

ABSTRACT
Measles virus (MV) infection is undergoing resurgence and remains one of the leading causes of death among young children worldwide despite the availability of an effective measles vaccine. MV infects its target cells by coordinated action of the MV hemagglutinin (H) and fusion (F) envelope glycoproteins; upon receptor engagement by H, the prefusion F undergoes a structural transition, extending and inserting into the target cell membrane and then refolding into a postfusion structure that fuses the viral and cell membranes. By interfering with this structural transition of F, peptides derived from the heptad repeat (HR) regions of F can inhibit MV infection at the entry stage. In previous work, we have generated potent MV fusion inhibitors by dimerizing the F-derived peptides and conjugating them to cholesterol. We have shown that prophylactic intranasal administration of our lead fusion inhibitor efficiently protects from MV infection in vivo. We show here that peptides tagged with lipophilic moieties self-assemble into nanoparticles until they reach the target cells, where they are integrated into cell membranes. The self-assembly feature enhances biodistribution and the half-life of the peptides, while integration into the target cell membrane increases fusion inhibitor potency. These factors together modulate in vivo efficacy. The results suggest a new framework for developing effective fusion inhibitory peptides.
IMPORTANCE Measles virus (MV) infection causes an acute illness that may be associated with infection of the central nervous system (CNS) and severe neurological disease. No specific treatment is available. We have shown that fusion-inhibitory peptides delivered intranasally provide effective prophylaxis against MV infection. We show here that specific biophysical properties regulate the in vivo efficacy of MV F-derived peptides.
KEYWORDS: Viral fusion inhibitor, self-assembling nanoparticles, lipid tagging, membrane insertion
Infection by measles virus (MV) remains one of the leading causes of death among young children worldwide (1) despite the availability of an effective measles vaccine. MV was considered to be eliminated in the United States in 2000 (defined as interruption of continuous transmission lasting ≥12 months) (2). However, the resurgence of measles in the United States in 2014 and 2015 made it clear that measles infection has yet to be eradicated. In 2014, MV caused 114,900 deaths, globally mostly in children under 5 years. As reported by the World Health Organization (WHO), the United States experienced more cases in 2014 than in any year since 1996 (3), and MV was added to the NIAID's list of emerging viruses. The 2015 outbreak that began at Disneyland in Southern California further highlights our vulnerability to MV, especially in situations where herd immunity has declined. Outbreaks in the United States are often attributed to the presence of unvaccinated individuals; however, a significant number of cases occur in previously vaccinated people (4, 5).
MV infection starts in the respiratory tract, where alveolar macrophages and dendritic cells are the primary CD150-expressing targets (6–9). The binding of the MV receptor binding protein hemagglutinin (H) to CD150, accompanied by membrane fusion mediated by the MV fusion (F) protein, leads to infection of these cells. The first MV-infected cells then transmit the virus to bronchus-associated lymphoid tissues and/or draining lymph nodes, where the virus proliferates in B and T lymphocytes (which also express CD150) and viremia ensues (6, 10). The adherent junction protein PVRL4 (or nectin-4) (11–15) MV receptor on the basolateral surface of the respiratory epithelial cells has been implicated in viral transmission at later stages of pathogenesis but is likely not involved at the early stages (10, 14).
Coordinated action of the MV H and F envelope glycoproteins is essential for viral entry. The trimeric F structure is kinetically trapped in a metastable conformation and primed for fusion activation upon engagement of H by a cell surface receptor, either CD150 (SLAM) or nectin-4 (11–17). After receptor engagement by H, the prefusion F undergoes a structural transition, extending and inserting its hydrophobic “fusion peptide” into the target cell membrane. The entry process is thought to be driven by the refolding of F into a six-helix bundle postfusion structure that brings together and fuses the viral and cell membranes (18–25). Peptides derived from the C-terminal heptad repeat (HRC) regions of F can potently inhibit MV infection at this early stage (26–28) by interfering with this structural transition of F. We have shown that the efficacy of F-inhibitory peptides is modulated by the H-receptor interaction; the peptide concentrations that inhibits fusion by 50% and 90% (IC50 and IC90, respectively) are increased in the presence of stronger H-receptor interaction (29). These relationships between fusion activation, receptor engagement, and inhibition are relevant to antiviral design and help explain some of the previous hurdles to developing effective entry inhibitors for MV (30–32). We contend that the dimeric form of the prototype lipid-conjugated fusion inhibitor (HRC4, shown in Table 1), either by targeting multiple N-terminal heptad repeat (HRN) domains and/or by targeting the HRN at multiple sites, has partially overcome this MV-specific issue, as evidenced by its in vivo efficacy (29).
TABLE 1.
MV HRC-derived peptides
| MV peptide | Sequence |
|---|---|
| HRC1a | Ac-PPISLERLDVGTNLGNAIAKLEDAKELLESSDQILR-GSGSG-C-(CH2CONH2)-NH2 |
| HRC2a | Ac-PPISLERLDVGTNLGNAIAKLEDAKELLESSDQILR-GSGSG-C-(PEG4-Chol)-NH2 |
| HRC3a | [Ac-PPISLERLDVGTNLGNAIAKLEDAKELLESSDQILR-GSGSG-C-(MAL-PEG11)-NH2]2 |
| HRC4a | [Ac-PPISLERLDVGTNLGNAIAKLEDAKELLESSDQILR-GSGSG-C-(MAL-PEG4)NH2]2-Chol |
| HRC5 | Ac-PPISLERLDVGTNLGNAIAKLEDAKELLESSDQILR-GSGSG-C-(PEG4-Toc)-NH2 |
| HRC6 | [Ac-PPISLERLDVGTNLGNAIAKLEDAKELLESSDQILR-GSGSG-C-(MAL-PEG4)NH2]2-Toc |
We show here that the addition of lipophilic moieties imparts self-assembling properties to fusion-inhibitory peptides. The kind of lipophilic moiety and the peptidic region both contribute to a balance between self-assembly and disaggregation. We propose that the propensity of a peptide to desegregate in the proximity of its target cell correlates with its in vivo efficacy. We speculate that an optimal balance between self-assembly and membrane integration regulates in vivo efficacy. Our results provide a framework for designing effective antiviral fusion-inhibitory peptides. This strategy, in which advances in nanotechnology are integrated with an understanding of the molecular mechanisms of viral entry and its in vivo inhibition, will permit a streamlined antiviral approach to existing and emerging viral pathogens.
RESULTS
Lipid-conjugated inhibitory peptides self-assemble into nanoparticles.
Peptides may exhibit high amphipathicity and a tendency to self-associate into higher-order structures, depending on the nature of their amino acid sequence and folding. Chemical conjugation of peptides with highly hydrophobic molecules such as cholesterol (Chol) and tocopherol (Toc) might affect peptide stability in aqueous solution. The MV HRC-derived fusion-inhibitory peptides described here (HRC1 to -6) (Table 1) contain a hydrophobic lipid binding domain connected to the virus-specific amino acid sequence by a flexible hydrophilic polyethylene glycol (PEG) linker, features resembling the building blocks of self-assembling nanoparticles such as albumin polymer-conjugated nanocarriers (33). We had hypothesized that the lipid-tagged peptides disperse into the target cell membrane (34) and therefore asked whether they self-assemble into nanoparticles (35, 36). Self-assembling properties could have implications for the peptides' mode of antiviral action. We propose that the in vivo antiviral activity we observed results from a combination of self-aggregation properties and integration into the target cell membrane, and we tested this hypothesis experimentally. We explored the relationship between physicochemical properties, aggregation, and membrane insertion kinetics of the conjugated peptides to identify features that correlate with inhibitory efficacy in vitro and in vivo. Specifically, the tendency of HRC peptides to self-assemble, the peptides' aggregation profiles, and their target membrane desegregation were assessed.
As an initial screen to examine the tendency of each peptide to self-assemble in aqueous solution, an 8-anilinonaphthalene-1-sulfonic acid (ANS) fluorescence emission-based methodology was used. ANS is a small probe (∼300 Da) which nonspecifically interacts with hydrophobic sites resulting from molecular aggregation (37). Upon binding to these sites, the probe fluorescence emission maximum λmax undergoes a blue shift and an intensity (IF) increase that correlates with peptide aggregation. In the presence of ANS, increasing concentrations of peptides HRC2, -4, -5, and -6 induced λmax blue shifts (Fig. 1A). The maximum observed shift was similar among peptides, averaging 65 (± 5) nm, and was constant for concentrations above 15 μM. Above this concentration the population of emitting molecules is predominantly bound to peptide aggregates. An increasing IF value as a function of the HRC2, -4, -5, and -6 peptide concentrations was also detected (Fig. 1B). At 40 μM each peptide, the ANS Σ[IF(λ)] was over 15-fold higher than the control value obtained in the absence of peptide. In the presence of the HRC1 and -3 peptides (without a lipid moiety), no significant ANS fluorescence emission variation was observed. These results suggest that peptides HRC2, -4, -5, and -6 aggregate in solution, forming local hydrophobic sites, while peptides HRC1 and -3 do not self-assemble.
FIG 1.

Aggregation of the MV HRC peptides evaluated by ANS fluorescence properties. ANS fluorescence emission properties are an indicator of peptide aggregation in aqueous solution. ANS (12.8 μM) was titrated with small volumes of each peptide up to final concentrations, [HRCX], where X represents the number of each peptide (HRC1, white circles; HRC2, red circles; HRC3, blue circles; HRC4, green circles; HRC5, gray circles; HRC6, black circles). The fluorescence maximum emission wavelength (λmax) (A) and the spectrum integral {Σ[IF(λ)]} (B) were plotted as a function of [HRCX]. Panels A1 and B1 correspond to zoomed representations of the dashed areas in panels A and B, respectively. Results correspond to the average of three independent replicates. Error bars represent the SD.
Given the potential of peptides HRC2, 4, -5, and -6 to self-assemble in aqueous solution, the resulting aggregates' dimensions were determined using dynamic light scattering (DLS). This technique is capable of nanoscale particle hydrodynamic radius (RH) measurements in solution and is suitable for peptide/protein assays (38). The number-averaged size distribution profiles of each peptide were used to retrieve the modal RH at different peptide concentrations (Fig. 2). Peptides HRC2 and -5 displayed number-averaged particle RHs between 31 and 47 nm and between 41 and 56 nm, respectively, irrespective of the peptide concentration used. Peptides HRC4 and -6 showed similar average particle RH values between 27 and 82 nm, but these values were significantly influenced by increasing peptide concentrations. These observations indicate that the conjugated HRC peptides self-assemble into stable aggregates in solution, even at low concentrations. For the peptides in the monomeric form, similar aggregates sizes were obtained even at different concentrations, which is indicative of organized aggregates with well-defined structures. In dimeric form, the peptides seem to aggregate into larger particles in a concentration-dependent fashion, which is indicative of loose aggregates of unspecific structure. Whether Chol or Toc was used as the lipid conjugate did not significantly affect the peptide's aggregation behavior.
FIG 2.

Aggregation of the MV HRC peptides evaluated by dynamic light scattering (DLS). DLS particle size determination of aggregated HRC2 (black), -4 (red), -5 (blue), and -6 (gray) peptides is shown. Size distribution profiles based on particle number were obtained from DLS autocorrelation functions. (A) Representative profile obtained for peptide HRC6 at 10 μM. (B) RH values were retrieved from the statistical mode (dark gray bar in panel A) of each peptide distribution at final concentrations, [HRCX]. The mean RH values were compared using Tukey's method to test pairwise differences between concentrations of the same peptide (comparison to 1 μM is shown). The presented results are the averages of three independent replicates. Error bars represent the SD. n.s., not significant; *, P ≤ 0.01; **, P ≤ 0.001; ***, P ≤ 0.0001.
Insertion of lipid-targeted peptides in lipid membranes.
Strategic conjugation of HRC peptides with either Chol or Toc moieties has considerably improved the efficacy of these peptides as antiviral fusion inhibitors (29). This effect was considered to be due to enhanced peptide-membrane interactions, as previously suggested for other antiviral fusion inhibitor peptides (39, 40). Because of the hydrophobicity of the conjugated groups, it is assumed that the peptides insert into the membrane surface. We propose that this insertion correlates with the antiviral activity. In order to test this hypothesis, we have used liposomes as membrane model systems for peptide-membrane interaction studies. The precise lipid composition of the MV envelope remains poorly understood, but it has been shown that MV virions emerge from high-cholesterol lipid raft domains (41, 42). 1-Palmitoyl-2-oleyl-sn-glycero-3-phosphocholine (POPC)–Chol (2:1) and POPC-Chol-chicken egg sphingomyelin (SM) (1:1:1) mixtures were used to mimic the MV envelope composition and raft domains, while POPC was used to mimic the cellular membrane composition. To study the peptide affinity toward different lipid membrane compositions, we used large unilamellar vesicles (LUVs) containing the lipophilic 4-(2-[6-(dioctylamino)-2-naphthalenyl]ethenyl)-1-(3-sulfopropyl)pyridinium inner salt (di-8-ANEPPS) fluorescent probe. The probe's excitation spectrum is sensitive to variations in the membrane dipole potential and undergoes positive or negative wavelength shifts as a result of protein/peptide interaction or insertion into the membrane. These shifts can be converted to amplitudes in differential fluorescence excitation spectra and analyzed through a semiquantitative ratiometric approach (43). The peptides that induced a larger di-8-ANEPPS spectral shift in the presence of the different lipid compositions were HRC2, -4, -5, and -6. This is evidenced by the decrease in the normalized excitation intensity ratio (Rnorm) as a function of the peptide concentration (Fig. 3). The Rnorm values may be higher or lower than one, depending on whether the spectral shift relative to the control spectra is to lower or higher wavelengths, respectively. HRC2 and -5, both conjugated monomers, seem to disturb Chol-containing membranes with greater magnitude. The HRC4 and -6 peptides display similar Rnorm values between the three lipid compositions. No changes in the di-8-ANEPPS spectra were observed in the presence of the HRC1 and -3 peptides, even at high concentrations. Note that it remains a formal possibility that these peptides interact with lipids without significantly disturbing the membrane dipole potential. The results suggest that Chol and Toc conjugation modified the peptides' behavior toward lipid membranes, allowing them to insert in lipid membranes and disturbing the lipid-water dipole equilibrium.
FIG 3.

Peptide-membrane interaction studies by ANEPPS excitation spectrum shift. Ratiometric analysis of di-8-ANEPPS excitation spectrum deviations induced by HRC peptide-membrane interactions is shown. Discrete concentrations of HRC1 to -6 (A to F, respectively) were incubated with di-8-ANEPPS-labeled POPC (white circles, black line), POPC-Chol (2:1) (red circles, red line) and POPC-Chol-SM (1:1:1) LUV (blue circles, blue line). Intensity ratios (Rnorm) were calculated based on the respective differential excitation spectrum peak wavelengths and normalized to the control in the absence of peptide. For each ratiometric graph, the respective di-8-ANEPPS differential excitation spectrum obtained at 40 μM peptide concentration is shown as a representative example. These were obtained by subtraction of di-8-ANEPPS normalized control spectra from the normalized spectra in the presence of each peptide. Ratiometric results correspond to the average of three independent replicates. The presented differential spectra are one of three independent replicates. Error bars represent the SD.
In order to complement the di-8-ANEPPS fluorescence-based assays and further understand how the HRC peptides interact and bind to lipid membranes, a surface plasmon resonance (SPR) approach using Biacore technology was applied. This flow-based technique allows real-time detection of bound molecules on a small unilamellar vesicle (SUV)-covered surface (44). Using the same lipid formulations that were used for the di-8-ANEPPS assay, kinetic profiles (sensorgrams) of each peptide binding to SUV surfaces were acquired. Each sensorgram is composed of sequential binding and unbinding phases, corresponding to the sample injection over the lipid vesicle surface followed by the removal of bound sample molecules through solvent flow, respectively. Based on the maximum peptide binding and SUV deposition response, the respective peptide-to lipid [mol(P)/mol(L)] ratios were calculated and plotted as a function of the injected peptide concentration (Fig. 4). HRC4 and -6 showed the highest binding toward each lipid composition tested, followed by HRC2. On the other hand, HRC1, -3, and -5 showed very low binding affinity to all the different lipid compositions used. In general, all the HRC peptides showed greater affinity toward deposited POPC SUVs. From the unbinding data for the HRC peptides (t > 200 s), we observed that HRC 2 remained tightly bound to the SUV surface after the binding phase. HRC4, -5, and -6 were removed from the surface slowly during unbinding, but faster than HRC2. HRC1 and -3 showed weaker binding and were quickly removed by solvent flow. Although HRC1 and -3 seem to transiently interact with lipids, Chol and Toc conjugation considerably improved the membrane interaction properties of the HRC peptides. Biacore SPR sensorgrams indicate that dimers have faster binding kinetics and achieve higher mol(P)/mol(L) values than the respective monomers. Additionally, our results suggest that a fraction of conjugated peptides remains bound to lipid membranes with increased stability, even after the unbinding phase.
FIG 4.

Sensorgram maximum binding response versus peptide concentration. SPR time-resolved sensorgrams of HRC1 to -6 (A to F) interacting with deposited POPC (left sensorgrams), POPC-Chol (2:1) (center sensorgrams), and POPC-Chol-SM (1:1:1) SUV (right sensorgrams). Discrete HRC-peptide concentrations were injected over the deposited vesicles at a 5-μl/min flow speed for a total of 200 s. Each sample was allowed a 600-s unbinding time. (G) Concentration-dependent SPR peptide-to-lipid ratios [mol(P)/mol(L)] of HRC1 (white circles), -2 (red circles), -3 (blue circles), -4 (green circles), -5 (gray circles), and -6 (black circles) peptides interacting with deposited POPC (left), POPC-Chol (2:1) (center), and POPC-Chol-SM (1:1:1) SUV (right). Peptide binding and SUV capture response values were retrieved from individual sensorgrams at specific reporting time points. mol(P)/mol(L) values were calculated based on the relationship 1 RU ∼ 1 pg/mm2 of bound peptide or lipid and plotted as a function of the injected peptide concentration. Error bars represent the SD of two independent replicates.
Lipid-conjugated inhibitory peptides inhibit cell-to-cell fusion.
We have previously identified a strong correlation between efficiency of inhibiting cell-to-cell fusion in vitro and in vivo potency (45). We have attributed the enhanced in vivo efficacy of specific peptides to their membrane insertion properties identified in vitro (34). Here we describe the biophysical properties of monomeric and dimeric peptides conjugated with different lipophilic moieties and peptide valencies, which resulted in different propensities for membrane insertion or self-association. For the dimeric peptides, these properties are also concentration dependent. We therefore asked which properties best correlate with fusion-inhibitory activity of the peptides studied here. We considered that the dimeric form of the fusion inhibitor peptides (HRC3, HRC4, and HRC6), by simultaneously targeting multiple N-terminal heptad repeat (HRN) domains, may have an inherent advantage over the monomeric peptides. However, the presence of the two peptidic sequences alters the membrane association. To determine how these factors affect in vitro potency we measured fusion inhibition using a semiquantitative β-galactosidase (β-gal) complementation assay (46). This assay measures the fusion of cells that express viral envelope glycoproteins (MV IC323 H/F) with cells that express the MV receptor SLAM or nectin-4 (Table 2). Table 2 shows the peptide concentrations that blocked 50% (IC50) and 90% (IC90) of fusion. As anticipated, the addition of a lipophilic moiety increased the antiviral potency of the HRC peptides compared to that of the untagged peptides. However, the difference in potency between the monomeric/dimeric peptides with Toc was not as dramatic as for the Chol-tagged peptides. When fusion-inhibitory potency was assessed using CD150 as a receptor on the target cells, the fusion-inhibitory potency was higher than that with target cells expressing the nectin-4 receptor. The H–nectin-4 receptor interaction is stronger than the interaction between H and CD150 (11–15), and the data presented here are in agreement with our previous findings that correlate peptide efficacy versus F protein-mediated fusion to the strength of interaction between the cellular receptor and the receptor binding protein (H) (29). The most potent peptide under these conditions remains the dimeric peptide conjugated to Chol.
TABLE 2.
In vitro efficacy of MV HRC-derived peptides
| MV peptide | Fusion inhibitiona at 4 h |
|||
|---|---|---|---|---|
| SLAM |
Nectin-4 |
|||
| IC50 (μM) | IC90 (μM) | IC50 (μM) | IC90 (μM) | |
| HRC1 | >10 | >10 | >10 | >10 |
| HRC2 | 0.05 ± 0.01 | ∼10 | 4.6 ± 0.6 | ∼10 |
| HRC3 | ND | ND | ND | ND |
| HRC4 | 0.01 ± 0.005 | ∼0.05 | 0.1 ± 0.04 | ∼0.5 |
| HRC5 | 0.25 ± 0.09 | ∼1 | 1.4 ± 0.5 | >10 |
| HRC6 | 0.30 ± 0.10 | ∼1 | 1.3 ± 0.5 | ∼10 |
Values are means and standard deviations. ND, not determined.
Lipid-targeted inhibitory peptides cross the HAE differentially.
To assess the potential for lung delivery of the lipid-conjugated HRC peptides, we used an ex vivo mucosal tissue model consisting of normal, human-derived tracheal/bronchial epithelial cells that have been cultured to form a highly differentiated model that closely resembles the human airway epithelium (HAE) tissue of the respiratory tract. We have used HAE for evaluating entry inhibitors (47–51). In previous work, we found that the monomeric (HRC2) and dimeric (HRC4) peptides conjugated to cholesterol both reach the basolateral face in similar concentrations (29). Based on the data in Fig. 4, we hypothesized that the HRC6 peptide may remain localized on the luminal side of the lung with minimal spillover into the circulation. If this hypothesis is correct, then the peptides, added to the apical surface, would remain on the apical surface of the airway epithelium with minimal transfer to the basolateral lumen.
HRC4 and HRC6 peptides were added to the apical side of the HAE to determine when and at what concentration they would reach the basolateral lumen (Fig. 5). The amount of peptide (20 μl at 250 μM) was sufficient to reach concentrations of 5 μM if added directly to the basolateral reservoir (which contains 1 ml of medium). The presence of peptide basolaterally was detected by enzyme-linked immunosorbent assay (ELISA) at 8, 24, 48, and 72 h after treatment, with peak concentrations for MV HRC4 (∼0.02 μM) at 24 h, indicating that both peptides reach the basolateral reservoir. Detectable levels of HRC6 peptide were observed only at 48 and 72 h and at two-log-lower concentrations. The results indicate that the biophysical properties imparted by the lipophilic (Toc) moiety altered the peptide permeability to the HAE.
FIG 5.

Peptides cross the human airway epithelium (HAE). Medium (liquid interface) was collected at each time point for quantification of peptide by ELISA. Kinetic analysis (ELISA) of the peptide concentration in the liquid reservoir of the culture reflects minimal transit through the HAE for HRC4 (A) and HRC6 (B).
Intranasally delivered lipid-conjugated dimer peptides inhibit MV infection in vivo.
MV infection in cotton rats (CR) is a natural infection model (52). MV replicates in CR lung tissue with peak titers occurring on day 4 or 5, and infection is overcome by day 8. We have previously shown that HRC2 and HRC4 delivered intranasally (i.n.) remain in the lungs (29, 45). Here we compared the biodistributions of HRC4 and HRC6 in the CR. Peptides were delivered intranasally (6 mg/kg, 3 animals per peptide), and the animals were euthanized 8 h after administration. Serum, lungs, and brain were collected for each animal, and peptide concentrations were measured by ELISA (29, 45) (Fig. 6). HRC4 and HRC6 remain in the lungs after intranasal delivery (HRC4, 0.44 μM; HRC6, 2.00 μM). Following i.n. delivery the serum concentrations of HRC4 and HRC6 were 0.03 μM and 0.003 μM, respectively. Thus, HRC4 crosses the airway epithelium of the CR in vivo, while HRC6 remains localized to the lungs. HRC4 attains a brain concentration of 0.002 μM; in contrast, HRC6 is below the limit of detection in brain tissue. The finding that most of the Toc-conjugated peptide (HRC6) that is delivered i.n. remains localized in the respiratory tract suggests that this peptide could be used for i.n. prophylaxis against viruses transmitted via the respiratory route.
FIG 6.
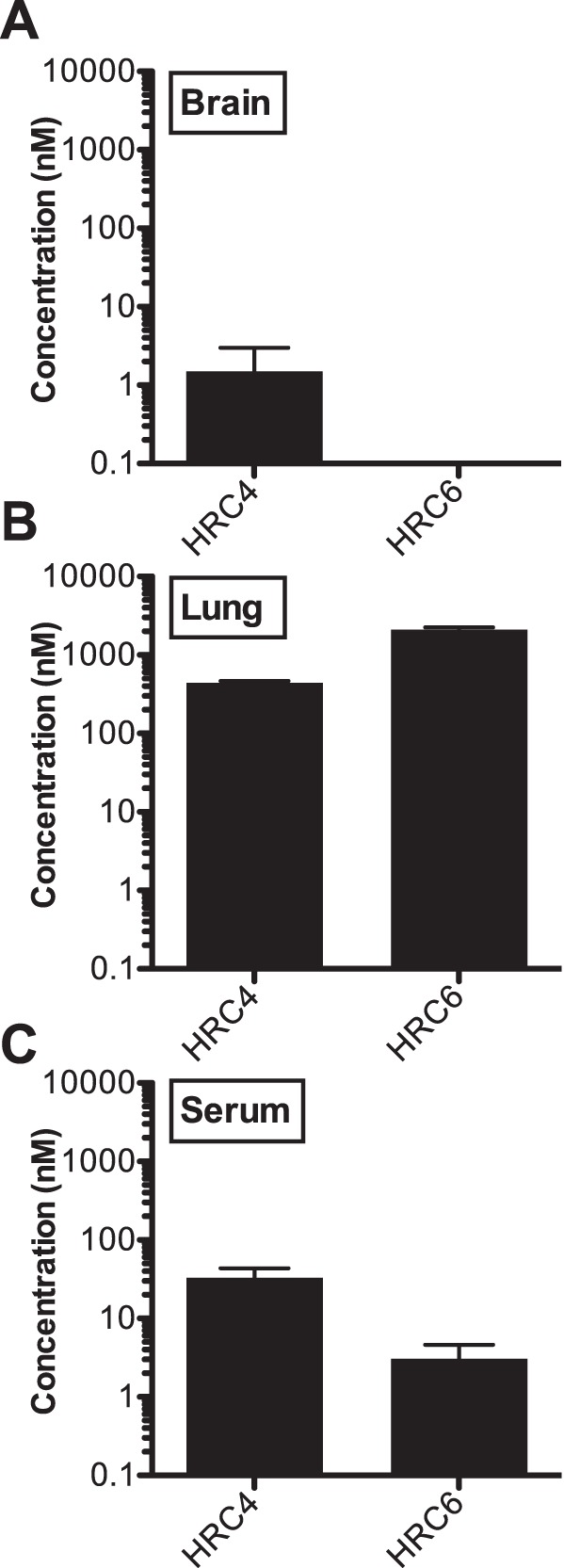
Bioavailability of MV HRC-derived peptides in cotton rats at 8 h after intranasal administration. Animals were inoculated with 100 μl per nare (200 μl total, 6 mg/kg, 3 animals) and then were euthanized 8 h after administration. Peptide concentrations in serum, lung, and brain were measured by ELISA (shown in log scale as in reference 29). Data are presented as an average from three animals ± SD. Data are on a log scale.
The peptides that were delivered intranasally have no detectable undesirable effects in the cotton rats. HRC6 (dimeric form conjugated with Toc) reached the highest lung concentration (Fig. 6) (29), with minimal presence in the serum and none detectable in the central nervous system (CNS). In HAE (Fig. 5), apically delivered HRC6 is found in the basolateral reservoir only 72 h after administration (and at a 10-fold lower concentration than the HRC4 cholesterol-conjugated peptide), indicating that the dimer with Toc is retained on the apical side of the human airway, in agreement with the biodistribution results. Our data indicate that the lipid moiety greatly influences in vivo biodistribution.
We next assessed the antiviral prophylactic efficacy of the peptides after intranasal delivery in the CR. We compared the two peptides whose biodistribution was assessed as described above (HRC4 and HRC6), and we added the other HRC peptides (Table 1) as controls (29, 50, 53).
The animals were given the fusion-inhibitory peptides i.n. twice (5 mg/kg each) at 24 and 12 h before infection (Fig. 7). Four days after infection, the viral titer in the lungs of untreated animals was 104 50% tissue culture infectious doses (TCID50)/g lung tissue (±1 × 103); the HRC5 peptide-treated animals had viral titers with no significant differences from those of the untreated animals (Fig. 7), despite the in vitro potency of this peptide (Table 2). After prophylaxis with the MV HRC1-derived peptides, the titer was reduced to 9 × 103 TCID50/g lung tissue (±2 × 103), and after prophylaxis with the other MV-derived peptides (HRC2, HRC3, HRC4, and HRC6), no MV virus at all was detected in the lungs. Statistical analysis using a Mann-Whitney U test (***, P < 0.001) revealed the strong significance of the MV peptides' inhibitory effect compared to either the untreated group or the group treated with HRC5-derived peptides. The i.n. delivered MV peptides (with the exception of the monomeric untagged HRC1 and the Toc-tagged HRC5) efficiently blocked MV infection in the CR.
FIG 7.
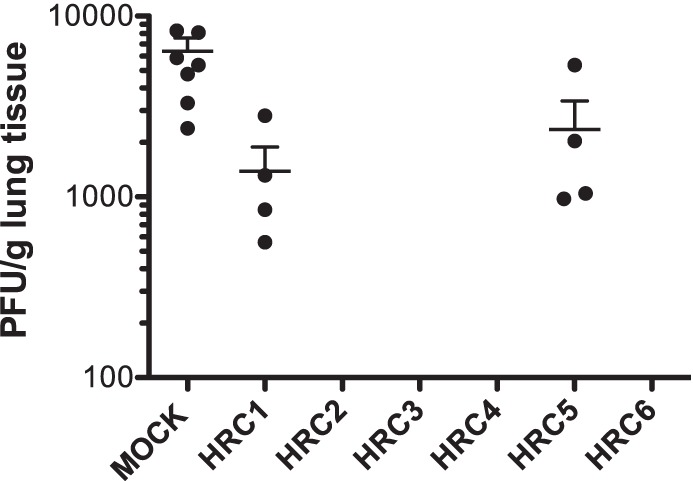
Intranasal administration of MV-derived peptides protects cotton rats from MV infection. Cotton rats (n = 4) were infected i.n. with MV at 24 h after the first peptide treatment and were euthanized at 4 days postinfection. MV titration of lung homogenates showed that HRC2, HRC3, HRC4, and HRC6 block infection in CR (***, P = 0.001 by the Mann-Whitney U test). The limit of viral detection was 102 PFU/g.
We previously showed that i.n. delivery of MV HRC4 peptides alone is sufficient to protect from fatal measles encephalitis. We now assessed whether the HRC5 and HRC6 peptides protect mice from a lethal MV challenge. Animals (SLAM×IFNARKO mice as used previously [29, 45]) were treated with HRC5 or HRC6 intranasally (6 mg/kg) at 24 h and 4 h before infection (10 μl of peptide-containing vehicle). A mock-infected group received vehicle at the same time. Animals were followed for 28 days after the infection. The survival median for the mock-infected group was 9.5 days. For the animals treated with HRC5, the survival median was 18 days (***, P = 0.0007; Mantel-Cox test); the peptide significantly enhanced the median survival time but did not fully protect. The i.n. administration of HRC6 completely protected SLAM×IFNARKO mice from fatal CNS infection. (Fig. 8).
FIG 8.
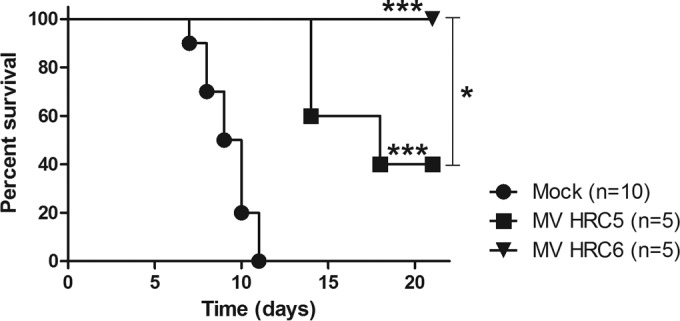
Intranasal administration of MV-derived peptides protects transgenic mice from MV infection. SLAM×IFNARKO mice were infected intranasally with 104 PFU of MV G954. Animals were treated with HRC5 or HRC6 intranasally (6 mg/kg) at 24 h and 4 h before infection. The mock-infected group only received the vehicle at the same time. the Mantel-Cox test was used for the survival comparison analysis: ***, P = 0.0007; *, P = 0.0486 (between HRC5 and -6).
DISCUSSION
Measles virus (MV) induces respiratory infection and can cause a profound suppression of the immune system that may permit opportunistic infections. CNS complications of MV infection may occur soon after infection in the case of acute encephalomyelitis or years after infection as a result of viral persistence in subacute sclerosing panencephalitis (SSPE) and progressive infectious encephalitis or measles inclusion body encephalitis (MIBE). There are no specific therapies for acute complications of MV or for persistent MV infections (54–58). While vaccine compliance is a major problem in MV eradication, important groups of vulnerable patients cannot be protected by vaccination, and vaccine failure also contributes to the susceptible population (59–65). A 2011 outbreak in New York City originated with a fully vaccinated young adult and spread to several vaccinated individuals with documented markers of immunity. Of the 98 cases of MV in a 2011 measles outbreak in Canada, over half had received two doses of MV vaccine (5, 60), and a significant number of vaccinated individuals contracted MV. Up to 10% of people do not develop adequate protective antibodies (even after the recommended two doses of vaccine), in contrast to the longstanding immunity elicited by the natural infection. Vaccine-elicited immunity also likely wanes over time (63, 64, 66), and other factors can affect the response (65, 67). Antiviral prophylaxis could protect unvaccinated individuals and prevent spread of virus in the population. In infants under 9 months who do not respond to live attenuated MV vaccine and in immunocompromised people, antiviral treatment could prevent severe consequences of MV infection such as pneumonia and encephalitis. Since we expect our proposed fusion-inhibitory peptide antivirals to be host factor independent, they will fill a specific need for immunocompromised people at risk for MV infection who cannot be vaccinated or do not respond adequately to vaccine.
Our results point consistently to the conclusions that the most efficient antiviral peptides are lipid-conjugated dimers, self-assemble into loose aggregates, and bind extensively to membranes with high residence times. Several examples of engineering polyvalent biological interactions have been described (68), and specific examples where polyvalency increases the potency of fusion inhibitors (69, 70) or entry inhibitors (17) have been reported. For HRC fusion inhibitors, dimerization/multimerization (45, 53) reduces the koff of the inhibitor-fusion protein complex formation in an effect that has been termed the “avidity effect” (71). The lipophilic tag, on the other hand, increases the kon for complex formation (45) by targeting the peptide to the cell membrane where the virus fuses, increasing the local peptide concentration. We asked whether the two modifications would work in concert, and indeed we demonstrate that the interplay between the lipid moiety and the peptidic portion affects two important properties of the peptides: self-assembly and membrane insertion. The hydrophobic lipid moiety confers the self-aggregation properties; however, the membrane insertion efficiency is influenced by the lipophilic moiety as well as by the peptide valency. Dimeric peptides have steric restrictions to organized packing in a micelle-like arrangement; monomeric peptides, however, do not have these restrictions and form organized micelle-like structures, which are stable in solution and therefore less prone to interact with lipids. In fact we found that only the dimeric HRC4 and HRC6 peptides insert efficiently into membranes, probably as a consequence of the assembly into unstable aggregates in aqueous solution. In contrast to the other peptides, HRC4 and HRC6 release slowly from membranes. We propose that these features of unstable self-aggregation in solution combined with the capacity to insert in target cell membranes with prolonged residence are key for the in vitro and in vivo potencies that we observe for the dimeric lipid-conjugated peptides.
The lack of efficacy of the monomeric peptide conjugated to tocopherol (HRC5), despite its in vitro potency, was a surprise. This peptide's in vitro activity was similar to that of the dimeric form (HRC6) (Table 2), but it was totally ineffective in cotton rats (Fig. 7). However, HRC5 improved survival in the mouse model (Fig. 8). We suggest that the specific biophysical properties of these two peptides (Fig. 4) led to different membrane association, which may explain this difference in efficacy and the relative inactivity of the monomer in vivo.
We considered the possibility that by targeting the lung, and particularly if in fact lung-resident dendritic cells and alveolar macrophages are targeted, the peptides might elicit an immune response that could affect their efficacy during subsequent exposures to MV (72, 73). This would be a drawback for our proposed antiviral strategy, which is designed for people who cannot be efficiently vaccinated. To explore this possibility, we performed a preliminary experiment using MV HRC4 and the corresponding human parainfluenza virus type 3 (HPIV3) F dimeric peptide as a control (29). Cotton rats (CR) were administered either peptide i.n. for 3 weeks (5 times per week, once a day), and sera were collected before and after peptide administration. After 3 weeks of treatment the animals—either peptide exposed, mock exposed, or exposed to the control HPIV3-derived peptide—were kept for a week without treatment and then challenged with virus. Pretreatment with HRC4 did not affect the efficacy of the subsequent treatment in terms of reducing viral titers, and the sera tested negative for the presence of antipeptide antibodies (data not shown). These preliminary results suggest that several peptide administrations are possible without eliciting a detrimental antipeptide response.
In this study, we confirmed our previous findings that the fusion-inhibitory potential of peptides is influenced by the nature of the target cell receptor (Table 2). To block fusion between effector cells and target cells expressing nectin-4 requires more peptides than when the target cells are expressing CD150. The affinity constant (Kd) between CD150 and H has been reported to be 93.5 nM, and that between nectin-4 and H has been reported to be 20.1 nM (11–15). Based on the different Kd values, we inferred that peptide efficacy inversely correlates with H receptor affinity (e.g., nectin-4 versus CD150). We considered the possibility that the overall number of receptor molecules present on the target cell surface may also alter the overall apparent avidity, and this difference may also contribute to the different antiviral activities of the peptides. A potential role of avidity effect would be important for in vivo application of the fusion inhibitor peptides, and therefore future work will assess the receptor molecule density ex vivo and in vivo and correlate these values with antiviral potency.
The biophysical analyses described here can be used to guide the design of effective antiviral fusion inhibitory peptides, in light of the coherent evidence here for the effects on peptide efficacy of specific lipid conjugation, dimerization, self-assembly properties, and membrane binding properties. These biophysical characteristics form a solid background to the pharmacological and biological results, correlating well with in vivo efficacy of the antiviral peptides for preventing or treating MV infection. By modulating the lipid moiety and/or the PEG linkers, we improved peptide antiviral potency through modulation of aggregation and lipid affinity, and we suggest that these properties may also be used to regulate in vivo biodistribution. The data presented here should in the future be confirmed in animal models that more closely reflect human infection. Nonhuman primates (NHP) mimic several aspects of MV infection in humans (6, 74), and future work will include assessment of optimized fusion inhibitors in NHP. This approach should readily translate into a streamlined development plan for safe and highly effective fusion inhibitors that can be aerosolized/inhaled for anti-MV prophylaxis in high-risk unvaccinated populations and vulnerable hosts.
MATERIALS AND METHODS
Chemicals and reagents.
The lipids 1-palmitoyl-2-oleyl-sn-glycero-3-phosphocholine (POPC) and chicken egg sphingomyelin (SM) were purchased from Avanti Polar Lipids (Alabaster, AL, USA), and cholesterol (Chol) was purchased from Sigma-Aldrich (St. Louis, MO, USA). HEPES, NaCl, NaOH, dimethyl sulfoxide (DMSO), ethanol, and chloroform (the last three with spectroscopic grade) were obtained from Merck (Darmstadt, Germany). The fluorescent probes 8-anilino-1-naphthalenesulfonic acid (ANS) and 4-(2-[6-(dioctylamino)-2-naphthalenyl]ethenyl)-1-(3-sulfopropyl)pyridinium inner salt (di-8-ANEPPS) were purchased from Sigma. Biacore sensor chip regeneration reagents, namely, 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS) and methanol, were also purchased from Sigma. Sodium dodecyl sulfate (SDS) was from GE Healthcare (Little Chalfont, United Kingdom). Large unilamellar vesicle (LUV) and small unilamellar vesicle (SUV) suspensions were prepared as previously described (75). The lipid mixture was first solubilized in chloroform in a round-bottom flask. The solvent was evaporated under nitrogen flow until a thin lipid film was formed on the flask walls. The lipid film was dried under vacuum overnight. A multilamellar vesicle (MLV) suspension was obtained after rehydration with the sample buffer and a series of 10 freeze-thaw cycles. The MLV suspension was extruded through a 50- or 100-nm-pore-size Nuclepore polycarbonate membrane purchased from Whatman/GE Healthcare (Maidstone, United Kingdom) using a LiposoFast-Basic plus Stabilizer setup from Avestin (Mannheim, Germany). This allowed the reorganization of MLVs into SUVs or LUVs, respectively. POPC, POPC-Chol (2:1), and POPC-Chol-SM (1:1:1) liposomes were prepared.
Plasmids and reagents.
The genes of MV IC323 wild-type (wt) H and wt F were codon optimized, synthesized, and subcloned into the mammalian expression vector pCAGGS. The constructs for SLAM and nectin 4 were commercially acquired.
Peptide synthesis.
The measles virus (MV) HRC-derived fusion inhibitor peptides (HRC1 to -6) (≥95% purity) were produced by standard 9-fluorenylmethoxy carbonyl (Fmoc) solid-phase methods. The lipid moieties were attached to the peptides via chemoselective reaction between the thiol group of an extra cysteine residue, added C terminally to the sequence, and a bromoacetyl derivative of cholesterol/tocopherol or a bis-maleimide functionalized cholesterol/tocopherol core, as previously described (24–26). Peptides were stored as powder, and before use they were dissolved in DMSO to 5 mM stock solutions that were kept at −80°C for in vitro fusion assays. For biophysical studies, lyophilized peptides were solubilized in 100% DMSO up to 40 mg/ml, sonicated in a water bath for 5 min, and stored at −20°C. Working peptide samples at defined final concentrations were solubilized from stock solutions in 10 mM HEPES–150 mM NaCl buffer, pH 7.4. The final DMSO content was maintained at 2% (vol/vol) in all experiments. Working peptide samples were sonicated in an ultrasonic bath for 2 to 5 min before use.
ANS fluorescence studies.
For the peptide aggregation studies, a 12.8 μM 8-anilinonaphthalene-1-sulfonic acid (ANS) solution (A369 of ∼0.1 to avoid inner filter effects) was titrated with each HRC peptide up to final concentrations between 0 and 40 μM. For each sample, fluorescence emission spectra were collected between 400 and 600 nm (excitation wavelength [λexc] = 369 nm) in an Edinburgh FLS920 spectrofluorometer (Livingston, UK). Excitation and emission slits were 5 and 10 nm, respectively. A 10-min incubation time was allowed before each measurement. The fluorescence emission intensity values were corrected for dilution and background noise. The maximum emission wavelength, λmax, and the spectrum integral, Σ[IF(λ)], were determined for each spectrum.
DLS.
For dynamic light scattering (DLS), HRC peptide solutions with concentrations ranging from 1 to 30 μM were prepared as described above and incubated at 25°C for 5 min before each measurement of particle size. Measurements were performed in a Malvern Zetasizer Nano ZS (Worcestershire, UK) and consisted of 15 individual runs, each corresponding to an averaged autocorrelation curve obtained from at least 12 repeated sample scans. The diffusion coefficient (D) values were calculated from autocorrelation curves using a CONTIN-like method (76). D values were used to determine number-averaged hydrodynamic radius (RH) profiles through the Stokes-Einstein-Sutherland equation (77).
Membrane dipole potential sensing.
To prepare di-8-ANEPPS-labeled LUVs, POPC, POPC-Chol (2:1), and POPC-Chol-SM (1:1:1), lipid mixtures were cosolubilized with a di-8-ANEPPS solution in chloroform for a final probe/lipid ratio of 1:200 (0.5 mol% di-8-ANEPPS relative to lipid). The subsequent LUV preparation steps were carried out as described in “Chemicals and Reagents” above. The variations in membrane dipole potential in the presence of the HRC peptides were monitored by di-8-ANEPPS fluorescence excitation spectrum shifts. The emission wavelength was fixed at 670 nm to avoid membrane fluidity artifacts (78), and fluorescence excitation spectra were collected between 380 and 580 nm in an FLS920 spectrofluorometer. Excitation and emission slits were 10 and 3 nm, respectively. Fixed concentrations of each HRC peptide were incubated with 200 μM labeled LUVs for 10 min before the measurements were performed. Fluorescence excitation spectra were corrected for background noise. The di-8-ANEPPS fluorescence spectra in the absence of peptide were used as controls. The normalized intensity ratios (Rnorm) were used to quantitatively analyze di-8-ANEPPS spectral shifts (43).
Initially, the excitation intensity (Iexc) ratios (R) were calculated based on the relationship R = [Iexc(λa)]/[Iexc(λb)], where λa and λb correspond to the wavelengths with the highest absolute intensity difference in each differential excitation spectrum (spectrum maximum or minimum). For accurate interpretation, λa and λb should be lower and higher than the maximum excitation wavelength, respectively. R values were normalized to the respective excitation spectrum area.
SPR.
For surface plasmon resonance (SPR), 10 mM HEPES–150 mM NaCl (pH 7.4) containing 2% (vol/vol) DMSO was used as running buffer to match the peptide and lipid sample buffer composition. Experiments were performed in a GE Healthcare Biacore X100. The system was primed at least three times with running buffer before starting an experiment. The L1 sensor chip, which was designed for lipid binding assays, was used in all experiments. The L1 sensor chip surface was first rinsed with three injections of 20 mM CHAPS before starting an assay. To prepare the lipid surface, a 1 mM POPC, POPC-Chol (2:1) or POPC-Chol-SM (1:1:1) SUV sample was injected over the sensor chip at a 2-μl/min flow speed for 2,400 s. Loosely bound vesicles were removed with a 36-s injection of 10 mM NaOH at 50 μl/min. HRC peptide samples with concentrations between 1 and 30 μM were injected over the deposited SUV surface at a 5-μl/min flow speed during 200 s. Each sample was allowed a 600-s unbinding time after injection stopped. After each run, the sensor chip surface was regenerated with sequential injections of 20 mM CHAPS (5 μl/min for 60 s), 0.5% (wt/vol) SDS (5 μl/min for 60 s), 10 mM NaOH containing 20% (vol/vol) methanol (50 μl/min for 36 s), and 10 mM NaOH (50 μl/min for 36 s). Response values were monitored to ensure effective surface regeneration. To analyze the SPR membrane interaction data, the SUV deposition on the sensor chip surface and the response values for HRC peptide binding to lipid were converted into units of moles per area and used to calculate the respective peptide-to-lipid ratio [mol(P)/mol(L)]. To perform these calculations, we considered 1 response unit (RU) to be approximately 1 pg/mm2 of peptide or lipid, as previously described in the literature (79). The SUV deposition response values were collected from sensorgrams at 100 s after rinsing with 10 mM NaOH. The HRC peptide binding response values were retrieved from sensorgrams at t = 200 s of the binding phase.
Transient expression of H and F genes.
Transfections were performed in 293T cells according to the Lipofectamine 2000 manufacturer's protocols (Invitrogen).
Cells and viruses.
293T cells (human kidney epithelial cells), BHK cells (baby hamster kidney cells), and Vero-SLAM cells (African green monkey kidney cells) were grown in Dulbecco's modified Eagle's medium (DMEM) (GIBCO, Invitrogen) supplemented with 10% fetal bovine serum and antibiotics in 5% CO2. The Vero-SLAM culture medium was supplemented with Geneticin. The WTFb strain for cotton rat infection was described previously 80. Wild-type MV strain G954 (genotype B3.2) was isolated in Gambia in 1993 (BB-0033-00053) (81). All virus strains were propagated and titrated in Vero SLAM cells.
β-Gal complementation-based fusion assay.
The β-galactosidase (β-gal) complementation-based fusion assay was performed as described previously (46, 82). Briefly, 293T cells transiently transfected with SLAM and the omega reporter subunit were incubated with cells coexpressing viral glycoproteins (MV H and MV F) and the alpha reporter subunit.
ELISA.
For biodistribution studies, each organ was weighed and mixed in phosphate-buffered saline (PBS) (1:1, wt/vol) using an Ultra Turrax homogenizer. Samples were then treated with acetonitrile–1% trifluoroacetic acid (1:4, vol/vol) for 1 h on a rotor at 4°C and then centrifuged for 10 min at 6,000 × g. Supernatant fluids were collected, and the peptide concentration was determined using an enzyme-linked immunosorbent assay (ELISA). MaxiSorp 96-well plates (Nunc) were coated overnight with purified rabbit anti-MV F HRC antibodies (5 μg/ml) in carbonate-bicarbonate buffer pH 7.4. Plates were washed twice using PBS, followed by incubation with 3% bovine serum albumin (BSA) in PBS (blocking buffer) for 30 min. The blocking buffer was replaced with 2 dilutions of each sample in 3% PBS-BSA in duplicate and incubated for 90 min at room temperature (RT). After multiple washes in PBS, the peptide was detected using a horseradish peroxidase (HRP)-conjugated rabbit custom-made anti-MV F HRC antibody (1:1,500) in blocking buffer for 2 h at RT. HRP activity was recorded as absorbance at 492 nm on the SigmaFast o-phenylenediamine dihydrochloride (OPD) substrate system (Sigma-Aldrich) after adding the stop solution. Standard curves were established for each peptide (using the same ELISA conditions as for the test samples), and the detection limit was determined to be 0.15 nM.
HAE cultures.
The EpiAirway AIR-100 system (MatTek Corporation) used in these experiments consists of normal, human-derived tracheobronchial epithelial cells that have been cultured to form a highly differentiated mucociliary epithelium. Upon receipt from the manufacturer, human airway epithelium (HAE) cultures were handled as previously described (47, 48, 83); we have used HAE for assessment of entry inhibitors (47–51). To assess the potential for delivery of the HRC peptides via the airway to the basolateral compartment, 20 μl of 250 μM peptide inhibitor was applied to the apical surface. At the indicated time points, aliquots of the basolateral supernatant fluid were collected to quantify the amount of peptide delivered to the basolateral reservoir. The 20 μl of 250 μM peptide inhibitor in the 1 ml of reservoir would have had an upper-limit final concentration of 5 μM.
Biodistribution analysis and infection of CR.
Inbred cotton rats (CR) (Sigmodon hispidus) were purchased from Harlan Laboratories, Inc., Indianapolis, IN, USA. Both male and female cotton rats aged 5 to 7 weeks were used. For biodistribution experiments in CR, the animals received the indicated peptides (6 mg/kg) intranasally (i.n.) in 100 μl of water for injection. After 8 h, blood was collected by intracardiac puncture in EDTA Vacutainer tubes, and plasma was conserved at −20°C until use in ELISA. Organs from each animal were collected, snap-frozen in liquid nitrogen, and conserved at −80°C. For i.n. infection, 105 TCID50 of MV WTFb was inoculated in phosphate-buffered saline to isoflurane-anesthetized cotton rats in a volume of 100 μl. To evaluate the effect of HRC peptides, animals were inoculated i.n. with peptide (5 mg/kg in 100 μl of water) at 24 h and 12 h before infection. Four days after infection, the animals were asphyxiated using CO2, and their lungs were collected and weighed. Lung tissue was minced with scissors and homogenized with a Dounce glass homogenizer. Serial 10-fold dilutions of supernatant fluids were assessed for the presence of infectious virus in a 48-well plates using cytopathic effect (CPE) in Vero SLAM cells as the endpoint. Plates were scored for CPE microscopically after 7 days. The amounts of virus in inocula were expressed as fold dilutions that resulted in the infection of 50% of inoculated tissue culture monolayers (TCID50). The TCID50 was calculated as described previously (84). The animal experiments were approved by the Institutional Animal Care and Use Committee of Ohio State University.
Infection of mice.
Three- to 4-week-old SLAM×IFNARKO mice, which are highly susceptible to MV infection (29, 45), were infected i.n., with 10 μl of DMEM containing 104 PFU of MV. Animals were treated with HRC5 or HRC6 i.n. (6 mg/kg) at 24 h and 4 h before infection (10 μl of peptide-containing vehicle). The mock-infected group received only the vehicle at the same time (Aguettant water containing 10% DMSO). All animals were observed daily for 28 days, and those showing clinical signs (neurological symptoms, ataxia, or lethargy) were euthanized. The protocol was approved by the Regional Ethical Committee (CECCAPP protocol no. ENS-2011-003 and ENS-2012-041).
Statistical analysis.
Data are expressed as mean and standard deviation (SD). Statistical analyses were performed using a Mann-Whitney U test, a Mantel-Cox test, and a Tukey range test with GraphPad Prism 5 software.
Data analysis.
Fitting of the equations mentioned in this article to the experimental data was done by nonlinear regression using GraphPad Prism. Error bars on data presentation represent the SD or standard error (SE).
ACKNOWLEDGMENTS
This work was supported by NIH grants NS091263 (NINDS) and AI119762 and AI109050 (NIAID) to M.P. and AI101333 and AI114736-01 (NIAID) to A.M., Fundação para a Ciência e Tecnologia-Ministério da Ciência, Tecnologia e Ensino Superior (FCT-MCTES, Portugal) projects VIH/SAU/0047/2011 to A.S.V. and PTDC/BBB-BQB/3494/2014 to N.C.S., and INSERM and the LABEX ECOFECT (ANR-11-LABX-0048) of Lyon University, within the program “Investissements d'Avenir” (ANR-11-IDEX-0007) operated by the French National Research Agency (ANR), to B.H. T.N.F. and A.S.V. acknowledge FCT for Ph.D. fellowship SFRH/BD/52383/2013 and fellowship IF/00803/2012 under the FCT Investigator Programme, respectively.
We thank Justine Girard for excellent technical support. We are grateful to Lawrence Golub for his support.
REFERENCES
- 1.Moss WJ, Griffin DE. 2012. Measles. Lancet 379:153–164. doi: 10.1016/S0140-6736(10)62352-5. [DOI] [PubMed] [Google Scholar]
- 2.Katz SL, Hinman AR. 2004. Summary and conclusions: measles elimination meeting, 16 to 17 March 2000. J Infect Dis 189(Suppl 1):S43–S47. doi: 10.1086/377696. [DOI] [PubMed] [Google Scholar]
- 3.Orenstein W, Seib K. 2014. Mounting a good offense against measles. N Engl J Med 371:1661–1663. doi: 10.1056/NEJMp1408696. [DOI] [PubMed] [Google Scholar]
- 4.Seward JF, Orenstein WA. 2012. Editorial commentary: a rare event: a measles outbreak in a population with high 2-dose measles vaccine coverage. Clin Infect Dis 55:403–405. doi: 10.1093/cid/cis445. [DOI] [PubMed] [Google Scholar]
- 5.De Serres G, Boulianne N, Defay F, Brousseau N, Benoit M, Lacoursiere S, Guillemette F, Soto J, Ouakki M, Ward BJ, Skowronski DM. 2012. Higher risk of measles when the first dose of a 2-dose schedule of measles vaccine is given at 12-14 months versus 15 months of age. Clin Infect Dis 55:394–402. doi: 10.1093/cid/cis439. [DOI] [PubMed] [Google Scholar]
- 6.de Vries RD, Mesman AW, Geijtenbeek TB, Duprex WP, de Swart RL. 2012. The pathogenesis of measles. Curr Opin Virol 2:248–255. doi: 10.1016/j.coviro.2012.03.005. [DOI] [PubMed] [Google Scholar]
- 7.Ferreira CS, Frenzke M, Leonard VH, Welstead GG, Richardson CD, Cattaneo R. 2010. Measles virus infection of alveolar macrophages and dendritic cells precedes spread to lymphatic organs in transgenic mice expressing human signaling lymphocytic activation molecule (SLAM, CD150). J Virol 84:3033–3042. doi: 10.1128/JVI.01559-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Avota E, Koethe S, Schneider-Schaulies S. 2013. Membrane dynamics and interactions in measles virus dendritic cell infections. Cell Microbiol 15:161–169. doi: 10.1111/cmi.12025. [DOI] [PubMed] [Google Scholar]
- 9.Koethe S, Avota E, Schneider-Schaulies S. 2012. Measles virus transmission from dendritic cells to T cells: formation of synapse-like interfaces concentrating viral and cellular components. J Virol 86:9773–9781. doi: 10.1128/JVI.00458-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lemon K, de Vries RD, Mesman AW, McQuaid S, van Amerongen G, Yuksel S, Ludlow M, Rennick LJ, Kuiken T, Rima BK, Geijtenbeek TB, Osterhaus AD, Duprex WP, de Swart RL. 2011. Early target cells of measles virus after aerosol infection of non-human primates. PLoS Pathog 7:e1001263. doi: 10.1371/journal.ppat.1001263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yanagi Y, Ono N, Tatsuo H, Hashimoto K, Minagawa H. 2002. Measles virus receptor SLAM (CD150). Virology 299:155–161. doi: 10.1006/viro.2002.1471. [DOI] [PubMed] [Google Scholar]
- 12.Yanagi Y, Takeda M, Ohno S, Hashiguchi T. 2009. Measles virus receptors. Curr Top Microbiol Immunol 329:13–30. [DOI] [PubMed] [Google Scholar]
- 13.Hashiguchi T, Maenaka K, Yanagi Y. 2011. Measles virus hemagglutinin: structural insights into cell entry and measles vaccine. Front Microbiol 2:247. doi: 10.3389/fmicb.2011.00247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Muhlebach MD, Mateo M, Sinn PL, Prufer S, Uhlig KM, Leonard VH, Navaratnarajah CK, Frenzke M, Wong XX, Sawatsky B, Ramachandran S, McCray PB Jr, Cichutek K, von Messling V, Lopez M, Cattaneo R. 2011. Adherens junction protein nectin-4 is the epithelial receptor for measles virus. Nature 480:530–533. doi: 10.1038/nature10639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Noyce RS, Bondre DG, Ha MN, Lin LT, Sisson G, Tsao MS, Richardson CD. 2011. Tumor cell marker PVRL4 (nectin 4) is an epithelial cell receptor for measles virus. PLoS Pathog 7:e1002240. doi: 10.1371/journal.ppat.1002240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tatsuo H, Ono N, Tanaka K, Yanagi Y. 2000. SLAM (CDw150) is a cellular receptor for measles virus. Nature 406:893–897. doi: 10.1038/35022579. [DOI] [PubMed] [Google Scholar]
- 17.Keeffe JR, Gnanapragasam PN, Gillespie SK, Yong J, Bjorkman PJ, Mayo SL. 2011. Designed oligomers of cyanovirin-N show enhanced HIV neutralization. Proc Natl Acad Sci U S A 108:14079–14084. doi: 10.1073/pnas.1108777108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yin HS, Paterson RG, Wen X, Lamb RA, Jardetzky TS. 2005. Structure of the uncleaved ectodomain of the paramyxovirus (hPIV3) fusion protein. Proc Natl Acad Sci U S A 102:9288–9293. doi: 10.1073/pnas.0503989102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lamb RA, Paterson RG, Jardetzky TS. 2006. Paramyxovirus membrane fusion: lessons from the F and HN atomic structures. Virology 344:30–37. doi: 10.1016/j.virol.2005.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yin HS, Wen X, Paterson RG, Lamb RA, Jardetzky TS. 2006. Structure of the parainfluenza virus 5 F protein in its metastable, prefusion conformation. Nature 439:38–44. doi: 10.1038/nature04322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harrison SC. 2008. Viral membrane fusion. Nat Struct Mol Biol 15:690–698. doi: 10.1038/nsmb.1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chang A, Dutch RE. 2012. Paramyxovirus fusion and entry: multiple paths to a common end. Viruses 4:613–636. doi: 10.3390/v4040613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Plemper RK, Brindley MA, Iorio RM. 2011. Structural and mechanistic studies of measles virus illuminate paramyxovirus entry. PLoS Pathog 7:e1002058. doi: 10.1371/journal.ppat.1002058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.White JM, Delos SE, Brecher M, Schornberg K. 2008. Structures and mechanisms of viral membrane fusion proteins: multiple variations on a common theme. Crit Rev Biochem Mol Biol 43:189–219. doi: 10.1080/10409230802058320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sapir A, Avinoam O, Podbilewicz B, Chernomordik LV. 2008. Viral and developmental cell fusion mechanisms: conservation and divergence. Dev Cell 14:11–21. doi: 10.1016/j.devcel.2007.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lambert DM, Barney S, Lambert AL, Guthrie K, Medinas R, Davis DE, Bucy T, Erickson J, Merutka G, Petteway SR Jr. 1996. Peptides from conserved regions of paramyxovirus fusion (F) proteins are potent inhibitors of viral fusion. Proc Natl Acad Sci U S A 93:2186–2191. doi: 10.1073/pnas.93.5.2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eckert DM, Kim PS. 2001. Mechanisms of viral membrane fusion and its inhibition. Annu Rev Biochem 70:777–810. doi: 10.1146/annurev.biochem.70.1.777. [DOI] [PubMed] [Google Scholar]
- 28.Berkhout B, Eggink D, Sanders RW. 2012. Is there a future for antiviral fusion inhibitors? Curr Opin Virol 2:50–59. doi: 10.1016/j.coviro.2012.01.002. [DOI] [PubMed] [Google Scholar]
- 29.Mathieu C, Huey D, Jurgens E, Welsch JC, DeVito I, Talekar A, Horvat B, Niewiesk S, Moscona A, Porotto M. 2015. Prevention of measles virus infection by intranasal delivery of fusion inhibitor peptides. J Virol 89:1143–1155. doi: 10.1128/JVI.02417-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sun A, Prussia A, Zhan W, Murray EE, Doyle J, Cheng LT, Yoon JJ, Radchenko EV, Palyulin VA, Compans RW, Liotta DC, Plemper RK, Snyder JP. 2006. Nonpeptide inhibitors of measles virus entry. J Med Chem 49:5080–5092. doi: 10.1021/jm0602559. [DOI] [PubMed] [Google Scholar]
- 31.Doyle J, Prussia A, White LK, Sun A, Liotta DC, Snyder JP, Compans RW, Plemper RK. 2006. Two domains that control prefusion stability and transport competence of the measles virus fusion protein. J Virol 80:1524–1536. doi: 10.1128/JVI.80.3.1524-1536.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Plemper RK, Doyle J, Sun A, Prussia A, Cheng LT, Rota PA, Liotta DC, Snyder JP, Compans RW. 2005. Design of a small-molecule entry inhibitor with activity against primary measles virus strains. Antimicrob Agents Chemother 49:3755–3761. doi: 10.1128/AAC.49.9.3755-3761.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jiang Y, Liang M, Svejkar D, Hart-Smith G, Lu H, Scarano W, Stenzel MH. 2014. Albumin-micelles via a one-pot technology platform for the delivery of drugs. Chem Commun (Camb) 50:6394–6397. doi: 10.1039/c4cc00616j. [DOI] [PubMed] [Google Scholar]
- 34.Porotto M, Yokoyama CC, Palermo LM, Mungall B, Aljofan M, Cortese R, Pessi A, Moscona A. 2010. Viral entry inhibitors targeted to the membrane site of action. J Virol 84:6760–6768. doi: 10.1128/JVI.00135-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hadinoto K, Sundaresan A, Cheow WS. 2013. Lipid-polymer hybrid nanoparticles as a new generation therapeutic delivery platform: a review. Eur J Pharm Biopharm 85:427–443. doi: 10.1016/j.ejpb.2013.07.002. [DOI] [PubMed] [Google Scholar]
- 36.Fang DL, Chen Y, Xu B, Ren K, He ZY, He LL, Lei Y, Fan CM, Song XR. 2014. Development of lipid-shell and polymer core nanoparticles with water-soluble salidroside for anti-cancer therapy. Int J Mol Sci 15:3373–3388. doi: 10.3390/ijms15033373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hawe A, Sutter M, Jiskoot W. 2008. Extrinsic fluorescent dyes as tools for protein characterization. Pharm Res 25:1487–1499. doi: 10.1007/s11095-007-9516-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Molla MR, Prasad P, Thayumanavan S. 2015. Protein-induced supramolecular disassembly of amphiphilic polypeptide nanoassemblies. J Am Chem Soc 137:7286–7289. doi: 10.1021/jacs.5b04285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Veiga AS, Santos NC, Loura LM, Fedorov A, Castanho MA. 2004. HIV fusion inhibitor peptide T-1249 is able to insert or adsorb to lipidic bilayers. Putative correlation with improved efficiency. J Am Chem Soc 126:14758–14763. [DOI] [PubMed] [Google Scholar]
- 40.Veiga S, Henriques S, Santos NC, Castanho M. 2004. Putative role of membranes in the HIV fusion inhibitor enfuvirtide mode of action at the molecular level. Biochem J 377:107–110. doi: 10.1042/bj20031350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Manie SN, de Breyne S, Vincent S, Gerlier D. 2000. Measles virus structural components are enriched into lipid raft microdomains: a potential cellular location for virus assembly. J Virol 74:305–311. doi: 10.1128/JVI.74.1.305-311.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vincent S, Gerlier D, Manie SN. 2000. Measles virus assembly within membrane rafts. J Virol 74:9911–9915. doi: 10.1128/JVI.74.21.9911-9915.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Matos PM, Franquelim HG, Castanho MA, Santos NC. 2010. Quantitative assessment of peptide-lipid interactions. Ubiquitous fluorescence methodologies. Biochim Biophys Acta 1798:1999–2012. doi: 10.1016/j.bbamem.2010.07.012. [DOI] [PubMed] [Google Scholar]
- 44.Besenicar M, Macek P, Lakey JH, Anderluh G. 2006. Surface plasmon resonance in protein-membrane interactions. Chem Phys Lipids 141:169–178. doi: 10.1016/j.chemphyslip.2006.02.010. [DOI] [PubMed] [Google Scholar]
- 45.Welsch JC, Talekar A, Mathieu C, Pessi A, Moscona A, Horvat B, Porotto M. 2013. Fatal measles virus infection prevented by brain-penetrant fusion inhibitors. J Virol 87:13785–13794. doi: 10.1128/JVI.02436-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moosmann P, Rusconi S. 1996. Alpha complementation of LacZ in mammalian cells. Nucleic Acids Res 24:1171–1172. doi: 10.1093/nar/24.6.1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Palmer SG, Porotto M, Palermo LM, Cunha LF, Greengard O, Moscona A. 2012. Adaptation of human parainfluenza virus to airway epithelium reveals fusion properties required for growth in host tissue. mBio 3:e00137-12. doi: 10.1128/mBio.00137-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moscona A, Porotto M, Palmer S, Tai C, Aschenbrenner L, Triana-Baltzer G, Li QX, Wurtman D, Niewiesk S, Fang F. 2010. A recombinant sialidase fusion protein effectively inhibits human parainfluenza viral infection in vitro and in vivo. J Infect Dis 202:234–241. doi: 10.1086/653621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Farzan SF, Palermo LM, Yokoyama CC, Orefice G, Fornabaio M, Sarkar A, Kellogg GE, Greengard O, Porotto M, Moscona A. 2011. Premature activation of the paramyxovirus fusion protein before target cell attachment with corruption of the viral fusion machinery. J Biol Chem 286:37945–37954. doi: 10.1074/jbc.M111.256248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Porotto M, Rockx B, Yokoyama CC, Talekar A, Devito I, Palermo LM, Liu J, Cortese R, Lu M, Feldmann H, Pessi A, Moscona A. 2010. Inhibition of Nipah virus infection in vivo: targeting an early stage of paramyxovirus fusion activation during viral entry. PLoS Pathog 6:e1001168. doi: 10.1371/journal.ppat.1001168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Palermo L, Porotto M, Yokoyama C, Palmer S, Mungall B, Greengard O, Niewiesk S, Moscona A. 2009. Human parainfluenza virus infection of the airway epithelium: the viral hemagglutinin-neuraminidase regulates fusion protein activation and modulates infectivity. J Virol 83:6900–6908. doi: 10.1128/JVI.00475-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Niewiesk S. 2009. Current animal models: cotton rat animal model. Curr Top Microbiol Immunol 330:89–110. [DOI] [PubMed] [Google Scholar]
- 53.Pessi A, Langella A, Capito E, Ghezzi S, Vicenzi E, Poli G, Ketas T, Mathieu C, Cortese R, Horvat B, Moscona A, Porotto M. 2012. A general strategy to endow natural fusion-protein-derived peptides with potent antiviral activity. PLoS One 7:e36833. doi: 10.1371/journal.pone.0036833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.O'Donnell LA, Rall GF. 2010. Blue moon neurovirology: the merits of studying rare CNS diseases of viral origin. J Neuroimmune Pharmacol 5:443–455 doi: 10.1007/s11481-010-9200-4. [DOI] [PubMed] [Google Scholar]
- 55.Young VA, Rall GF. 2009. Making it to the synapse: measles virus spread in and among neurons. Curr Top Microbiol Immunol 330:3–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Makhortova NR, Askovich P, Patterson CE, Gechman LA, Gerard NP, Rall GF. 2007. Neurokinin-1 enables measles virus trans-synaptic spread in neurons. Virology 362:235–244. doi: 10.1016/j.virol.2007.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Reuter D, Schneider-Schaulies J. 2010. Measles virus infection of the CNS: human disease, animal models, and approaches to therapy. Med Microbiol Immunol 199:261–271. doi: 10.1007/s00430-010-0153-2. [DOI] [PubMed] [Google Scholar]
- 58.Chiu MH, Meatherall B, Nikolic A, Cannon K, Fonseca K, Joseph JT, MacDonald J, Pabbaraju K, Tellier R, Wong S, Koch MW. 2016. Subacute sclerosing panencephalitis in pregnancy. Lancet Infect Dis 16:366–375. doi: 10.1016/S1473-3099(15)00524-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jacobson RM, Ovsyannikova IG, Vierkant RA, Pankratz VS, Poland GA. 2012. Independence of measles-specific humoral and cellular immune responses to vaccination. Hum Immunol 73:474–479. doi: 10.1016/j.humimm.2012.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Poland GA, Jacobson RM. 2012. The re-emergence of measles in developed countries: time to develop the next-generation measles vaccines? Vaccine 30:103–104. doi: 10.1016/j.vaccine.2011.11.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Haralambieva IH, Ovsyannikova IG, Pankratz VS, Kennedy RB, Jacobson RM, Poland GA. 2013. The genetic basis for interindividual immune response variation to measles vaccine: new understanding and new vaccine approaches. Expert Rev Vaccines 12:57–70. doi: 10.1586/erv.12.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.LeBaron CW, Beeler J, Sullivan BJ, Forghani B, Bi D, Beck C, Audet S, Gargiullo P. 2007. Persistence of measles antibodies after 2 doses of measles vaccine in a postelimination environment. Arch Pediatr Adolesc Med 161:294–301. doi: 10.1001/archpedi.161.3.294. [DOI] [PubMed] [Google Scholar]
- 63.Kontio M, Jokinen S, Paunio M, Peltola H, Davidkin I. 2012. Waning antibody levels and avidity: implications for MMR vaccine-induced protection. J Infect Dis 206:1542–1548. doi: 10.1093/infdis/jis568. [DOI] [PubMed] [Google Scholar]
- 64.Haralambieva IH, Ovsyannikova IG, O'Byrne M, Pankratz VS, Jacobson RM, Poland GA. 2011. A large observational study to concurrently assess persistence of measles specific B-cell and T-cell immunity in individuals following two doses of MMR vaccine. Vaccine 29:4485–4491. doi: 10.1016/j.vaccine.2011.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Haralambieva IH, Kennedy RB, Ovsyannikova IG, Whitaker JA, Poland GA. 2015. Variability in humoral immunity to measles vaccine: new developments. Trends Mol Med 21:789–801. doi: 10.1016/j.molmed.2015.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chen CJ, Lee PI, Hsieh YC, Chen PY, Ho YH, Chang CJ, Liu DP, Chang FY, Chiu CH, Huang YC, Lee CY, Lin TY. 2012. Waning population immunity to measles in Taiwan. Vaccine 30:6721–6727. doi: 10.1016/j.vaccine.2012.05.019. [DOI] [PubMed] [Google Scholar]
- 67.Bochennek K, Allwinn R, Langer R, Becker M, Keppler OT, Klingebiel T, Lehrnbecher T. 2014. Differential loss of humoral immunity against measles, mumps, rubella and varicella-zoster virus in children treated for cancer. Vaccine 32:3357–3361. doi: 10.1016/j.vaccine.2014.04.042. [DOI] [PubMed] [Google Scholar]
- 68.Varner CT, Rosen T, Martin JT, Kane RS. 2015. Recent advances in engineering polyvalent biological interactions. Biomacromolecules 16:43–55. doi: 10.1021/bm5014469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Welch BD, VanDemark AP, Heroux A, Hill CP, Kay MS. 2007. Potent D-peptide inhibitors of HIV-1 entry. Proc Natl Acad Sci U S A 104:16828–16833. doi: 10.1073/pnas.0708109104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Welch BD, Francis JN, Redman JS, Paul S, Weinstock MT, Reeves JD, Lie YS, Whitby FG, Eckert DM, Hill CP, Root MJ, Kay MS. 2010. Design of a potent D-peptide HIV-1 entry inhibitor with a strong barrier to resistance. J Virol 84:11235–11244. doi: 10.1128/JVI.01339-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mack ET, Snyder PW, Perez-Castillejos R, Bilgicer B, Moustakas DT, Butte MJ, Whitesides GM. 2012. Dependence of avidity on linker length for a bivalent ligand-bivalent receptor model system. J Am Chem Soc 134:333–345. doi: 10.1021/ja2073033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liu H, Moynihan KD, Zheng Y, Szeto GL, Li AV, Huang B, Van Egeren DS, Park C, Irvine DJ. 2014. Structure-based programming of lymph-node targeting in molecular vaccines. Nature 507:519–522. doi: 10.1038/nature12978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Liu H, Irvine DJ. 2015. Guiding principles in the design of molecular bioconjugates for vaccine applications. Bioconjug Chem 26:791–801. doi: 10.1021/acs.bioconjchem.5b00103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.de Vries RD, McQuaid S, van Amerongen G, Yuksel S, Verburgh RJ, Osterhaus AD, Duprex WP, de Swart RL. 2012. Measles immune suppression: lessons from the macaque model. PLoS Pathog 8:e1002885. doi: 10.1371/journal.ppat.1002885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Vemuri S, Rhodes CT. 1995. Preparation and characterization of liposomes as therapeutic delivery systems: a review. Pharm Acta Helv 70:95–111. doi: 10.1016/0031-6865(95)00010-7. [DOI] [PubMed] [Google Scholar]
- 76.Provencher S. 1982. Contin—a general-purpose constrained regularization program for inverting noisy linear algebraic and integral-equations. Comp Phys Commun 27:13. [Google Scholar]
- 77.Berne BJ, Pecora R. 1990. Dynamic light scattering: with applications to chemistry, biology, and physics. R.E. Krieger Publishing Co., Malabar, FL. [Google Scholar]
- 78.Clarke RJ, Kane DJ. 1997. Optical detection of membrane dipole potential: avoidance of fluidity and dye-induced effects. Biochim Biophys Acta 1323:223–239. doi: 10.1016/S0005-2736(96)00188-5. [DOI] [PubMed] [Google Scholar]
- 79.Craik DJ, Henriques ST, Mylne JS, Wang CK. 2012. Cyclotide isolation and characterization. Methods Enzymol 516:37–62. doi: 10.1016/B978-0-12-394291-3.00024-1. [DOI] [PubMed] [Google Scholar]
- 80.Rima BK, Earle JA, Baczko K, ter Meulen V, Liebert UG, Carstens C, Carabana J, Caballero M, Celma ML, Fernandez-Munoz R. 1997. Sequence divergence of measles virus haemagglutinin during natural evolution and adaptation to cell culture. J Gen Virol 78:97–106. doi: 10.1099/0022-1317-78-1-97. [DOI] [PubMed] [Google Scholar]
- 81.Kouomou DW, Nerrienet E, Mfoupouendoun J, Tene G, Whittle H, Wild TF. 2002. Measles virus strains circulating in Central and West Africa: Geographical distribution of two B3 genotypes. J Med Virol 68:433–440. doi: 10.1002/jmv.10222. [DOI] [PubMed] [Google Scholar]
- 82.Porotto M, Fornabaio M, Kellogg GE, Moscona A. 2007. A second receptor binding site on human parainfluenza virus type 3 hemagglutinin-neuraminidase contributes to activation of the fusion mechanism. J Virol 81:3216–3228. doi: 10.1128/JVI.02617-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Xu R, Palmer SG, Porotto M, Palermo LM, Niewiesk S, Wilson IA, Moscona A. 2013. Interaction between the hemagglutinin-neuraminidase and fusion glycoproteins of human parainfluenza virus type III regulates viral growth in vivo. mBio 4:e00803-13. doi: 10.1128/mBio.00803-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pfeuffer J, Puschel K, Meulen V, Schneider-Schaulies J, Niewiesk S. 2003. Extent of measles virus spread and immune suppression differentiates between wild-type and vaccine strains in the cotton rat model (Sigmodon hispidus). J Virol 77:150–158. doi: 10.1128/JVI.77.1.150-158.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]