

Abstract
Type I interferons (IFN-I) have long been heralded as key contributors to effective antiviral responses. More widely understood in the context of acute viral infection, the role of this pleiotropic cytokine has been characterized as triggering antiviral states in cells and potentiating adaptive immune responses. Upon induction in the innate immune response, IFN-I triggers the expression of interferon-stimulated genes (ISGs), which upregulate the effector function of immune cells (e.g., dendritic cells, B cells, and T cells) toward successful resolution of infections. However, emerging lines of evidence reveal that viral persistence in the course of chronic infections could be driven by deleterious immunomodulatory effects upon sustained IFN-I expression. In this setting, elevation of IFN-I and ISGs is directly correlated to viral persistence and elevated viral loads. It is important to note that the correlation among IFN-I expression, ISGs, and viral persistence may be a cause or effect of chronic infection and this is an important distinction to make toward establishing the dichotomous nature of IFN-I responses. The aim of this mini review is to (i) summarize the interaction between IFN-I and downstream effector responses and therefore (ii) delineate the function of this cytokine on positive and negative immunoregulation in chronic infection. This is a significant consideration given the current therapeutic administration of IFN-I in chronic viral infections whose therapeutic significance is projected to continue despite emergence of increasingly efficacious antiviral regimens. Furthermore, elucidation of the interplay between virus and the antiviral response in the context of IFN-I will elucidate avenues toward more effective therapeutic and prophylactic measures against chronic viral infections.
Keywords: type-I interferon, chronic viral infection, immunopathology, IFNAR, immunoregulation
Introduction
Upon viral infection, the immune response comprises a multi-layered coordination of effector functions broadly characterized as a progression from innate to adaptive immunity. Within the immunological milieu, Type I interferons (IFN-I) play a central role in driving an antiviral state in non-immune cells as well as orchestrating antiviral immune responses through: (i) inhibiting viral replication in infected cells in the innate stage of the immune response; (ii) activating and enhancing antigen presentation in the “early induced” immune response, and (iii) triggering the adaptive immune response through direct and indirect action on T and B cells that make up the memory response [reviewed in Ref. (1)]. Therefore, this cytokine acts as a master regulator whose induction in the early stages of viral infection modulates downstream signaling cascades that promote both pro-inflammatory and anti-inflammatory responses depending on the context of activation as discussed below. Whereas the protective role of IFNs has been widely characterized, emerging lines of evidence illustrate a deleterious effect borne by IFN-associated immunopathology (2, 3). These characterizations bear particular importance given the historic use and ongoing studies on IFN therapy in the treatment of chronic viral infections [e.g., HCV (4) and HIV (5–8)], autoimmune diseases [e.g., systemic lupus erythematosus (9)], and cancer (10–13). Whereas the advent of new therapies has spurred a trend toward IFN-free treatments in HCV, HIV, and oncology, IFN therapy is still considered to be a significant therapeutic agent due to its efficacy against HCV-associated complications [e.g., hepatocellular carcinoma (4)] and combinatorial effect in cancer therapy (14). In addition, cost restriction due to the price of emergent therapies also sustains the use of IFN-based therapies (15).
Described here in the context of viral infections, this review focuses on the course of IFN-I upon (i) elicitation; (ii) downstream signaling in various cell types, and (iii) the consequent binary effect on immunity. Collectively, we discuss the development of IFN-driven antiviral responses and key features that highlight potential targets toward effective treatment measures against chronic viral infections.
Diversity in IFN-Associated Immune Responses
Type I interferons can be broadly characterized into three groups: IFN-I, Type II (IFN-II), and Type III (IFN-III) with subcategories therein based on gene loci of the IFN transcribing genes as well as difference in their cognate receptors. IFN-I is the largest and most well-characterized group with seven classes: IFNα, IFNβ, IFNδ, IFNϵ, IFNκ, IFNω, and IFNτ whereas IFN-II comprises IFNγ. IFN-I and IFN-II signal through IFNαR1/R2 (IFNAR) and IFNγR1/R2 (IFNGR), respectively. The last class IFN-III, otherwise classified as “IFN-like cytokines,” consists of interleukin (IL)-28A (IFNλ2), IL28B (IFNλ3), and IL29 (IFNλ1) and signals through IL-28RI/IL10R2 receptor chains [reviewed in Ref. (16)].
Upon pathogen-encounter, a plethora of cells are induced into IFN-I expression through recognition of pathogen-associated molecular patterns by putative pattern recognition receptors (PRRs), such as (i) toll-like receptors (TLRs) (17–24), (ii) retinoic-acid inducible gene I (RIG-I) (20, 25), (iii) melanoma differentiation-associated gene 5 (26), and (iv) nucleotide-binding oligomerization domain-containing protein (27). Consequent to PRR activation, signal transduction occurs through downstream transcription regulators called IFN regulatory factors (IRFs). This family of nine members, IRF1–IRF9 [see Table 1 in Ref. (28) for summary] offers yet another layer of diversity in the IFN response; convergence to and transcription by different sets of IRFs is determined by the nature of the sensing PRR, which resultantly determines the nature of the subsequent IFN responses.
The third layer of diversity entails the initiation of transcription by IRFs, which is facilitated by the variety of signal transduction pathways triggered upon elicitation of IFNs. Upon ligation of IFNAR, signal transducer and activator of transcription 1 (STAT1) and STAT2 are induced through phosphorylation by the tyrosine kinase 2 (TYK2) and Janus kinase (JAK1). Thereafter, STAT1 and STAT2 form a trimeric transcription factor, IFN-stimulated gene factor 3, by assembling with IRF9 that subsequently migrates into the nucleus to initiate transcription of IFN-stimulated genes (ISGs) by binding to the promoter regions known as IFN-stimulated response elements (ISRE) (29). Within this signal cascade lies combinatorial differences through which IFNs foster both proinflammatory and anti-inflammatory responses. For example, while signaling by IFN α/β through IFNAR typically leads to heterodimerization of STAT1 and STAT2, homodimerization between STAT1 and STAT3 may occur concurrently or alternatively upon IFNAR signaling. This different pairing of downstream STAT dimers therefore results in (i) the aforementioned engagement of ISRE toward antiviral responses (STAT1/3 heterodimers), (ii) the induction of pro-inflammatory responses by binding to IFNγ response elements (GAS) (STAT1 homodimers), or (iii) binding of STAT3-binding elements (SBE) to trigger an anti-inflammatory response (STAT3 homodimers) [reviewed in Ref. (30)].
Importantly, whereas STAT1 drives a pro-inflammatory, pro-apoptotic response, STAT3 dimerization favors an anti-inflammatory response that negatively regulates the action of STAT1 (31); we surmise that this is likely a homeostatic mechanism to counter the immunopathological effects of sustained IFN-associated pro-inflammatory responses. However, in the context of IL-6 cytokine signaling, the anti-inflammatory effect of STAT3 upon IFNAR signaling can also be counteracted through a negative feedback loop as well; this further underscores the multiplicity of interactions that govern IFN-I-associated signaling and its downstream effects (31). Lastly, in addition to the plethora of molecular interactions, the presence of IFN-I receptors on various cell types [e.g., hematopoietic stem cells (32, 33), macrophages (34–36), dendritic cells (DCs) (37–43), and natural killer (NK) cells (35, 44–47)] further enhances the impact of IFN-I upon induction.
IFN-I Responses in Chronic Infection
It is important to consider that the antiviral effects of IFN-I have been primarily made in the framework of an acute infection in which the intricate interplay of well-timed and tightly regulated IFN responses functions optimally toward resolution of an infection. What are the effects of prolonged IFN-I production such as in the case of chronic infections? This is an open question that is gaining increasing traction based on emerging data on the deleterious effects of IFN-I in the chronic setting. Importantly, various combinations of IFN-I are used as therapeutic measures particularly in chronic infections. Given the historical and continued use in clinical applications, this is a crucial factor to consider given the multifaceted ways in which IFN elicitation and response are regulated in a fine balance whose perturbation bears impact ranging from hematopoiesis to mature differentiated adaptive immune responses.
IFN-I Responses in Lymphocytic Choriomeningitis Virus (LCMV) Infection
The deleterious effect of IFN-I responses has been brought into sharper focus more recently by two independent studies using a chronic (LCMV-Clone 13) versus acute (LCMV-Armstrong) infection model, which revealed that viral persistence was diminished by in vivo IFNAR blockade (2, 3). In their analyses, Teijaro et al. illustrated that IFNAR blockade led to the rescue of IFNγ+ CD4 T cells, which as discussed comprise the T helper 1 (TH1) cellular subsets that potentiate cytotoxic T lymphocyte (CTL) responses. Strikingly, this study revealed that the size of the CTL subpopulation was not changed despite the enhanced viral clearance observed; thus, functional quiescence (similar to exhaustion) in the face of sustained IFN-I signaling partially facilitates impairment of viral clearance by CTLs. A significant finding in these studies was that in addition to the net detrimental effects of sustained IFN-I, elicitation of high concentration of the cytokine early in the course of infection correlated with viral persistence.
As outlined, IFN-related mechanisms are governed by feedback loops to ascertain homeostasis and prevent immunopathology. An example of these coordinate measures is observed in the switch from TH1 responses toward T follicular helper (TFH) cells. Fahey et al. originally depicted this transition using LCMV. By comparing LCMV-Armstrong versus LCMV-Clone 13, they observed that while mice infected with an acute strain of the virus did not bear any aberrant elevation of TFH markers, the chronic phase of LCMV-Clone 13 infection exhibited increased proportions of TFH cells depicted by putative markers such as (i) CXCR5; a B cell homing chemokine receptor; (ii) ICOS; an inducible T cell costimulatory molecule; and (iii) inducible T cell costimulatory OX40, also known as TNFRSF4. A significant distinction to make here is that TFH cells were also present in the acute infection but these abated upon resolution of the infection (48). In follow-up analyses, Osokine and colleagues revealed that this switch occurred in an IFN-I-dependent manner wherein the absence of IFN signaling, TH1 responses were maintained; in the presence of IFN-I, the cytokine actively suppressed the emergence of de novo TH1 cells in a pre-programed function that occurred early in the priming stages of the infection (49). The underlying principle behind this transition is to curb the TH1 response, which triggers IFNγ expression that in turn activates CTLs and NK cells. From a homeostatic point of view, prolonged effector function of these cells may lead to excessive cytotoxicity and other detrimental effects resulting in host tissue damage.
However, in the event of viral persistence, this skew toward TFH responses results in a number of aberrant responses that hinder viral clearance. Decades-long characterization of CTL exhaustion has been at the forefront of chronic-infection immune response perturbations [(50), reviewed in Ref. (51)]. Initially characterized in LCMV infection as well, exhausted CTLs were observed to be refractory to activation signals, prone to apoptosis, and feature an upregulation of inhibitory markers (52–56). Notably, the aforementioned switch to TFH from TH1 results in diminished activation of CTLs based on the resultant reduction of the second activation signal required to fully activate naïve CTLs. As shown by Fuller et al., the absence of TH1 licensing (57) along with the reduction of IFNγ due to contraction of TH1 cell populations as infection progresses toward chronicity leaves CTLs in a pseudoactivated state characterized as exhaustion.
That the TFH subpopulation is atypically expanded in chronic infections (48, 49) also imposes dysregulation on their close immunological counterparts, the B cells. In the context of a chronic infection, perturbations such as atypical B-cell subpopulations, hypergammaglobulinemia (HGG), and polyspecificity are well characterized (58–66). Along with others, we observed the extensive impact of IFN-mediated responses on humoral immunity both directly and indirectly in the context of viral persistence. In our study, we found that in addition to the indirect TFH-associated humoral response perturbation, there was a direct IFN-I-mediated effect on B cells (67). Comparing LCMV-Clone 13 versus LCMV-WE (acute), we observed sustained ablation of antigen specificity against a secondary immunogen, nitrophenylacetyl-chicken gamma globulin (NP-CGG), in the former whereas the latter only showed transient impact on antigen specificity. Furthermore, we also evaluated antigen specificity of NP-CGG in the context of vesicular stomatitis virus (an acute infection), which remained unchanged. Remarkably, we observed the rescue of antigen specificity upon IFNAR blockade in addition to a recovery of lymphoid architecture similar to previous studies (2, 3, 67, 68). Most importantly, we also assessed the humoral response using a chimeric mouse model comprising reconstitution of irradiated B6 mice with a mix of bone marrow cells from JHT (B-cell deficient) (69) and IFNAR−/− mice. Here, we observed that in the absence of IFNAR signaling in B cells, neutralizing antibodies (nAbs) against LCMV were elicited more robustly and earlier than in wildtype mice and control JHT/B6 chimeras. These results are in agreement with previous findings by Price et al. who also showed that in the absence of IFN-I signaling, nAb responses against influenza virus developed more efficiently (70). Recently, the direct effect of IFN signaling on B cells has also been illustrated using Leishmania donovani, which is the etiological agent of the chronic disease, visceral leishmaniasis. In this study, Silva-Barrios et al. illustrated that B-cell activation occurred in an IFN-associated, TLR-dependent manner that culminated in disruption of the humoral immune response that typifies other chronic infections. Similar to our findings, they also observed the reduction of HGG upon B-cell-specific IFNAR knockout in mice (71), which further supports the role played by IFN signaling toward this phenomenon.
IFN-I Responses in HCV Infection
In the perspective of human infection, the role of IFN responses is particularly important based on the widespread use of IFN therapy against chronic viral diseases such as HCV (4), HIV (5–8, 72), and more broadly in clinical setting such as systemic lupus erythematosus (9), melanoma, and other neoplastic indications [(11–13), reviewed in Ref. (10)]. It is important to state that the standard of care in HCV is slowly moving away from IFN-based therapy, whereas HIV anti-retroviral therapy is almost entirely IFN-free except in impoverished regions. Of note, although some of these conditions are non-viral infections, they all feature antigenic persistence and therefore resemble chronic viral infections despite different etiologies. Given the pervasive influence of IFN-I responses and data revealing both positive as well as negative effects of the cytokine, it is also imperative to critically delineate the effect of IFN-I in chronic disease settings.
Generally, the immunopathology associated with IFN-I, e.g., aberrant cellular populations, inadequate immune responses, and disrupted cytokine environments are also observed in HCV. On a molecular level, most characterizations of IFN cellular responses have been made using in vitro models, e.g., HCV pseudoparticles (73) and HCV cell culture (74, 75) systems whereby the impact of IFN is observed in the context of both endogenous expression in cell culture and exogenous supplementation akin to administration of therapy. Detection of viral RNA occurs through typical PRR-recognition pathways [(76, 77), reviewed in Ref. (78)], upon which upregulation of ISGs occurs (79). Interestingly, researchers observed a coincidence between low response rates to IFN treatment in patients with high baseline levels of IFN in their plasma (80). In this study, Sarasin-Filipowicz and colleagues revealed that hepatocytes obtained from chronically infected, non-responder patients bore non-responsive signaling to IFN treatment ex vivo. Similarly, evidence of attenuation in IFN responses in the chronic phase of HCV is also suggested by the prevalence of ineffective CTL responses upon delayed induction of IFNα-therapy, whereas functional effector activity was maintained or restored in spontaneous resolvers or responders, respectively (81). At the transcriptional level, clues toward IFN-resistance are posited by the discovery of proviral ISGs whereby recent work has shown that some ISGs work to promote the HCV resistance in cell culture. For example, overexpression of ubiquitin-specific protease 18 (USP-18), which functions as a negative regulator of IFN signaling drives, a proviral response highlighted by evidence of up regulation in HCV patients who do not respond to IFN treatment (82). Conversely, USP18−/− mice are resistant to viral infection (83). Here, USP18 works in concert with ISG-15, therefore inhibiting effective JAK/STAT signaling; based on the significance of this signaling pathway toward effective IFN signaling, the expression of these ISGs results in diminished IFN responses and counterintuitively facilitate HCV replication (84, 85). Important to note here is that transcription of both antiviral and proviral ISGs are driven by ligation of IFN receptors. Similarly, the presence of “negative regulators” such as these is therefore likely a negative feedback mechanism, which when functioning optimally reverts the host immunological milieu to “steady-state”. However, against chronic infection, the presence of such processes also contributes to desensitization to therapeutic IFN-administration in HCV patients with high levels of IFN expression (86). In this setting, the consequent evocation of ISGs such as USP-18 and ISG-15 renders the patients non-responsive to therapy (87, 88). This feature also underscores the possibility that efficacious virologic responses against persistent infection are blunted over time due to the presence of proviral ISGs. Along with the IFN-led dysregulation described in the LCMV model, the presence of dysregulation at the ISG level further renders the immune response in a state of flux and incapable of clearing the infection.
IFN Responses in HIV Infection
The progression of the HIV-associated IFN-I response closely mirrors that observed upon HCV infection. This evolution has been elegantly laid out using a simian immunodeficiency virus (SIV) model in rhesus macaques. In this study, Sandler et al. observed that IFN blockade in vivo accelerated advancement to AIDS with unchecked SIV replication whereas IFNα administration conferred resistance to the host upon challenge (89). However, in line with the observation of desensitization discussed in HCV, they also observed that sustained IFN administration led to a reversal of host resistance to infection and conversely, resembled the IFN blockade scenario in which the SIV reservoir was enlarged along with CD4+ T-cell depletion and AIDS. Notably, CD4+ T-cell depletion in this setting could be a function of the cellular tropism of the virus rather than solely the direct effect of IFN-mediated effects.
Furthermore, a wealth of research has also underscored the elevated IFN signature observed in the chronic stage of HIV infection, which correlates with high levels of viral load and thus, failed viremia control. Following transcriptome analyses on CD4+ T cells, Rotger et al. found that ISGs were upregulated in untreated patients relative to patients on therapy and healthy controls. In addition, upon induction of antiretroviral therapy and reduction of viremia, the ISG profiles in patient T cells reverted to those observed in the cohorts of HIV-infected individuals who maintain a CD4 T cell count of ≥500 (elite controllers) whose IFN level, and resultantly ISG expression is at a lower baseline (90). These findings were supported by previous findings of ISG upregulation in vitro and in vivo in CD4+ T cells from chronically infected HIV+ patients relative to healthy controls (91). Furthermore, despite similarity in expression levels in the acute phase of infection, the absence of hyperactivated IFN expression is a distinctive factor between pathogenic and non-pathogenic forms of SIV; while pathogenic SIVmac in rhesus macaques features an elevated IFN signature and resultant disease and the non-pathogenic SIVagm and SIVsmm in African green monkeys and Sootey mangabeys, respectively, neither exhibit aberrant IFN upregulation nor immune activation (92–94).
Lastly, the differences between pathogenic and non-pathogenic forms of SIV are partially driven by distinct signaling potentials through PRRs in pDCs (94); strong signaling through TLRs is observed in pathogenic SIV, which results in a surge of IFN that further propagates an immunopathogenic response as outlined in the various scenarios described above.
Closing Remarks and Outlook
It is important to note that causality between prolonged IFN expression and viral persistence is yet to be fully determined: does prolonged IFN diminish the immune response leading to viral persistence or does persistent infection lead to prolonged IFN expression whose dysregulation of immune responses is misconstrued as cause rather than effect? Nevertheless, the dizzying network of IFN-activating and IFN-inhibiting responses highlights the complexity in elucidating the exact nature of the IFN-related immunopathology in chronic infection (summarized in Figure 1). Intuitively, disruption of the delicate balance using exogenous IFN may result in less efficacious responses and adverse event profiles in therapeutic administration of IFN (95, 96). On the contrary, the multiplicity of pathways and molecules offers avenues that can be useful toward more effective therapeutic approaches by specific targeting of the deleterious moieties. For example, targeting proviral ISGs may offer an incisive approach toward triggering effective IFN responses and through their rescue, obviate exogenous IFN administration. From a prophylactic perspective, induction of nAbs in the absence of IFN signaling in B cells offers insight into the mechanisms that drive the delayed effective humoral response in diseases such as HIV and HCV. Given that the emergence of broadly nAbs against these chronic infections is delayed and in a highly altered immunological milieu, delineating the role of IFN-I facilitates a more comprehensive understanding of the conditions present during elicitation of broadly nAbs. In this regard, it is tempting to speculate that perhaps modulation of the IFN response along with the appropriate immunogen may advance vaccine work in these chronic infections along with other prophylactic measures as well. Altogether, these emergent insights bear significant impact on our understanding of the role of IFN-I in the immune response and importantly, its use in therapeutic settings. Guided by these findings, future work will more clearly determine the delicate balance that tips IFN responses from friend to foe.
Figure 1.
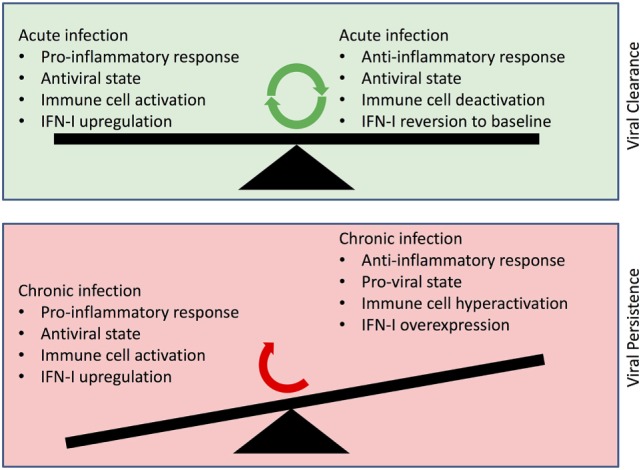
Depiction of changes in Type I interferon (IFN-I) roles in viral clearance vs. viral persistence. (Upper panel) Balance of functional IFN-I signaling, which results in viral clearance within the context of an acute infection. (Lower panel) Dysfunctional IFN-I signaling in the face of a chronic infection resulting in aberrant immune cell activation and viral persistence.
Author Contributions
AM contributed to the conceptualization of the subject, literature search, and writing the manuscript. AL contributed to the conceptualization of the subject, critical review of compiled literature, and writing the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. The reviewers MR and NF and handling Editor declared their shared affiliation, and the handling Editor states that the process nevertheless met the standards of a fair and objective review.
Funding
This work was supported by the Canadian Network on Hepatitis C (CanHepC) (AM) and the Natural Sciences and Engineering Research Council of Canada (NSERC), grant 355979-2013, the Canadian Institutes of Health Research (CIHR), grant MOP-89797, and the Jeanne and J.-Louis Lévesque Research Chair in Immunovirology from the J.-Louis Lévesque Foundation (AL).
References
- 1.McNab F, Mayer-Barber K, Sher A, Wack A, O’Garra A. Type I interferons in infectious disease. Nat Rev Immunol (2015) 15(2):87–103. 10.1038/nri3787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wilson EB, Yamada DH, Elsaesser H, Herskovitz J, Deng J, Cheng G, et al. Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science (2013) 340(6129):202–7. 10.1126/science.1235208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Teijaro JR, Ng C, Lee AM, Sullivan BM, Sheehan KC, Welch M, et al. Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science (2013) 340(6129):207–11. 10.1126/science.1235214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Enomoto H, Nishiguchi S. Factors associated with the response to interferon-based antiviral therapies for chronic hepatitis C. World J Hepatol (2015) 7(26):2681–7. 10.4254/wjh.v7.i26.2681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Azzoni L, Foulkes AS, Papasavvas E, Mexas AM, Lynn KM, Mounzer K, et al. Pegylated interferon alfa-2a monotherapy results in suppression of HIV type 1 replication and decreased cell-associated HIV DNA integration. J Infect Dis (2013) 207(2):213–22. 10.1093/infdis/jis663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lane HC, Davey V, Kovacs JA, Feinberg J, Metcalf JA, Herpin B, et al. Interferon-alpha in patients with asymptomatic human immunodeficiency virus (HIV) infection. A randomized, placebo-controlled trial. Ann Intern Med (1990) 112(11):805–11. 10.7326/0003-4819-112-11-805 [DOI] [PubMed] [Google Scholar]
- 7.Pillai SK, Abdel-Mohsen M, Guatelli J, Skasko M, Monto A, Fujimoto K, et al. Role of retroviral restriction factors in the interferon-alpha-mediated suppression of HIV-1 in vivo. Proc Natl Acad Sci U S A (2012) 109(8):3035–40. 10.1073/pnas.1111573109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tavel JA, Huang CY, Shen J, Metcalf JA, Dewar R, Shah A, et al. Interferon-alpha produces significant decreases in HIV load. J Interferon Cytokine Res (2010) 30(7):461–4. 10.1089/jir.2009.0090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Crow MK. Interferon-alpha: a therapeutic target in systemic lupus erythematosus. Rheum Dis Clin North Am (2010) 36(1):173–86,x. 10.1016/j.rdc.2009.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kirkwood J. Cancer immunotherapy: the interferon-alpha experience. Semin Oncol (2002) 29(3 Suppl 7):18–26. 10.1053/sonc.2002.33078 [DOI] [PubMed] [Google Scholar]
- 11.Kirkwood JM, Ibrahim JG, Sosman JA, Sondak VK, Agarwala SS, Ernstoff MS, et al. High-dose interferon alfa-2b significantly prolongs relapse-free and overall survival compared with the GM2-KLH/QS-21 vaccine in patients with resected stage IIB-III melanoma: results of intergroup trial E1694/S9512/C509801. J Clin Oncol (2001) 19(9):2370–80. [DOI] [PubMed] [Google Scholar]
- 12.Kirkwood JM, Strawderman MH, Ernstoff MS, Smith TJ, Borden EC, Blum RH. Interferon alfa-2b adjuvant therapy of high-risk resected cutaneous melanoma: the Eastern Cooperative Oncology Group Trial EST 1684. J Clin Oncol (1996) 14(1):7–17. [DOI] [PubMed] [Google Scholar]
- 13.Tarhini AA, Gogas H, Kirkwood JM. IFN-alpha in the treatment of melanoma. J Immunol (2012) 189(8):3789–93. 10.4049/jimmunol.1290060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Talpaz M, Hehlmann R, Quintas-Cardama A, Mercer J, Cortes J. Re-emergence of interferon-alpha in the treatment of chronic myeloid leukemia. Leukemia (2013) 27(4):803–12. 10.1038/leu.2012.313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hoofnagle JH, Sherker AH. Therapy for hepatitis C – the costs of success. N Engl J Med (2014) 370(16):1552–3. 10.1056/NEJMe1401508 [DOI] [PubMed] [Google Scholar]
- 16.Pestka S, Krause CD, Walter MR. Interferons, interferon-like cytokines, and their receptors. Immunol Rev (2004) 202:8–32. 10.1111/j.0105-2896.2004.00204.x [DOI] [PubMed] [Google Scholar]
- 17.Blasius AL, Beutler B. Intracellular toll-like receptors. Immunity (2010) 32(3):305–15. 10.1016/j.immuni.2010.03.012 [DOI] [PubMed] [Google Scholar]
- 18.Gantier MP, Tong S, Behlke MA, Xu D, Phipps S, Foster PS, et al. TLR7 is involved in sequence-specific sensing of single-stranded RNAs in human macrophages. J Immunol (2008) 180(4):2117–24. 10.4049/jimmunol.180.4.2117 [DOI] [PubMed] [Google Scholar]
- 19.Gorden KB, Gorski KS, Gibson SJ, Kedl RM, Kieper WC, Qiu X, et al. Synthetic TLR agonists reveal functional differences between human TLR7 and TLR8. J Immunol (2005) 174(3):1259–68. 10.4049/jimmunol.174.3.1259 [DOI] [PubMed] [Google Scholar]
- 20.Guo Z, Garg S, Hill KM, Jayashankar L, Mooney MR, Hoelscher M, et al. A distal regulatory region is required for constitutive and IFN-beta-induced expression of murine TLR9 gene. J Immunol (2005) 175(11):7407–18. 10.4049/jimmunol.175.11.7407 [DOI] [PubMed] [Google Scholar]
- 21.Heil F, Hemmi H, Hochrein H, Ampenberger F, Kirschning C, Akira S, et al. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science (2004) 303(5663):1526–9. 10.1126/science.1093620 [DOI] [PubMed] [Google Scholar]
- 22.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol (2010) 11(5):373–84. 10.1038/ni.1863 [DOI] [PubMed] [Google Scholar]
- 23.Molteni M, Gemma S, Rossetti C. The role of toll-like receptor 4 in infectious and noninfectious inflammation. Mediators Inflamm (2016) 2016:6978936. 10.1155/2016/6978936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zahringer U, Lindner B, Inamura S, Heine H, Alexander C. TLR2 – promiscuous or specific? A critical re-evaluation of a receptor expressing apparent broad specificity. Immunobiology (2008) 213(3–4):205–24. 10.1016/j.imbio.2008.02.005 [DOI] [PubMed] [Google Scholar]
- 25.Kim N, Now H, Nguyen NT, Yoo JY. Multilayered regulations of RIG-I in the anti-viral signaling pathway. J Microbiol (2016) 54(9):583–7. 10.1007/s12275-016-6322-2 [DOI] [PubMed] [Google Scholar]
- 26.McCartney SA, Thackray LB, Gitlin L, Gilfillan S, Virgin HW, Colonna M. MDA-5 recognition of a murine norovirus. PLoS Pathog (2008) 4(7):e1000108. 10.1371/journal.ppat.1000108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kufer TA, Banks DJ, Philpott DJ. Innate immune sensing of microbes by Nod proteins. Ann N Y Acad Sci (2006) 1072:19–27. 10.1196/annals.1326.020 [DOI] [PubMed] [Google Scholar]
- 28.Tamura T, Yanai H, Savitsky D, Taniguchi T. The IRF family transcription factors in immunity and oncogenesis. Annu Rev Immunol (2008) 26:535–84. 10.1146/annurev.immunol.26.021607.090400 [DOI] [PubMed] [Google Scholar]
- 29.Aaronson DS, Horvath CM. A road map for those who don’t know JAK-STAT. Science (2002) 296(5573):1653–5. 10.1126/science.1071545 [DOI] [PubMed] [Google Scholar]
- 30.Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol (2005) 5(5):375–86. 10.1038/nri1604 [DOI] [PubMed] [Google Scholar]
- 31.Hu X, Chakravarty SD, Ivashkiv LB. Regulation of interferon and Toll-like receptor signaling during macrophage activation by opposing feedforward and feedback inhibition mechanisms. Immunol Rev (2008) 226:41–56. 10.1111/j.1600-065X.2008.00707.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Essers MA, Offner S, Blanco-Bose WE, Waibler Z, Kalinke U, Duchosal MA, et al. IFNalpha activates dormant haematopoietic stem cells in vivo. Nature (2009) 458(7240):904–8. 10.1038/nature07815 [DOI] [PubMed] [Google Scholar]
- 33.Sato T, Onai N, Yoshihara H, Arai F, Suda T, Ohteki T. Interferon regulatory factor-2 protects quiescent hematopoietic stem cells from type I interferon-dependent exhaustion. Nat Med (2009) 15(6):696–700. 10.1038/nm.1973 [DOI] [PubMed] [Google Scholar]
- 34.Baranek T, Manh TP, Alexandre Y, Maqbool MA, Cabeza JZ, Tomasello E, et al. Differential responses of immune cells to type I interferon contribute to host resistance to viral infection. Cell Host Microbe (2012) 12(4):571–84. 10.1016/j.chom.2012.09.002 [DOI] [PubMed] [Google Scholar]
- 35.Gill N, Chenoweth MJ, Verdu EF, Ashkar AA. NK cells require type I IFN receptor for antiviral responses during genital HSV-2 infection. Cell Immunol (2011) 269(1):29–37. 10.1016/j.cellimm.2011.03.007 [DOI] [PubMed] [Google Scholar]
- 36.Guan J, Miah SM, Wilson ZS, Erick TK, Banh C, Brossay L. Role of type I interferon receptor signaling on NK cell development and functions. PLoS One (2014) 9(10):e111302. 10.1371/journal.pone.0111302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Berghöfer B, Haley G, Frommer T, Bein G, Hackstein H. Natural and synthetic TLR7 ligands inhibit CpG-A- and CpG-C-oligodeoxynucleotide-induced IFN-alpha production. J Immunol (2007) 178(7):4072–9. 10.4049/jimmunol.178.7.4072 [DOI] [PubMed] [Google Scholar]
- 38.Fitzgerald-Bocarsly P, Dai J, Singh S. Plasmacytoid dendritic cells and type I IFN: 50 years of convergent history. Cytokine Growth Factor Rev (2008) 19(1):3–19. 10.1016/j.cytogfr.2007.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hochrein H, Schlatter B, O’Keeffe M, Wagner C, Schmitz F, Schiemann M, et al. Herpes simplex virus type-1 induces IFN-alpha production via Toll-like receptor 9-dependent and -independent pathways. Proc Natl Acad Sci U S A (2004) 101(31):11416–21. 10.1073/pnas.0403555101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ito T, Amakawa R, Inaba M, Ikehara S, Inaba K, Fukuhara S. Differential regulation of human blood dendritic cell subsets by IFNs. J Immunol (2001) 166(5):2961–9. 10.4049/jimmunol.166.5.2961 [DOI] [PubMed] [Google Scholar]
- 41.Kadowaki N, Antonenko S, Lau JY, Liu YJ. Natural interferon alpha/beta-producing cells link innate and adaptive immunity. J Exp Med (2000) 192(2):219–26. 10.1084/jem.192.2.219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Libri NA, Barker SJ, Rosenberg WM, Semper AE. A class C CpG toll-like receptor 9 agonist successfully induces robust interferon-alpha production by plasmacytoid dendritic cells from patients chronically infected with hepatitis C. J Viral Hepat (2009) 16(5):315–24. 10.1111/j.1365-2893.2008.01011.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Longman RS, Braun D, Pellegrini S, Rice CM, Darnell RB, Albert ML. Dendritic-cell maturation alters intracellular signaling networks, enabling differential effects of IFN-alpha/beta on antigen cross-presentation. Blood (2007) 109(3):1113–22. 10.1182/blood-2006-05-023465 [DOI] [PubMed] [Google Scholar]
- 44.Martinez J, Huang X, Yang Y. Direct action of type I IFN on NK cells is required for their activation in response to vaccinia viral infection in vivo. J Immunol (2008) 180(3):1592–7. 10.4049/jimmunol.180.3.1592 [DOI] [PubMed] [Google Scholar]
- 45.Matikainen S, Paananen A, Miettinen M, Kurimoto M, Timonen T, Julkunen I, et al. IFN-alpha and IL-18 synergistically enhance IFN-gamma production in human NK cells: differential regulation of Stat4 activation and IFN-gamma gene expression by IFN-alpha and IL-12. Eur J Immunol (2001) 31(7):2236–45. 10.1002/1521-4141(200107)31:7<2236::AID-IMMU2236>3.3.CO;2-7 [DOI] [PubMed] [Google Scholar]
- 46.Nguyen KB, Salazar-Mather TP, Dalod MY, Van Deusen JB, Wei XQ, Liew FY, et al. Coordinated and distinct roles for IFN-alpha beta, IL-12, and IL-15 regulation of NK cell responses to viral infection. J Immunol (2002) 169(8):4279–87. 10.4049/jimmunol.169.8.4279 [DOI] [PubMed] [Google Scholar]
- 47.Zhu J, Martinez J, Huang X, Yang Y. Innate immunity against vaccinia virus is mediated by TLR2 and requires TLR-independent production of IFN-beta. Blood (2007) 109(2):619–25. 10.1182/blood-2006-06-027136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fahey LM, Wilson EB, Elsaesser H, Fistonich CD, McGavern DB, Brooks DG. Viral persistence redirects CD4 T cell differentiation toward T follicular helper cells. J Exp Med (2011) 208(5):987–99. 10.1084/jem.20101773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Osokine I, Snell LM, Cunningham CR, Yamada DH, Wilson EB, Elsaesser HJ, et al. Type I interferon suppresses de novo virus-specific CD4 Th1 immunity during an established persistent viral infection. Proc Natl Acad Sci U S A (2014) 111(20):7409–14. 10.1073/pnas.1401662111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Moskophidis D, Lechner F, Pircher H, Zinkernagel RM. Virus persistence in acutely infected immunocompetent mice by exhaustion of antiviral cytotoxic effector T cells. Nature (1993) 362(6422):758–61. 10.1038/362758a0 [DOI] [PubMed] [Google Scholar]
- 51.Wherry EJ. T cell exhaustion. Nat Immunol (2011) 12(6):492–9. 10.1038/ni.2035 [DOI] [PubMed] [Google Scholar]
- 52.Fuller MJ, Khanolkar A, Tebo AE, Zajac AJ. Maintenance, loss, and resurgence of T cell responses during acute, protracted, and chronic viral infections. J Immunol (2004) 172(7):4204–14. 10.4049/jimmunol.172.7.4204 [DOI] [PubMed] [Google Scholar]
- 53.Fuller MJ, Zajac AJ. Ablation of CD8 and CD4 T cell responses by high viral loads. J Immunol (2003) 170(1):477–86. 10.4049/jimmunol.170.1.477 [DOI] [PubMed] [Google Scholar]
- 54.Wherry EJ, Blattman JN, Murali-Krishna K, van der Most R, Ahmed R. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J Virol (2003) 77(8):4911–27. 10.1128/JVI.77.8.4911-4927.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zajac AJ, Blattman JN, Murali-Krishna K, Sourdive DJ, Suresh M, Altman JD, et al. Viral immune evasion due to persistence of activated T cells without effector function. J Exp Med (1998) 188(12):2205–13. 10.1084/jem.188.12.2205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wherry EJ, Ha SJ, Kaech SM, Haining WN, Sarkar S, Kalia V, et al. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity (2007) 27(4):670–84. 10.1016/j.immuni.2007.09.006 [DOI] [PubMed] [Google Scholar]
- 57.Fuller MJ, Hildeman DA, Sabbaj S, Gaddis DE, Tebo AE, Shang L, et al. Cutting edge: emergence of CD127high functionally competent memory T cells is compromised by high viral loads and inadequate T cell help. J Immunol (2005) 174(10):5926–30. 10.4049/jimmunol.174.10.5926 [DOI] [PubMed] [Google Scholar]
- 58.Charles ED, Green RM, Marukian S, Talal AH, Lake-Bakaar GV, Jacobson IM, et al. Clonal expansion of immunoglobulin M+CD27+ B cells in HCV-associated mixed cryoglobulinemia. Blood (2008) 111(3):1344–56. 10.1182/blood-2007-07-101717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mayo MJ. Extrahepatic manifestations of hepatitis C infection. Am J Med Sci (2003) 325(3):135–48. 10.1097/00000441-200303000-00006 [DOI] [PubMed] [Google Scholar]
- 60.Racanelli V, Frassanito MA, Leone P, Galiano M, De Re V, Silvestris F, et al. Antibody production and in vitro behavior of CD27-defined B-cell subsets: persistent hepatitis C virus infection changes the rules. J Virol (2006) 80(8):3923–34. 10.1128/JVI.80.8.3923-3934.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.De Milito A, Nilsson A, Titanji K, Thorstensson R, Reizenstein E, Narita M, et al. Mechanisms of hypergammaglobulinemia and impaired antigen-specific humoral immunity in HIV-1 infection. Blood (2004) 103(6):2180–6. 10.1182/blood-2003-07-2375 [DOI] [PubMed] [Google Scholar]
- 62.Cagigi A, Du L, Dang LV, Grutzmeier S, Atlas A, Chiodi F, et al. CD27(-) B-cells produce class switched and somatically hyper-mutated antibodies during chronic HIV-1 infection. PLoS One (2009) 4(5):e5427. 10.1371/journal.pone.0005427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.He B, Qiao X, Klasse PJ, Chiu A, Chadburn A, Knowles DM, et al. HIV-1 envelope triggers polyclonal Ig class switch recombination through a CD40-independent mechanism involving BAFF and C-type lectin receptors. J Immunol (2006) 176(7):3931–41. 10.4049/jimmunol.176.7.3931 [DOI] [PubMed] [Google Scholar]
- 64.Moir S, Ho J, Malaspina A, Wang W, DiPoto AC, O’Shea MA, et al. Evidence for HIV-associated B cell exhaustion in a dysfunctional memory B cell compartment in HIV-infected viremic individuals. J Exp Med (2008) 205(8):1797–805. 10.1084/jem.20072683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Carbonari M, Caprini E, Tedesco T, Mazzetta F, Tocco V, Casato M, et al. Hepatitis C virus drives the unconstrained monoclonal expansion of VH1-69-expressing memory B cells in type II cryoglobulinemia: a model of infection-driven lymphomagenesis. J Immunol (2005) 174(10):6532–9. 10.4049/jimmunol.174.10.6532 [DOI] [PubMed] [Google Scholar]
- 66.Qiao X, He B, Chiu A, Knowles DM, Chadburn A, Cerutti A. Human immunodeficiency virus 1 Nef suppresses CD40-dependent immunoglobulin class switching in bystander B cells. Nat Immunol (2006) 7(3):302–10. 10.1038/ni1302 [DOI] [PubMed] [Google Scholar]
- 67.Daugan M, Murira A, Mindt BC, Germain A, Tarrab E, Lapierre P, et al. Type I interferon impairs specific antibody responses early during establishment of LCMV infection. Front Immunol (2016) 7:564. 10.3389/fimmu.2016.00564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ng CT, Sullivan BM, Teijaro JR, Lee AM, Welch M, Rice S, et al. Blockade of interferon beta, but not interferon alpha, signaling controls persistent viral infection. Cell Host Microbe (2015) 17(5):653–61. 10.1016/j.chom.2015.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen J, Trounstine M, Alt FW, Young F, Kurahara C, Loring JF, et al. Immunoglobulin gene rearrangement in B cell deficient mice generated by targeted deletion of the JH locus. Int Immunol (1993) 5(6):647–56. 10.1093/intimm/5.6.647 [DOI] [PubMed] [Google Scholar]
- 70.Price GE, Gaszewska-Mastarlarz A, Moskophidis D. The role of alpha/beta and gamma interferons in development of immunity to influenza A virus in mice. J Virol (2000) 74(9):3996–4003. 10.1128/JVI.74.9.3996-4003.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Silva-Barrios S, Smans M, Duerr CU, Qureshi ST, Fritz JH, Descoteaux A, et al. Innate immune B cell activation by Leishmania donovani exacerbates disease and mediates hypergammaglobulinemia. Cell Rep (2016) 15(11):2427–37. 10.1016/j.celrep.2016.05.028 [DOI] [PubMed] [Google Scholar]
- 72.Asmuth DM, Murphy RL, Rosenkranz SL, Lertora JJ, Kottilil S, Cramer Y, et al. Safety, tolerability, and mechanisms of antiretroviral activity of pegylated interferon Alfa-2a in HIV-1-monoinfected participants: a phase II clinical trial. J Infect Dis (2010) 201(11):1686–96. 10.1086/652420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bartosch B, Dubuisson J, Cosset FL. Infectious hepatitis C virus pseudo-particles containing functional E1-E2 envelope protein complexes. J Exp Med (2003) 197(5):633–42. 10.1084/jem.20021756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Steinmann E, Pietschmann T. Cell culture systems for hepatitis C virus. Curr Top Microbiol Immunol (2013) 369:17–48. 10.1007/978-3-642-27340-7_2 [DOI] [PubMed] [Google Scholar]
- 75.Wilson GK, Stamataki Z. In vitro systems for the study of hepatitis C virus infection. Int J Hepatol (2012) 2012:292591. 10.1155/2012/292591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li K, Li NL, Wei D, Pfeffer SR, Fan M, Pfeffer LM. Activation of chemokine and inflammatory cytokine response in hepatitis C virus-infected hepatocytes depends on Toll-like receptor 3 sensing of hepatitis C virus double-stranded RNA intermediates. Hepatology (2012) 55(3):666–75. 10.1002/hep.24763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wang N, Liang Y, Devaraj S, Wang J, Lemon SM, Li K. Toll-like receptor 3 mediates establishment of an antiviral state against hepatitis C virus in hepatoma cells. J Virol (2009) 83(19):9824–34. 10.1128/JVI.01125-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Horner SM, Gale M, Jr. Regulation of hepatic innate immunity by hepatitis C virus. Nat Med (2013) 19(7):879–88. 10.1038/nm.3253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.de Veer MJ, Holko M, Frevel M, Walker E, Der S, Paranjape JM, et al. Functional classification of interferon-stimulated genes identified using microarrays. J Leukoc Biol (2001) 69(6):912–20. [PubMed] [Google Scholar]
- 80.Sarasin-Filipowicz M, Oakeley EJ, Duong FH, Christen V, Terracciano L, Filipowicz W, et al. Interferon signaling and treatment outcome in chronic hepatitis C. Proc Natl Acad Sci U S A (2008) 105(19):7034–9. 10.1073/pnas.0707882105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Abdel-Hakeem MS, Bédard N, Badr G, Ostrowski M, Sékaly RP, Bruneau J, et al. Comparison of immune restoration in early versus late alpha interferon therapy against hepatitis C virus. J Virol (2010) 84(19):10429–35. 10.1128/JVI.01094-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Frankova S, Jirsa M, Merta D, Neroldova M, Urbanek P, Senkerikova R, et al. USP18 downregulation in peripheral blood mononuclear cells predicts nonresponse to interferon-based triple therapy in patients with chronic hepatitis C, genotype 1: a pilot study. Ther Clin Risk Manag (2015) 11:1853–61. 10.2147/TCRM.S94010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ritchie KJ, Hahn CS, Kim KI, Yan M, Rosario D, Li L, et al. Role of ISG15 protease UBP43 (USP18) in innate immunity to viral infection. Nat Med (2004) 10(12):1374–8. 10.1038/nm1133 [DOI] [PubMed] [Google Scholar]
- 84.Francois-Newton V, Magno de Freitas Almeida G, Payelle-Brogard B, Monneron D, Pichard-Garcia L, Piehler J, et al. USP18-based negative feedback control is induced by type I and type III interferons and specifically inactivates interferon alpha response. PLoS One (2011) 6(7):e22200. 10.1371/journal.pone.0022200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Potu H, Sgorbissa A, Brancolini C. Identification of USP18 as an important regulator of the susceptibility to IFN-alpha and drug-induced apoptosis. Cancer Res (2010) 70(2):655–65. 10.1158/0008-5472.CAN-09-1942 [DOI] [PubMed] [Google Scholar]
- 86.Dill MT, Duong FH, Vogt JE, Bibert S, Bochud PY, Terracciano L, et al. Interferon-induced gene expression is a stronger predictor of treatment response than IL28B genotype in patients with hepatitis C. Gastroenterology (2011) 140(3):1021–31. 10.1053/j.gastro.2010.11.039 [DOI] [PubMed] [Google Scholar]
- 87.Chen L, Borozan I, Sun J, Guindi M, Fischer S, Feld J, et al. Cell-type specific gene expression signature in liver underlies response to interferon therapy in chronic hepatitis C infection. Gastroenterology (2010) 138(3):.e1–3. 10.1053/j.gastro.2009.10.046 [DOI] [PubMed] [Google Scholar]
- 88.Randall G, Chen L, Panis M, Fischer AK, Lindenbach BD, Sun J, et al. Silencing of USP18 potentiates the antiviral activity of interferon against hepatitis C virus infection. Gastroenterology (2006) 131(5):1584–91. 10.1053/j.gastro.2006.08.043 [DOI] [PubMed] [Google Scholar]
- 89.Sandler NG, Bosinger SE, Estes JD, Zhu RT, Tharp GK, Boritz E, et al. Type I interferon responses in rhesus macaques prevent SIV infection and slow disease progression. Nature (2014) 511(7511):601–5. 10.1038/nature13554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Rotger M, Dang KK, Fellay J, Heinzen EL, Feng S, Descombes P, et al. Genome-wide mRNA expression correlates of viral control in CD4+ T-cells from HIV-1-infected individuals. PLoS Pathog (2010) 6(2):e1000781. 10.1371/journal.ppat.1000781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sedaghat AR, German J, Teslovich TM, Cofrancesco J, Jr, Jie CC, Talbot CC, Jr, et al. Chronic CD4+ T-cell activation and depletion in human immunodeficiency virus type 1 infection: type I interferon-mediated disruption of T-cell dynamics. J Virol (2008) 82(4):1870–83. 10.1128/JVI.02228-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Jacquelin B, Mayau V, Targat B, Liovat AS, Kunkel D, Petitjean G, et al. Nonpathogenic SIV infection of African green monkeys induces a strong but rapidly controlled type I IFN response. J Clin Invest (2009) 119(12):3544–55. 10.1172/JCI40093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Harris LD, Tabb B, Sodora DL, Paiardini M, Klatt NR, Douek DC, et al. Downregulation of robust acute type I interferon responses distinguishes nonpathogenic simian immunodeficiency virus (SIV) infection of natural hosts from pathogenic SIV infection of rhesus macaques. J Virol (2010) 84(15):7886–91. 10.1128/JVI.02612-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bosinger SE, Li Q, Gordon SN, Klatt NR, Duan L, Xu L, et al. Global genomic analysis reveals rapid control of a robust innate response in SIV-infected sooty mangabeys. J Clin Invest (2009) 119(12):3556–72. 10.1172/JCI40115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Manns MP, McHutchison JG, Gordon SC, Rustgi VK, Shiffman M, Reindollar R, et al. Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: a randomised trial. Lancet (2001) 358(9286):958–65. 10.1016/S0140-6736(01)06102-5 [DOI] [PubMed] [Google Scholar]
- 96.Hadziyannis SJ, Sette H, Jr, Morgan TR, Balan V, Diago M, Marcellin P, et al. Peginterferon-alpha2a and ribavirin combination therapy in chronic hepatitis C: a randomized study of treatment duration and ribavirin dose. Ann Intern Med (2004) 140(5):346–55. 10.7326/0003-4819-140-5-200403020-00010 [DOI] [PubMed] [Google Scholar]