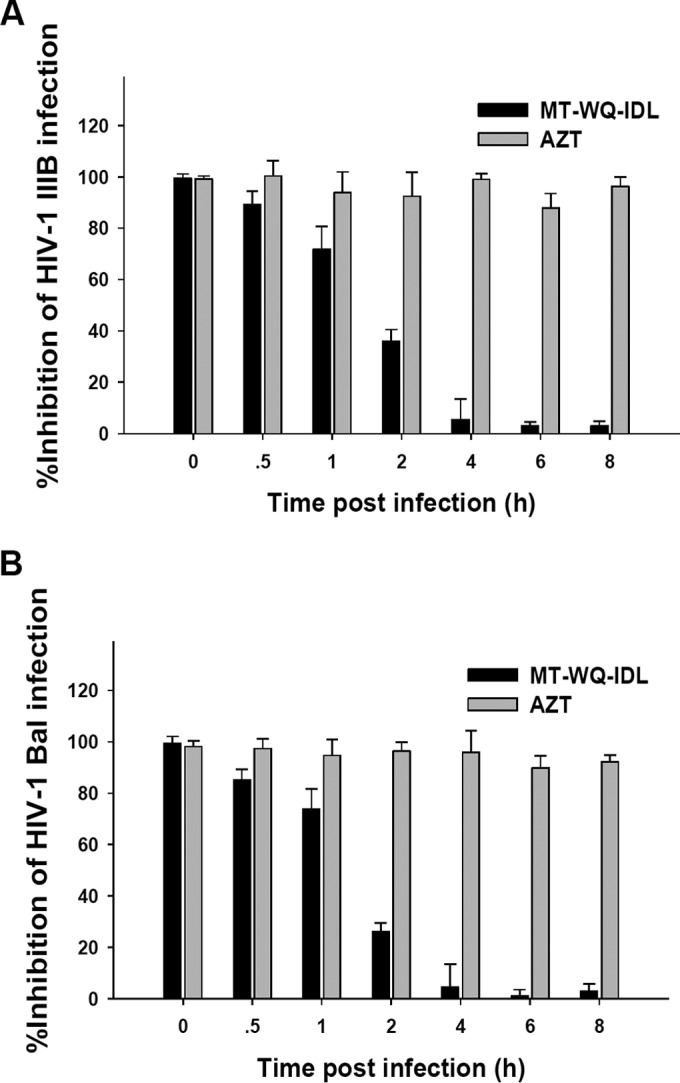
MT-WQ-IDL targets the early steps of HIV-1 replication. A time-of-addition assay was performed to evaluate the inhibitory activities of MT-WQ-IDL and AZT (an HIV reverse transcriptase inhibitor used as a control) against infection by HIV-1 IIIB, an X4 virus (A), or Bal, an R5 virus (B), at different intervals postinfection. MT-WQ-IDL could not inhibit infection by either X4 or R5 HIV-1 when it was added after 2 h postinfection. Error bars show standard deviations.
To determine whether MT-WQ-IDL blocks the attachment of HIV-1 to CD4-expressing cells (U87 CD4+ CCR5+ cells), we performed a washout assay as previously described (23, 24). MT-WQ-IDL peptide was incubated with target cells and virus at 4°C for 1 h, followed by washes with cold phosphate-buffered saline (PBS) to remove the unbound peptide and virions. If the peptide can bind to receptor on cells or gp120, no HIV-1 infection is expected (25). As shown by the results in Fig. 4A, HIV-1 infection was not affected after this treatment, suggesting that MT-WQ-IDL-mediated inhibition of HIV-1 replication is not associated with inhibition of the HIV-1 attachment to CD4-expressing cells. In contrast, heparin, a nonspecific HIV-1 attachment inhibitor (26, 27), could inhibit HIV-1 infection with or without washing (Fig. 4B).
FIG 4.
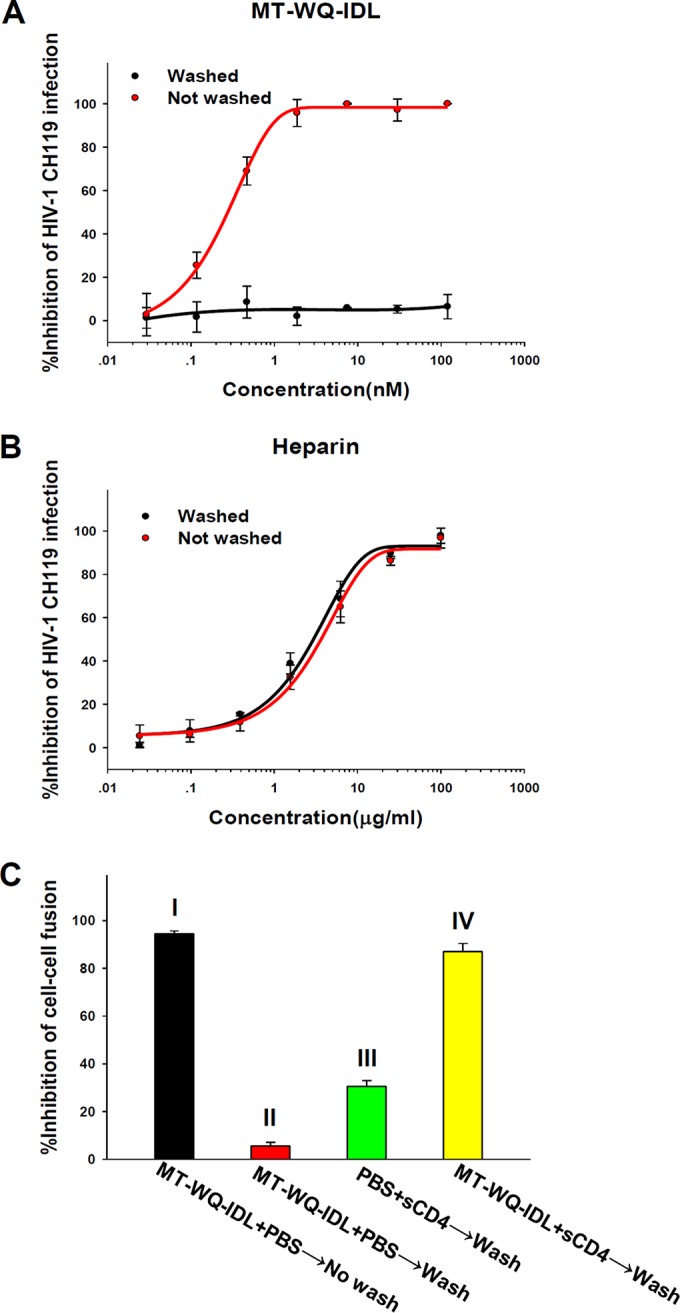
MT-WQ-IDL does not block the attachment of HIV-1 to cells expressing CD4 receptor but may interact with the CD4-triggered prehairpin intermediate of the HIV-1 gp41. (A, B) A washout assay was performed to determine whether MT-WQ-IDL (A) and heparin, a nonspecific HIV-1 attachment inhibitor (B), can block the attachment of HIV-1 to CD4-expressing cells (U87 CD4+ CCR5+ cells). (C) An HIV-1 Env-mediated cell-cell fusion washout assay was performed to determine the Env engagement of CD4 receptor in the MT-WQ-IDL-mediated inhibition of HIV-1 entry. Inhibition rates were as follows: MT-WQ-IDL alone without wash, 93.8%; MT-WQ-IDL alone with wash, 5.6%; sCD4 alone with wash, 30.6%; MT-WQ-IDL and sCD4 with wash, 87.1%. Error bars show standard deviations.
To determine the Env engagement of CD4 receptor in peptide-mediated inhibition of HIV-1 entry, we performed an HIV-1 Env-mediated cell-cell fusion washout assay as previously described (28). H9/HIV-1IIIB cells, used as effector cells, were preincubated with MT-WQ-IDL and/or soluble CD4 (sCD4) at 37°C for 30 min and then washed with PBS to remove the unbound peptide and sCD4 before the addition of MT-2 cells, used as target cells, for the cell-cell fusion assay. As shown by the results in Fig. 4C, MT-WQ-IDL could effectively block HIV-1 Env-mediated cell-cell fusion when the H9/HIV-1IIIB cells preincubated with peptide were not washed, while there was no inhibition when the H9/HIV-1IIIB cells preincubated with MT-WQ-IDL only were washed. However, the H9/HIV-1IIIB cells pretreated with both sCD4 and MT-WQ-IDL lost their ability to fuse with MT-2 cells after the unbonded sCD4 and MT-WQ-IDL were removed by washing, whereas pretreatment of H9/HIV-1IIIB cells with sCD4 only led to a partial inhibition of the HIV-1 Env-mediated cell-cell fusion. These results suggest that MT-WQ-IDL inhibits HIV-1 entry by interacting with the prehairpin intermediate of gp41 triggered by sCD4 or cell-associated CD4 receptor, which is consistent with the mechanism of action of most other HIV-1 CHR peptides (29–32).
IDL-conjugated peptide formed stable 6-HB with the NHR peptides and blocked 6-HB formation between the NHR and CHR peptides.
Since the above-described data indicated that IDL-conjugated peptide inhibited HIV-1 Env-mediated membrane fusion, probably by targeting viral gp41, we then analyzed the secondary structure of the complexes of the conjugated peptides and the NHR peptide by circular dichroism (CD) spectroscopy. After these CHR peptides were incubated with equal molar concentrations of the NHR peptide N46 (amino acids [aa] 536 to 581), the CD spectra of the mixture were measured. Similar to the WQ-LDL, WQ-IDI, WQ-LDI, and MT-WQ-IDL conjugates, WQ-IDL could interact with N46 to form a 6-HB with a helicity similar to that of the 6-HB formed between C34 or WQ and N46 (Fig. 5 and Table 1), suggesting that the addition of the artificial tails does not change the overall conformation of the CHR peptides.
FIG 5.

Secondary structures of CHR peptide-NHR peptide complexes. (A to E) Circular dichroism (CD) spectroscopy results for the complexes formed by N46 and WQ, WQ-LDL, WQ-IDI, WQ-LDI, WQ-IDL, and MT-WQ-IDL are shown. The circular dichroism spectra of these complexes displayed typical double minima at 208 and 222 nm for the α-helical feature. C34 was used as a positive control. deg, degrees. (F) Inhibitory activities of WQ-IDL, MT-WQ, MT-WQ-IDL, and WQ against 6-HB formation between N46 and C34. C34 and T20 were included as controls. Error bars show standard deviations.
Next, a thermal denaturation experiment was conducted to compare the thermostability (melting temperature [Tm] value) of the 6-HBs formed between the CHR peptide C34 or the WQ peptide with different artificial tails and the NHR peptide N46. As shown by the results in Fig. 6 and Table 1, the Tm value of the 6-HB formed between N46 and WQ-IDL (81.8°C) was much higher than that of the 6-HB formed between N46 and C34 (67.9°C) or WQ (65.1°C), but it was similar to those of the 6-HBs formed between N46 and WQ-LDL, WQ-IDI, WQ-LDI, and MT-WQ-IDL, respectively. These results suggested that the binding affinities between N46 and the WQ peptides with artificial tail anchors were much higher than that between N46 and C34. Therefore, these conjugated peptides can bind the viral gp41 NHR to form more stable heterogeneous 6-HBs and, hence, more effectively block homologous 6-HB formation between viral gp41 NHR and CHR domains, finally resulting in the inhibition of viral fusion and entry.
FIG 6.

Thermostability of the complexes formed by a CHR peptide and N46. (A to D) Melting curves of the complexes formed by N46 and WQ, WQ-IDL, and MT-WQ-IDL. C34 was used as a control. (E) Comparison of melting curves of the complexes formed by N46 and WQ, WQ-IDL, and MT-WQ-IDL.
Interestingly, the Tm value of the 6-HB formed between MT-WQ-IDL and N46 (90.7°C) was even higher than that of the 6-HB formed between WQ-IDL and N46, again suggesting that the addition of the MT hook to the N terminus of WQ-IDL further increases the binding affinity of WQ-IDL with N46.
We then determined whether WQ-IDL, MT-WQ, and MT-WQ-IDL could block 6-HB formation between N46 and C34. As shown by the results in Fig. 5F, WQ-IDL, MT-WQ, and MT-WQ-IDL exhibited much stronger inhibition of 6-HB formation than WQ, confirming that an artificial tail at both the N and C terminus of a CHR peptide can strongly anchor into the N-terminal pocket in the NHR domain, thus enhancing the inhibitory activity of the CHR peptide on 6-HB formation between the native NHR and CHR sequences in gp41.
Most recently, a new model of viral fusion was proposed. In this model, the NHR trimer at the prehairpin intermediate state dissociates and becomes embedded in the viral membranes, pulling the target cell membranes into juxtaposition. The fusogenic prebundle is formed, promoting the formation of mature, postfusion 6-HB trimers (33, 34). To accommodate this model, we hypothesize that MT-WQ-IDL peptide may bind to the viral NHR trimer at the prehairpin intermediate state to form homologous 6-HB, preventing the viral NHR trimer from dissociating and embedding into the viral membranes because of the high stability of the 6-HB and shielding of the hydrophobic residues on the NHR trimer that are responsible for binding to the lipid membrane by CHR peptide. However, it is questionable whether MT-WQ-IDL is able to bind to the viral NHR trimer formed at the fusogenic prebundle state. Certainly, approval of these hypotheses requires further investigations.
The IDL-conjugated peptide was more effective than the CHR peptides without an IDL tail in inhibiting infection by HIV-1 clinical strains, Chinese epidemic strains, and T20- and T2635-resistant variants.
We then compared the antiviral activities of the CHR peptides containing the IDL tail (WQ-IDL and MT-WQ-IDL) to those of CHR peptides without the IDL tail (WQ and T20), testing them against a panel of HIV-1 clinical isolates of different subtypes (A, B, D, F, O, and A/E) and tropisms (X4, R5, and X4/R5) and a panel of Chinese epidemic strains. As shown by the results in Table 2, WQ-IDL and MT-WQ-IDL exhibited much more potent activities than WQ or T20 against all clinical isolates and Chinese epidemic strains tested, with IC50s at low nM levels, demonstrating that these CHR peptides with the IDL tail possess improved antiviral activity against a broad spectrum of HIV-1 isolates of different subtypes and tropisms.
TABLE 2.
Inhibitory activities of conjugated peptides and control peptides against HIV-1 clinical strains and pseudovirus
| Virus (subtype, tropism) | Tier | Mean IC50 ± SD (nM)a |
|||
|---|---|---|---|---|---|
| WQ | WQ-IDL | MT-WQ-IDL | T20 | ||
| HIV-1 clinical isolates | |||||
| 92UG029 (A, X4) | —b | 81.2 ± 1.9 | 3.0 ± 1.0 | 0.6 ± 0.1 | 45.5 ± 3.2 |
| MN/H9 (A, X4) | 1 | 383.3 ± 13.0 | 20.6 ± 2.9 | 4.1 ± 0.9 | 43.4 ± 4.1 |
| US4GS007 (B, R5) | — | >500 | 86.9 ± 12.0 | 12.5 ± 4.0 | 157.3 ± 18.0 |
| BK132/GS009 (B, X4) | — | 125.8 ± 12.0 | 18.3 ± 2.8 | 3.0 ± 0.6 | 30.5 ± 2.7 |
| BZ167/GS010 (B, X4) | 1 | >500 | 90.7 ± 18.0 | 15.1 ± 3.7 | 150.3 ± 12.0 |
| TZA68/125A (C, R5) | — | 360.5 ± 29.0 | 8.4 ± 0.8 | 3.9 ± 0.8 | 115.0 ± 33.0 |
| 93IN101 (C, R5) | — | 115.1 ± 4.4 | 3.5 ± 1.1 | 1.1 ± 0.8 | 20.4 ± 12.0 |
| 93BR020 (F, X4/R5) | — | 326.5 ± 11.0 | 4.1 ± 0.8 | 0.5 ± 0.1 | 37.2 ± 4.1 |
| BCF02 (O, R5) | — | 283.5 ± 28.0 | 2.6 ± 1.6 | 0.6 ± 0.1 | 38.3 ± 2.1 |
| NP1525 (A/E, X4/R5) | — | 138.5 ± 19.0 | 2.0 ± 0.2 | 0.6 ± 0.1 | 46.2 ± 7.4 |
| Pseudoviruses bearing Env of Chinese epidemic strains | |||||
| MW965.26 (C, R5) | 1 | 31.9 ± 4.2 | 12.4 ± 1.0 | 2.1 ± 1.2 | 16.2 ± 1.6 |
| 255-34 (02_AG, R5) | 2 | 46.6 ± 6.6 | 1.0 ± 0.4 | 0.1 ± 0.1 | 17.7 ± 2.1 |
| 235-47 (02_AG, R5) | 2 | 22.6 ± 1.3 | 0.9 ± 1.5 | 0.1 ± 0.1 | 18.1 ± 3.1 |
| 211-9 (02_AG, R5) | 2 | 56.8 ± 9.3 | 1.7 ± 0.7 | 0.1 ± 0.2 | 27.8 ± 4.8 |
| AE03 (A/E, R5) | 2 | 235.2 ± 12.0 | 3.6 ± 1.6 | 1.3 ± 0.3 | 7.8 ± 0.6 |
| 257-31 (02_AG, R5) | 3 | 225.2 ± 9.3 | 6.8 ± 1.3 | 0.9 ± 0.2 | 29.3 ± 1.8 |
| 253-11 (02_AG, R5) | 3 | 168.8 ± 2.9 | 5.4 ± 0.9 | 0.1 ± 0.1 | 19.4 ± 2.7 |
| BC02 (B/C, R5) | — | 190.2 ± 20.0 | 1.6 ± 1.0 | 0.8 ± 0.5 | 27.4 ± 5.6 |
| HB5-3 (B/C, R5) | — | 54.2 ± 9.1 | 2.4 ± 0.8 | 0.7 ± 0.2 | 7.9 ± 3.6 |
| GX11.13 (A/E, R5) | — | 615.8 ± 13.0 | 27.5 ± 2.4 | 9.0 ± 1.1 | 58.0 ± 5.4 |
| GX2010.36 (A/E, R5) | — | 686.2 ± 33.0 | 37.4 ± 1.6 | 5.2 ± 1.3 | 91.0 ± 2.4 |
| SHX335.24 (A/E, R5) | — | 13.5 ± 3.1 | 1.2 ± 0.6 | 0.2 ± 0.1 | 7.1 ± 1.4 |
| B02 (B', R5) | — | 207.2 ± 28.0 | 0.8 ± 0.3 | 0.3 ± 0.3 | 49.7 ± 2.4 |
| 43-22 (B', R5) | — | 52.8 ± 13.0 | 7.0 ± 0.6 | 2.1 ± 0.3 | 12.6 ± 2.4 |
IC50 data were derived from the results of three independent experiments.
—, the sensitivity to antibody-mediated neutralization is unknown.
We next compared the inhibitory activities of WQ-IDL and MT-WQ-IDL with those of WQ and T20 against HIV-1 variants with single or double mutations in gp41 that are resistant to T20 or T2635, the first and a modified HIV fusion inhibitor, respectively (35). As shown by the results in Table 3, except for wild-type and A6V strains, T20 was unable to inhibit any T20- or T2635-resistant strain at concentrations as high as 500 nM. WQ could inhibit both T20- and T2635-senstive and -resistant strains with IC50s in a range of 35 to 1,129 nM. WQ-IDL exhibited much higher potency against all T20- and T2635-resistant strains tested, with IC50s ranging from 1.6 to 62 nM, about 22-fold more potent than WQ on average. Impressively, MT-WQ-IDL showed even higher potency than WQ-IDL against T20- and T2635-resistant strains, with IC50s ranging from 0.1 to 12 nM, about 198-fold more potent than WQ on average. These results provide strong evidence that the addition of a C-terminal IDL tail anchor with or without an N-terminal MT hook results in significant improvement in CHR peptide potency against T20- and T2635-resistant strains. Also, the high and broad antiviral potency of MT-WQ-IDL made it a potential candidate for development as a new anti-HIV-1 drug.
TABLE 3.
Inhibitory activities of conjugated peptides and control peptides against HIV-1 T20- and T2635-resistant strains
| Virus description | Mean IC50 ± SD (nM)a |
|||
|---|---|---|---|---|
| WQ | WQ-IDL | MT-WQ-IDL | T20 | |
| T20-resistant strains | ||||
| Wild type | 160.8 ± 20.0 | 6.8 ± 1.9 | 2.2 ± 1.0 | 55.3 ± 2.6 |
| V38A | 433.1 ± 67.0 | 22.0 ± 2.9 | 4.4 ± 1.0 | >500 |
| V38A N42D | 34.8 ± 9.1 | 2.9 ± 0.2 | 1.0 ± 0.2 | >500 |
| V38E N42S | 65.2 ± 2.3 | 1.6 ± 0.6 | 0.3 ± 0.3 | >500 |
| V38A N42T | 118.0 ± 13.0 | 7.5 ± 2.4 | 0.1 ± 0.1 | >500 |
| T2635-resistant strains | ||||
| Wild type | 195.1 ± 20.0 | 17.8 ± 1.9 | 3.9 ± 1.0 | 98.9 ± 2.6 |
| A6V | 303.4 ± 67.0 | 18.3 ± 2.9 | 6.4 ± 1.0 | 66.2 ± 5.6 |
| Q66R | 434.8 ± 9.1 | 12.9 ± 0.2 | 6.0 ± 0.2 | >500 |
| Q79E | 965.2 ± 2.3 | 51.6 ± 0.6 | 11.3 ± 0.3 | >500 |
| K90E | 218.4 ± 13.0 | 8.5 ± 2.4 | 1.1 ± 0.1 | >500 |
| Q79E N126K | 1128.4 ± 33.0 | 61.4 ± 1.6 | 9.2 ± 1.3 | >500 |
IC50 data were derived from the results of three independent experiments.
It has been reported that an MT hook at the N terminus of CHR peptides can strengthen most of these CHR peptides only if it binds to the correct site at the N terminus, W628 (11). Therefore, the addition of IDL at the correct site of the C terminus, Q653, is expected to enhance the anti-HIV-1 activity of most CHR peptide fusion inhibitors (Fig. 7D). However, this approach is not applicable to T20. Unlike most other CHR peptides (e.g., C34 and SC22EK), T20 does not contain the pocket-binding domain (PBD) at its N terminus but has a long sequence at its C terminus, including the lipid-binding domain (LBD), which contains two Trp residues (Fig. 1A). Consequently, T20 cannot form stable a 6-HB with the NHR trimer (Fig. 5F) and has lower levels of anti-HIV activity than many other CHR peptides. Therefore, the addition of IDL to the C terminus of T20 is not expected to improve T20's anti-HIV-1 activity.
FIG 7.

Crystal structure of the 6-HB formed by MT-WQ-IDL and gp41 NHR. (A) Crystal structures of gp41 NHR+CHR (PDB 2X7R), NHR+MT-C34 (PDB 3VTP), and N46-MT-WQ-IDL (PDB 5H0N) are shown as cartoon representation or electrostatic surface. NHR is colored in gray, CHR in light green, MT-WQ-IDL in forest green, and both the MT and IDL tails in magenta. Important residues are enlarged as sticks and identified. (B) Enlargement of interactions between IDL tail and NHR. NHR is colored in gray, MT-WQ-IDL in forest green, and IDL tail in magenta. IDL tail is shown in stick or cartoon representation. (C, D) Electron density of IDL tail in the N46-MT-WQ-IDL crystal structure. The 2Fo–Fc electron density is drawn as blue meshes at a contour level of 0.8 σ, and Fo–Fc electron density is drawn as red and green meshes at a contour level of 2.6 σ. The omit map (C) was generated with the phases of a molecular replacement solution with preliminary refinement but without the IDL tail modeled from the beginning. (E) Schematic diagram showing the effect of MT-WQ-IDL on the process of HIV entry into host cell. MT-WQ-IDL can bind to the NHR more stably and block the formation of a homologous 6-helix bundle, thus inhibiting viral infection.
The crystal structure of the 6-HB formed by MT-WQ-IDL and N46 confirmed binding of the IDL tail anchor to the novel hydrophobic pocket.
To verify the mode of activity of these modified peptides, we determined the crystal structure of the 6-HB formed by MT-WQ-IDL and N46, as shown in Fig. 7A. 654IDL656 exhibited a hooklike tail stretching into the shallow pocket formed by L544, L545, I548, and V549 of the NHR trimer core. The hydrophobic side chains of I654 and L656 were buried in this pocket, and D655 was forced outward (Fig. 7B to D). To facilitate the IDL tail binding, Q652 and Q653 in WQ unwound from the helical conformation to form a prolonged linker. This conformation is expected to stabilize the binding between the inhibitor and the NHR better than in a traditional helical conformation by providing more interactions. This result indicates that, given the correct site of Q653 within the C terminus, IDL could be adopted for use in most HIV-1 CHR peptide fusion inhibitors to enhance their antiviral activity (Fig. 7D). In particular, since small-molecule fusion inhibitors binding to the deep C-terminal pocket in the hydrophobic groove are less effective than peptides (36), our strategy that targets the shallow pocket outside the hydrophobic groove could also be used to develop this type of HIV fusion inhibitor.
To further investigate whether this strategy could also be adopted to develop fusion inhibitors against other class I enveloped viruses for which crystal structures of their HR1/HR2 complexes are available, we took Middle East respiratory syndrome coronavirus (MERS-CoV), which is an enveloped virus with class I membrane fusion protein, as an example. Based on the crystal structure of the MERS-CoV fusion core (37), we searched for a potential pocket adjacent to the hydrophobic groove and designed an artificial anchor tail to target it. We added a sequence consisting of three amino acids (EAN) to the peptidic MERS-CoV fusion inhibitor M0, which is derived from the natural sequence of the HR2 domain of MERS-CoV spike protein, to generate a new peptide, M1 (Fig. 8A). The simulated interaction model showed that 1254EAN1256 formed a hooklike anchor and stretched into the pocket formed by A1032, L1033, K1035, and A1037 of the HR1 trimer core (Fig. 8B). In addition, the resultant peptide M1 exhibited significantly enhanced inhibition activity against MERS-CoV-mediated membrane fusion and infection of pseudovirus (Fig. 8C and D). These results suggest that our strategy can also be utilized to design or optimize peptidic fusion inhibitors against other enveloped viruses with class I membrane fusion proteins.
FIG 8.

Application of the artificial tail anchor strategy to optimize a peptidic MERS-CoV fusion inhibitor. (A) Schematic representation of the functional domains in the S2 subunit of MERS-CoV spike protein and sequences of M0 and M1 peptides. (B) Structural prediction for binding of M0 (a) or M1 (b) peptide with HR1 domain of MERS-CoV S protein. HR1 is colored in gray, the M0 peptide in green, and the EAN hook in magenta. The original sequence of MERS-CoV HR1 aa 1254 to 1256 is colored in cyan for comparison. The EAN hook is superposed on M0 in panel c and shown for residue interactions in panel d. (C) Inhibitory activities of M0 and M1 peptides against MERS-CoV S-mediated cell-cell fusion. (D) Inhibitory activities of M0 and M1 peptides against MERS pseudovirus infection. Error bars show standard deviations.
In conclusion, to optimize the current strategies for improving the efficacy and pharmaceutical properties of the CHR peptide-based HIV fusion inhibitors by targeting the deep hydrophobic pocket in the N-terminal region of the gp41 NHR trimer, we developed a new strategy to add an artificial tail anchor that targets the shallow pocket(s) in the C-terminal region of the gp41 NHR trimer. We found that the addition of residues IDL, forming an artificial tail anchor, to the C termini of the CHR WQ peptides resulted in significant increases of their anti-HIV-1 activities, possibly because the artificial tail anchor, IDL, binds to the C-terminal shallow pocket in the gp41 NHR trimer. Notably, the fusion inhibitor with both an MT hook and an IDL tail, at its N and C terminus, respectively, exhibits more potent anti-HIV-1 activity than those with the MT hook or IDL tail only, suggesting its potential to be further developed as a new anti-HIV drug to replace T20, the only U.S. FDA-approved HIV fusion inhibitor used in clinics. Furthermore, we have shown that this strategy could also be used for designing anti-MERS-CoV peptides with improved fusion inhibitory activity, confirming that this new approach can also be applied for developing fusion inhibitors against other enveloped viruses with class I membrane fusion proteins.
MATERIALS AND METHODS
Peptides.
Peptides (Fig. 1A) were synthesized by KareBay Biochem, Inc., using a standard solid-phase 9-fluorenylmethoxy carbonyl (FMOC) method, to a purity of >98% when tested by high-performance liquid chromatography (HPLC), as described previously (38). The peptides were solubilized using dimethyl sulfoxide (DMSO). The concentrations of the peptides were tested by UV absorbance and a theoretically calculated molar-extinction coefficient based on tryptophan and tyrosine residues. Peptides were diluted using PBS for experimental analysis.
Inhibition of HIV-1 infection by peptides.
The inhibitory activities of the peptides against infection by laboratory-adapted HIV-1 X4 strain IIIB and HIV-1 R5 strain Bal, HIV-1 clinical strains, and T20- and T2635-resistant strains were determined as described previously (39, 40). Amounts of 100 50% tissue culture infective doses (TCID50) of the HIV-1 viruses were used to infect 1 × 105/ml MT-2 cells or CEMx174 5.25 M7 cells in RPMI 1640 medium containing 10% fetal bovine serum (FBS) and graded concentrations of peptides. After overnight culture, the medium was replaced with fresh medium. Fifty-microliter amounts of the culture supernatant were collected from each well on the fourth day for MT-2 and CEMx174 5.25 M7 cells and mixed with equal volumes of 5% Triton X-100. The p24 antigen was detected by enzyme-linked immunosorbent assay (ELISA) as previously described (41). The maximum infectivity was determined in the absence of peptide, and the background was measured in the absence of virus. IC50s were calculated using Calcusyn software (Biosoft, Ferguson, MO) (15), and the lines of best fit were drawn using the SigmaPlot 10.1 program (Systat Software, Inc., Chicago, IL).
Time-of-addition assay.
The assay of the effect of time of addition on HIV-1 infection was performed as previously described (21, 22). Briefly, 10 nM MT-WQ-IDL or 2 μM AZT (an HIV reverse transcriptase inhibitor used as a control) was added to mixtures of MT-2 cells and HIV-1 IIIB or CEMx174 5.25 M7 cells and HIV-1 BaL at different intervals (0, 0.5, 1, 2, 4, 6, and 8 h) postinfection. At 4 days postinfection, the levels of p24 antigen in the culture supernatants were detected by ELISA as described above.
Washout assay.
The washout assay was conducted as previously described (23, 24). Briefly, U87 CD4+ CCR5+ cells (104/well) were incubated in 96-well plates at 37°C overnight before the culture plate was placed on ice. After incubation at 4°C for 1 h, the culture supernatants were removed and a peptide or heparin (as a control) at various concentrations was added to U87 CD4+ CCR5+ cells (104/well) along with 100 TCID50 of the HIV-1 CH119 strain. The mixture was incubated at 4°C for 1 h and then washed with cold PBS, followed by the addition of 200 μl DMEM containing 10% FBS. The 96-well plate was cultured at 37°C for 48 h, and the luciferase activity was measured using a luciferase kit (Promega, Madison, WI) and an ultra 384 luminometer (Tecan) as previously described (42).
HIV-1-mediated cell-cell fusion assay.
A dye transfer assay was used to detect HIV-1 Env-mediated cell-cell fusion as described previously (20). H9/HIV-1IIIB cells were labeled with a fluorescent reagent, calcein AM (Molecular Probes, Inc., Eugene, OR), and then incubated with MT-2 cells (ratio of 1:10) in 96-well plates at 37°C for 2 h in the presence or absence of the test peptide at graded concentrations. The sample at each concentration was tested in triplicate. The fused and unfused calcein-labeled HIV-1IIIB cells were counted under an inverted fluorescence microscope (Zeiss, Germany) with an eyepiece micrometer disc. Four fields per well were counted. The percentages of inhibition of cell fusion by peptides were calculated based on the data from all 12 fields observed, as described previously (20); IC50s were calculated by using the Calcusyn computer program (43), and lines of best fit were drawn using the Sigma Plot 10.1 program.
The HIV-1 Env-mediated cell-cell fusion washout assay was performed as previously described (28). In brief, MT-WQ-IDL (5 nM), sCD4 (100 nM), or a mixture of MT-WQ-IDL and sCD4 was preincubated with H9/HIV-1IIIB cells labeled with calcein AM at 37°C for 0.5 h. The treated H9/HIV-1IIIB cells were washed with PBS (unwashed H9/HIV-1IIIB cells in the presence of MT-WQ-IDL were used as a control) before the addition of MT-2 cells. After incubation at 37°C for 2 h, the fused and unfused calcein-labeled HIV-1IIIB cells were counted as described above. Each sample was tested in triplicate.
CD spectroscopy.
Circular dichroism (CD) spectroscopy was conducted as described previously (43). Briefly, a CHR peptide was incubated with an equal molar concentration of the NHR peptide N46 at 37°C for 30 min. The final concentration of each peptide was 10 μM in double-distilled water (ddH2O). The CD spectra were tested on a Jasco spectropolarimeter (model J-815; Jasco, Inc., Easton, MD), using a 1-nm bandwidth with a 1-nm step resolution from 195 to 260 nm at 4°C. The spectra were corrected with ddH2O. The helical content was calculated from the CD signal by dividing the mean residue ellipticity at 222 nm by the value expected for 100% helix formation (−3.3 × 10−4 degrees cm2 dmol−1). The thermal denaturation experiment was conducted by monitoring the changes in ellipticity at 222 nm at increasing temperatures (4 to 98°C), using a temperature controller. The temperature was increased at a rate of 1.2°C/min; data were acquired at 222 nm at a frequency of 0.25 Hz at a 1-nm bandwidth.
Inhibition of 6-HB formation by peptides in vitro.
The inhibitory activity of peptides on 6-HB formation was measured by a modified ELISA as previously described (44). Briefly, a 96-well polystyrene plate was coated with 50 μl NC-1 (a monoclonal antibody specific for HIV-1 gp41 6-HB formed by NHR and CHR [9, 10]) (4 μg/ml) in 0.1 M Tris buffer (pH 8.8). A tested peptide at graded concentrations was mixed with 50 μl N46 (1 μM) and incubated at 37°C for 30 min. Then, the mixture was incubated with 100 μl C34-biotin (0.5 μM) at 37°C for 30 min. Afterwards, 50 μl of the mixture was added to the wells of a plate coated with NC-1, followed by incubation for 1 h and washing with a washing buffer (PBS containing 0.1% Tween 20) three times. Finally, horseradish peroxidase (HRP)-labeled streptavidin (Jackson) and the substrate 3,3,5,5-tetramethylbenzidine (TMB; Sigma) were added sequentially. The absorbance at 450 nm (A450) was measured using an ELISA reader (ultra 384; Tecan).
Inhibition of pseudotyped HIV-1 infection.
HIV-1 pseudoviruses were generated as described previously (42). Briefly, 293T cells (ATCC, Manassas, VA) were cotransfected with envelope and a backbone plasmid (pNL4-3.Luc.R-E from the NIH AIDS Reagent Program) that encodes an Env-defective, luciferase-expressing HIV-1 genome using VigoFect reagent (Vigorous Biotech, Beijing, China). After 12 h, the culture supernatants were replaced with fresh DMEM–10% FBS. The culture supernatants were collected 48 h after the replacement, filtered using 0.45-μm-pore-size filters, and stored at −80°C before testing. The antiviral activities of C peptides were determined using U87 CD4+ CCR5+ cells. Briefly, the peptides were prepared at graded concentrations, mixed with 100 TCID50 virus and U87 CD4+ CCR5+ cells (104/well), and incubated at 37°C for 48 h, and the luciferase activity was measured using a luciferase kit (Promega, Madison, WI) and an ultra 384 luminometer (Tecan).
Crystallization, data collection, and structure determination.
To obtain the complex crystal of MT-WQ-IDL and NHR, synthesized MT-WQ-IDL was first mixed with peptide N46 at a 1:1 molar ratio and then applied onto a Superdex-75 gel filtration column (GE Healthcare, Piscataway, NJ) to isolate the 6-HB formed. Fractions containing N46-MT-WQ-IDL trimer were collected, concentrated to 20 mg/ml, and then crystallized at 16°C using the hanging-drop vapor diffusion method. The drops were set on a siliconized cover slip by equilibrating a mixture containing 1 μl protein solution (20 mM Tris-HCl, pH 8.0, and 150 mM NaCl) and 1 μl reservoir solution (0.2 M zinc acetate, 0.1 M imidazole–HCl [pH 8.0], 20% [vol/vol] 1,4-butanediol) against a 400-μl reservoir solution. After 3 days, single crystals formed and were flash frozen using liquid nitrogen for future data collection.
The diffraction data sets of N46-MT-WQ-IDL complex crystals were collected at beamline BL-19U1 of the Shanghai Synchrotron Radiation Facility, China. X-ray diffraction data were integrated and scaled using the HKL2000 program (45). The phasing problem was solved by the molecular replacement method using PHENIX.phaser (46) with a crystal structure of HIV gp41 NHR-CHR (Protein Data Bank [PDB] accession number 5CMZ) as a search model. The final model was manually adjusted in COOT (17) and refined with PHENIX.refine (47). The coordinates were deposited in the PDB under accession number 5H0N. The statistics of data collection and structure refinement are given in Table 4.
TABLE 4.
N46-MT-WQ-IDL diffraction data and refinement statistics
| Parameter | Valuea |
|---|---|
| Crystallographic data | |
| Resolution (Å) (range) | 39.60–2.80 (2.85–2.80) |
| Wavelength (Å) | 0.97853 |
| No. of reflections measured | 12,543 |
| Avg redundancy | 2.5 (2.45) |
| Mean I/σ(I) | 10.73 (1.79) |
| Completeness (%) | 89.3 (90.7) |
| Rmergeb | 0.079 (0.288) |
| Refinement statistics | |
| Space group | P21 |
| Cell parameters | |
| a (Å) | 55.42 |
| b (Å) | 79.24 |
| c (Å) | 70.94 |
| β (°) | 111.24 |
| No. of reflections in working set | 11,375 |
| No. of reflections in test set | 634 |
| Rcrystc | 0.2371 |
| Rfreed | 0.2759 |
| RMSD bonds (Å)e | 0.004 |
| RMSD angles (°) | 0.465 |
| Average B factor (Å2) | 54.40 |
| No. of waters | 7 |
| MolProbity results | |
| Ramachandran outliers (%) | 0 (0.00) |
| Ramachandran favored (%) | 404 (97.58) |
| Clash score | 6.94 |
Values in parentheses indicate the corresponding statistics in the highest-resolution shell.
Rmerge = (Ii − >Ii<|)/Ii|, where Ii is the integrated intensity of a given reflection.
Rcryst = (||Fo| − |Fc||)/|Fo|, where Fo and Fc denote observed and calculated structure factors, respectively.
Rfree is equivalent to Rcryst but calculated using randomly chosen 5% reflections, which were excluded from the refinement process,s as the test set.
RMSD, root mean square deviation.
Accession number(s).
The GenBank accession numbers of the HIV-1 strains are as follows: IIIB (KJ925006.1), Bal (DQ318210.1), 92UG029 (AF205862.1), MN/H9 (AY736819.1), US4GS007 (AY214062.1), BK132/GS009 (AH003229.2), BZ167/GS010 (L22087.1), 93IN101 (AY669738.1), 93BR020 (AF005494.1), BCF02 (AF458282.1), NP1525 (AY713420.1), HIV-1 backbone (AF324493.1), BC02 (EU363832), CH119.102 (EF117261), AE03 (EU363851), HB5-3 (HQ326124), GX11.13 (GU475042), GX2010.36 (GU475026), SHX335.24 (GU475033), B02 (EU363826), T20-sensitive strain, WT (EU236444), T20-resistant strain, V38A (EU236445), V38A N42D strain (EU236487), V38E N42S strain (JF957865), V38A N42T strain (JQ248223), MW965.26 (U08455), 255-34 (EU513184), 235-47 (EU513195), 211-9 (EU513187), 257-31 (EU513185), and 253-11 (EU513191). There are no data bank records available for HIV-1 strains 43-22 (19) and TZA68/125A (48) or the T2635-resistant strains (49). The crystal structure accession numbers are as follows: gp41 NHR trimer and NHR+CHR (PDB 2X7R), NHR+MT-C34 (PDB 3VTP), and N46-MT-WQ-IDL (PDB 5H0N).
ACKNOWLEDGMENTS
We thank Rogier W. Sanders at Weill Cornell Medical College, New York, NY, for providing the HIV-1 T2635-resistant strains, Yuxian He at the Center for AIDS Research, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China, for providing the plasmids of Chinese epidemic strains, and the NIH AIDS Reagent Program for H9/HIV-1IIIB, MT-2, and U87 CD4+ CCR5+ cells, HIV-1 IIIB and Bal, and T20-resistant strains, as well as some HIV-1 clinical isolates, shown in Table 2.
This study was supported by the National 863 Program of China (2015AA020930 to L.L.), National Natural Science Foundation of China (81361120378 and 81590762 to S.J. and 81373456 to L.L.), National Key Research and Development Program of China (2016YFC1202901 to L.L.), National Basic Science Talent Training Funds (J1210041 to Z.M.), and Shanghai Rising-Star Program (16QA1400300 to L.L.). The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Weissenhorn W, Dessen A, Harrison SC, Skehel JJ, Wiley DC. 1997. Atomic structure of the ectodomain from HIV-1 gp41. Nature 387:426–428. doi: 10.1038/387426a0. [DOI] [PubMed] [Google Scholar]
- 2.Lu M, Kim PS. 1997. A trimeric structural subdomain of the HIV-1 transmembrane glycoprotein. J Biomol Struct Dyn 15:465–471. doi: 10.1080/07391102.1997.10508958. [DOI] [PubMed] [Google Scholar]
- 3.Jiang S, Lin K, Strick N, Neurath AR. 1993. HIV-1 inhibition by a peptide. Nature 365:113. doi: 10.1038/365113a0. [DOI] [PubMed] [Google Scholar]
- 4.Jolly C. 2011. Cell-to-cell transmission of retroviruses: innate immunity and interferon-induced restriction factors. Virology 411:251–259. doi: 10.1016/j.virol.2010.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Galloway NL, Doitsh G, Monroe KM, Yang Z, Munoz-Arias I, Levy DN, Greene WC. 2015. Cell-to-cell transmission of HIV-1 is required to trigger pyroptotic death of lymphoid-tissue-derived CD4 T cells. Cell Rep 12:1555–1563. doi: 10.1016/j.celrep.2015.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mutnal MB, Schachtele SJ, Hu S, Lokensgard JR. 2013. T-cell reconstitution during murine acquired immunodeficiency syndrome (MAIDS) produces neuroinflammation and mortality in animals harboring opportunistic viral brain infection. J Neuroinflamm 10:98. doi: 10.1186/1742-2094-10-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chan DC, Chutkowski CT, Kim PS. 1998. Evidence that a prominent cavity in the coiled coil of HIV type 1 gp41 is an attractive drug target. Proc Natl Acad Sci U S A 95:15613–15617. doi: 10.1073/pnas.95.26.15613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lalezari JP, Henry K, O'Hearn M, Montaner JS, Piliero PJ, Trottier B, Walmsley S, Cohen C, Kuritzkes DR, Eron JJ Jr, Chung J, DeMasi R, Donatacci L, Drobnes C, Delehanty J, Salgo M. 2003. Enfuvirtide, an HIV-1 fusion inhibitor, for drug-resistant HIV infection in North and South America. N Engl J Med 348:2175–2185. doi: 10.1056/NEJMoa035026. [DOI] [PubMed] [Google Scholar]
- 9.He Y, Liu S, Li J, Lu H, Qi Z, Liu Z, Debnath AK, Jiang S. 2008. Conserved salt-bridge between the N- and C-terminal heptad repeat regions of HIV-1 gp41 core structure is critical for virus entry and inhibition. J Virol 82:11129–11139. doi: 10.1128/JVI.01060-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jiang S, Debnath AK. 2000. A salt bridge between an N-terminal coiled coil of gp41 and an antiviral agent targeted to the gp41 core is important for anti-HIV-1 activity. Biochem Biophys Res Commun 270:153–157. doi: 10.1006/bbrc.2000.2411. [DOI] [PubMed] [Google Scholar]
- 11.Otaka A, Nakamura M, Nameki D, Kodama E, Uchiyama S, Nakamura S, Nakano H, Tamamura H, Kobayashi Y, Matsuoka M, Fujii N. 2002. Remodeling of gp41-C34 peptide leads to highly effective inhibitors of the fusion of HIV-1 with target cells. Angew Chem Int Ed Engl 41:2937–2940. doi:. [DOI] [PubMed] [Google Scholar]
- 12.Stephens OM, Kim S, Welch BD, Hodsdon ME, Kay MS, Schepartz A. 2005. Inhibiting HIV fusion with a beta peptide foldamer. J Am Chem Soc 127:13126–13127. doi: 10.1021/ja053444+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tam JP, Yu Q. 2002. A facile ligation approach to prepare three-helix bundles of HIV fusion-state protein mimetics. Org Lett 4:4167–4170. doi: 10.1021/ol026932k. [DOI] [PubMed] [Google Scholar]
- 14.Naito T, Izumi K, Kodama E, Sakagami Y, Kajiwara K, Nishikawa H, Watanabe K, Sarafianos SG, Oishi S, Fujii N, Matsuoka M. 2009. SC29EK, a peptide fusion inhibitor with enhanced alpha-helicity, inhibits replication of human immunodeficiency virus type 1 mutants resistant to enfuvirtide. Antimicrob Agents Chemother 53:1013–1018. doi: 10.1128/AAC.01211-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qi Z, Shi W, Xue N, Pan C, Jing W, Liu K, Jiang S. 2008. Rationally designed anti-HIV peptides containing multifunctional domains as molecule probes for studying the mechanisms of action of the first and second generation HIV fusion inhibitors. J Biol Chem 283:30376–30384. doi: 10.1074/jbc.M804672200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chong H, Yao X, Qiu Z, Sun J, Qiao Y, Zhang M, Wang M, Cui S, He Y. 2014. The M-T hook structure increases the potency of HIV-1 fusion inhibitor sifuvirtide and overcomes drug resistance. J Antimicrob Chemother 69:2759–2769. doi: 10.1093/jac/dku183. [DOI] [PubMed] [Google Scholar]
- 17.Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 18.Buzon V, Natrajan G, Schibli D, Campelo F, Kozlov MM, Weissenhorn W. 2010. Crystal structure of HIV-1 gp41 including both fusion peptide and membrane proximal external regions. PLoS Pathog 6:e1000880. doi: 10.1371/journal.ppat.1000880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chong H, Yao X, Qiu Z, Sun J, Zhang M, Waltersperger S, Wang M, Liu SL, Cui S, He Y. 2013. Short peptide fusion inhibitors with high potency against wild-type and enfuvirtide-resistant HIV-1. FASEB J 27:1203–1213. doi: 10.1096/fj.12-222547. [DOI] [PubMed] [Google Scholar]
- 20.Wang Q, Bi W, Zhu X, Li H, Qi Q, Yu F, Lu L, Jiang S. 2015. Nonneutralizing antibodies induced by the HIV-1 gp41 NHR domain gain neutralizing activity in the presence of the HIV fusion inhibitor enfuvirtide: a potential therapeutic vaccine strategy. J Virol 89:6960–6964. doi: 10.1128/JVI.00791-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li L, Qiu J, Lu L, An S, Qiao P, Jiang S, Liu S. 2013. 3-Hydroxyphthalic anhydride-modified human serum albumin as a microbicide candidate inhibits HIV infection by blocking viral entry. J Antimicrob Chemother 68:573–576. doi: 10.1093/jac/dks458. [DOI] [PubMed] [Google Scholar]
- 22.Yang J, Li L, Tan S, Jin H, Qiu J, Mao Q, Li R, Xia C, Jiang ZH, Jiang S, Liu S. 2012. A natural theaflavins preparation inhibits HIV-1 infection by targeting the entry step: potential applications for preventing HIV-1 infection. Fitoterapia 83:348–355. doi: 10.1016/j.fitote.2011.11.016. [DOI] [PubMed] [Google Scholar]
- 23.Gallo SA, Clore GM, Louis JM, Bewley CA, Blumenthal R. 2004. Temperature-dependent intermediates in HIV-1 envelope glycoprotein-mediated fusion revealed by inhibitors that target N- and C-terminal helical regions of HIV-1 gp41. Biochemistry 43:8230–8233. doi: 10.1021/bi049957v. [DOI] [PubMed] [Google Scholar]
- 24.Douglas JL, Panis ML, Ho E, Lin KY, Krawczyk SH, Grant DM, Cai R, Swaminathan S, Cihlar T. 2003. Inhibition of respiratory syncytial virus fusion by the small molecule VP-14637 via specific interactions with F protein. J Virol 77:5054–5064. doi: 10.1128/JVI.77.9.5054-5064.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Henderson HI, Hope TJ. 2006. The temperature arrested intermediate of virus-cell fusion is a functional step in HIV infection. Virol J 3:36. doi: 10.1186/1743-422X-3-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baba M, Pauwels R, Balzarini J, Arnout J, Desmyter J, De Clercq E. 1988. Mechanism of inhibitory effect of dextran sulfate and heparin on replication of human immunodeficiency virus in vitro. Proc Natl Acad Sci U S A 85:6132–6136. doi: 10.1073/pnas.85.16.6132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kilby JM, Hopkins S, Venetta TM, DiMassimo B, Cloud GA, Lee JY, Alldredge L, Hunter E, Lambert D, Bolognesi D, Matthews T, Johnson MR, Nowak MA, Shaw GM, Saag MS. 1998. Potent suppression of HIV-1 replication in humans by T-20, a peptide inhibitor of gp41-mediated virus entry. Nature Med 4:1302–1307. doi: 10.1038/3293. [DOI] [PubMed] [Google Scholar]
- 28.Wang H, Qi Z, Guo A, Mao Q, Lu H, An X, Xia C, Li X, Debnath AK, Wu S, Liu S, Jiang S. 2009. ADS-J1 inhibits human immunodeficiency virus type 1 entry by interacting with the gp41 pocket region and blocking fusion-active gp41 core formation. Antimicrob Agents Chemother 53:4987–4998. doi: 10.1128/AAC.00670-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chan DC, Fass D, Berger JM, Kim PS. 1997. Core structure of gp41 from the HIV envelope glycoprotein. Cell 89:263–273. doi: 10.1016/S0092-8674(00)80205-6. [DOI] [PubMed] [Google Scholar]
- 30.Liu S, Lu H, Xu Y, Wu S, Jiang S. 2005. Different from the HIV fusion inhibitor C34, the anti-HIV drug Fuzeon (T-20) inhibits HIV-1 entry by targeting multiple sites in gp41 and gp120. J Biol Chem 280:11259–11273. doi: 10.1074/jbc.M411141200. [DOI] [PubMed] [Google Scholar]
- 31.Liu S, Jing W, Cheung B, Lu H, Sun J, Yan X, Niu J, Farmar J, Wu S, Jiang S. 2007. HIV gp41 C-terminal heptad repeat contains multifunctional domains: relation to mechanisms of action of anti-HIV peptides. J Biol Chem 282:9612–9620. doi: 10.1074/jbc.M609148200. [DOI] [PubMed] [Google Scholar]
- 32.He Y, Cheng J, Lu H, Li J, Hu J, Qi Z, Liu Z, Jiang S, Dai Q. 2008. Potent HIV fusion inhibitors against enfuvirtide-resistant HIV-1 strains. Proc Natl Acad Sci U S A 105:16332–16337. doi: 10.1073/pnas.0807335105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roche J, Louis JM, Grishaev A, Ying J, Bax A. 2014. Dissociation of the trimeric gp41 ectodomain at the lipid-water interface suggests an active role in HIV-1 Env-mediated membrane fusion. Proc Natl Acad Sci U S A 111:3425–3430. doi: 10.1073/pnas.1401397111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Roche J, Louis JM, Aniana A, Ghirlando R, Bax A. 2015. Complete dissociation of the HIV-1 gp41 ectodomain and membrane proximal regions upon phospholipid binding. J Biomol NMR 61:235–248. doi: 10.1007/s10858-015-9900-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dwyer JJ, Wilson KL, Davison DK, Freel SA, Seedorff JE, Wring SA, Tvermoes NA, Matthews TJ, Greenberg ML, Delmedico MK. 2007. Design of helical, oligomeric HIV-1 fusion inhibitor peptides with potent activity against enfuvirtide-resistant virus. Proc Natl Acad Sci U S A 104:12772–12777. doi: 10.1073/pnas.0701478104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cai L, Jiang S. 2010. Development of peptide and small-molecule HIV-1 fusion inhibitors that target gp41. ChemMedChem 5:1813–1824. doi: 10.1002/cmdc.201000289. [DOI] [PubMed] [Google Scholar]
- 37.Gao J, Lu G, Qi J, Li Y, Wu Y, Deng Y, Geng H, Li H, Wang Q, Xiao H, Tan W, Yan J, Gao GF. 2013. Structure of the fusion core and inhibition of fusion by a heptad repeat peptide derived from the S protein of Middle East respiratory syndrome coronavirus. J. Virol 87:13134–13140. doi: 10.1128/JVI.02433-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lu L, Tong P, Yu X, Pan C, Zou P, Chen YH, Jiang S. 2012. HIV-1 variants with a single-point mutation in the gp41 pocket region exhibiting different susceptibility to HIV fusion inhibitors with pocket- or membrane-binding domain. Biochim Biophys Acta 1818:2950–2957. doi: 10.1016/j.bbamem.2012.07.020. [DOI] [PubMed] [Google Scholar]
- 39.Li W, Wang Q, Li Y, Yu F, Liu Q, Qin B, Xie L, Lu L, Jiang S. 2015. A nanoparticle-encapsulated non-nucleoside reverse-transcriptase inhibitor with enhanced anti-HIV-1 activity and prolonged circulation time in plasma. Curr Pharm Des 21:925–935. doi: 10.2174/1381612820666141014125213. [DOI] [PubMed] [Google Scholar]
- 40.Sun Z, Zhu Y, Wang Q, Ye L, Dai Y, Su S, Yu F, Ying T, Yang C, Jiang S, Lu L. 2016. An immunogen containing four tandem 10E8 epitope repeats with exposed key residues induces antibodies that neutralize HIV-1 and activates an ADCC reporter gene. Emerg Microbes Infect 5:e65. doi: 10.1038/emi.2016.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tong P, Lu Z, Chen X, Wang Q, Yu F, Zou P, Yu X, Li Y, Lu L, Chen YH, Jiang S. 2013. An engineered HIV-1 gp41 trimeric coiled coil with increased stability and anti-HIV-1 activity: implication for developing anti-HIV microbicides. J Antimicrob Chemother 68:2533–2544. doi: 10.1093/jac/dkt230. [DOI] [PubMed] [Google Scholar]
- 42.Yao X, Chong H, Zhang C, Waltersperger S, Wang M, Cui S, He Y. 2012. Broad antiviral activity and crystal structure of HIV-1 fusion inhibitor sifuvirtide. J Biol Chem 287:6788–6796. doi: 10.1074/jbc.M111.317883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lu L, Pan C, Li Y, Lu H, He W, Jiang S. 2012. A bivalent recombinant protein inactivates HIV-1 by targeting the gp41 prehairpin fusion intermediate induced by CD4 D1D2 domains. Retrovirology 9:104. doi: 10.1186/1742-4690-9-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhu X, Zhu Y, Ye S, Wang Q, Xu W, Su S, Sun Z, Yu F, Liu Q, Wang C, Zhang T, Zhang Z, Zhang X, Xu J, Du L, Liu K, Lu L, Zhang R, Jiang S. 2015. Improved pharmacological and structural properties of HIV fusion inhibitor AP3 over enfuvirtide: highlighting advantages of artificial peptide strategy. Sci Rep 5:13028. doi: 10.1038/srep13028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Otwinowski Z, Minor W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 46.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. 2007. Phaser crystallographic software. J Appl Crystallogr 40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mehta N, Trzmielina S, Nonyane BA, Eliot MN, Lin R, Foulkes AS, McNeal K, Ammann A, Eulalievyolo V, Sullivan JL, Luzuriaga K, Somasundaran M. 2009. Low-cost HIV-1 diagnosis and quantification in dried blood spots by real time PCR. PLoS One 4:e5819. doi: 10.1371/journal.pone.0005819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Eggink D, Bontjer I, Langedijk JP, Berkhout B, Sanders RW. 2011. Resistance of human immunodeficiency virus type 1 to a third-generation fusion inhibitor requires multiple mutations in gp41 and is accompanied by a dramatic loss of gp41 function. J Virol 85:10785–10797. doi: 10.1128/JVI.05331-11. [DOI] [PMC free article] [PubMed] [Google Scholar]