

Abstract
Binuclear Cu proteins play vital roles in O2 binding and activation in biology and can be classified into coupled and noncoupled binuclear sites based on the magnetic interaction between the two Cu centers. Coupled binuclear Cu proteins include hemocyanin, tyrosinase, and catechol oxidase. These proteins have two Cu centers strongly magnetically coupled through direct bridging ligands that provide a mechanism for the 2-electron reduction of O2 to a μ-η2:η2 side-on peroxide bridged  species. This side-on bridged peroxo-CuII2 species is activated for electrophilic attack on the phenolic ring of substrates. Noncoupled binuclear Cu proteins include peptidylglycine α-hydroxylating monooxygenase and dopamine β-monooxygenase. These proteins have binuclear Cu active sites that are distant, that exhibit no exchange interaction, and that activate O2 at a single Cu center to generate a reactive CuII/O2 species for H-atom abstraction from the C–H bond of substrates. O2 intermediates in the coupled binuclear Cu enzymes can be trapped and studied spectroscopically. Possible intermediates in noncoupled binuclear Cu proteins can be defined through correlation to mononuclear CuII/O2 model complexes. The different intermediates in these two classes of binuclear Cu proteins exhibit different reactivities that correlate with their different electronic structures and exchange coupling interactions between the binuclear Cu centers. These studies provide insight into the role of exchange coupling between the Cu centers in their reaction mechanisms.
species. This side-on bridged peroxo-CuII2 species is activated for electrophilic attack on the phenolic ring of substrates. Noncoupled binuclear Cu proteins include peptidylglycine α-hydroxylating monooxygenase and dopamine β-monooxygenase. These proteins have binuclear Cu active sites that are distant, that exhibit no exchange interaction, and that activate O2 at a single Cu center to generate a reactive CuII/O2 species for H-atom abstraction from the C–H bond of substrates. O2 intermediates in the coupled binuclear Cu enzymes can be trapped and studied spectroscopically. Possible intermediates in noncoupled binuclear Cu proteins can be defined through correlation to mononuclear CuII/O2 model complexes. The different intermediates in these two classes of binuclear Cu proteins exhibit different reactivities that correlate with their different electronic structures and exchange coupling interactions between the binuclear Cu centers. These studies provide insight into the role of exchange coupling between the Cu centers in their reaction mechanisms.
Biological Cu centers play important roles in O2 binding, activation, and reduction to H2O (1). Hemocyanin (Hc), tyrosinase (Tyr), and catechol oxidase (CO) are well studied systems and contain similar binuclear Cu active sites in which the two Cu centers are close in distance (≈3.6 Å) with strong magnetic interactions (1, 2). The binuclear Cu center in Hc reversibly binds O2, whereas the binuclear Cu active sites in Tyr and CO activate O2 for substrate hydroxylation/oxidation. Upon O2 binding, these binuclear Cu proteins generate the same 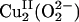 peroxide intermediate, where the
peroxide intermediate, where the 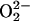 binds in a μ-η2:η2 side-on bridging fashion providing a direct orbital overlap pathway for the 2-electron (e–) reduction of O2 to peroxide (3). This side-on
binds in a μ-η2:η2 side-on bridging fashion providing a direct orbital overlap pathway for the 2-electron (e–) reduction of O2 to peroxide (3). This side-on 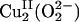 species has been extensively characterized, both in model systems and trapped protein intermediates, and exhibits characteristic spectral features, including an intense charge transfer (CT) transition at ≈350 nm (ε ≈ 20,000 M–1·cm–1) in its absorption spectrum and a very low vO–O vibrational frequency (≈750 cm–1) in its resonance Raman (rR) spectrum (1, 2). These spectral features result from the side-on bound peroxide
species has been extensively characterized, both in model systems and trapped protein intermediates, and exhibits characteristic spectral features, including an intense charge transfer (CT) transition at ≈350 nm (ε ≈ 20,000 M–1·cm–1) in its absorption spectrum and a very low vO–O vibrational frequency (≈750 cm–1) in its resonance Raman (rR) spectrum (1, 2). These spectral features result from the side-on bound peroxide 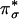 orbital charge donation and the σ* backbonding interactions with the CuII dx2–y2 orbitals [Fig. 1 A,
orbital charge donation and the σ* backbonding interactions with the CuII dx2–y2 orbitals [Fig. 1 A,  donor, lowest unoccupied molecular orbital (LUMO), and B, σ* acceptor, highest occupied molecular orbital (HOMO)]. These CuII-peroxide bonding interactions lead to a large energy splitting between the LUMO and HOMO of the side-on
donor, lowest unoccupied molecular orbital (LUMO), and B, σ* acceptor, highest occupied molecular orbital (HOMO)]. These CuII-peroxide bonding interactions lead to a large energy splitting between the LUMO and HOMO of the side-on  species and the strong antiferromagnetic coupling between its two CuII centers. In Tyr, the large peroxide
species and the strong antiferromagnetic coupling between its two CuII centers. In Tyr, the large peroxide 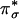 to Cu charge donation results in significant peroxide character in the LUMO (Fig. 1 A), which activates the side-on
to Cu charge donation results in significant peroxide character in the LUMO (Fig. 1 A), which activates the side-on  species for electrophilic attack at the phenolic ring of the substrate, leading to its hydroxylation (Fig. 1C). The σ* backbonding interaction (in Fig. 1B) weakens the O–O bond, facilitating its cleavage.
species for electrophilic attack at the phenolic ring of the substrate, leading to its hydroxylation (Fig. 1C). The σ* backbonding interaction (in Fig. 1B) weakens the O–O bond, facilitating its cleavage.
Fig. 1.

FMO of the side-on  species. (A and B) Contour plots of the LUMO (A) and HOMO (B) of the side-on
species. (A and B) Contour plots of the LUMO (A) and HOMO (B) of the side-on 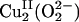 species showing peroxide
species showing peroxide 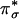 donation and σ* backbonding interactions with the CuII dx2–y2 orbitals. (C) Frontier molecular orbitals of the phenyl HOMO and side-on
donation and σ* backbonding interactions with the CuII dx2–y2 orbitals. (C) Frontier molecular orbitals of the phenyl HOMO and side-on 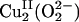 LUMO showing the orbital overlap for electrophilic attack.
LUMO showing the orbital overlap for electrophilic attack.
Based on the strong magnetic exchange interaction between the Cu centers [quantitated by the exchange coupling constant J (H = –2 JS1·S2)], Hc, Tyr, and CO have been classified as coupled binuclear Cu proteins (1). In contrast, noncoupled binuclear Cu proteins have two structurally inequivalent Cu centers largely separated in space (≈11 Å) with no direct bridging ligand and no observable magnetic interaction (4–6). This class of binuclear Cu proteins includes peptidylglycine α-hydroxylating monooxygenase (PHM) and dopamine β-monooxygenase (DβM), both of which catalyze substrate C–H bond hydroxylation (a Gly backbone C–H bond in PHM or a dopamine benzylic C–H bond in DβM) in a stereo- and regiospecific fashion by means of an H-atom abstraction mechanism (4). This C–H bond H-atom abstraction is performed by a reactive mononuclear CuII/O2 species at the CuM site [alternatively labeled CuB (6)]. The other CuH site [alternatively labeled CuA (6)] provides an additional electron through long-range electron transfer (ET) to the CuM site. Because the two Cu centers in PHM and DβM showed no electronic coupling (i.e., noncoupled), the mechanism for this intramolecular long-range ET was not clear. A superoxide-channeling mechanism (7) and a substrate-facilitated ET mechanism (8, 9) were proposed to account for this inter-Cu ET process.
In contrast to the well studied side-on Cu2 (O –2) species in coupled binuclear proteins, the nature of the reactive mononuclear CuII/O2 species in PHM and DβM was unclear. Structural information from crystallographic (6, 8) and EXAFS (extended x-ray absorption fine structure) studies (10–12), combined with recent advances in spectroscopic characterization coupled with density functional theory (DFT) calculations, have defined geometric and electronic structural models of the resting oxidized and reduced CuM and CuH sites (Fig. 2) (13). In correlation to detailed spectroscopic and electronic structure studies of related model complexes (14, 15), DFT calculations have been used to evaluate two possible mononuclear CuII/O2 species proposed to be the intermediates in the H-atom abstraction reactivity in PHM DβM (16): a 2-e– reduced 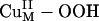 species and an 1-e– reduced CuM–superoxide species. These studies generated a reasonable reaction coordinate for the H-atom abstraction reaction in PHM and DβM, involving the mononuclear
species and an 1-e– reduced CuM–superoxide species. These studies generated a reasonable reaction coordinate for the H-atom abstraction reaction in PHM and DβM, involving the mononuclear  species. The
species. The 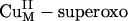 intermediate is consistent with and further supported by recent kinetic studies on DβM (17). These studies on the non-coupled binuclear Cu enzymes PHM and DβM reveal a very different reaction mechanism from that of the coupled binuclear Cu proteins and define important contributions from the differences in exchange coupling to variation in O2 activation by binuclear Cu enzymes.
intermediate is consistent with and further supported by recent kinetic studies on DβM (17). These studies on the non-coupled binuclear Cu enzymes PHM and DβM reveal a very different reaction mechanism from that of the coupled binuclear Cu proteins and define important contributions from the differences in exchange coupling to variation in O2 activation by binuclear Cu enzymes.
Fig. 2.

Geometry-optimized structures of resting oxidized (A) and reduced (B) CuM site and resting oxidized (C) and reduced (D)CuH site.
Electronic Structure Descriptions of Possible Mononuclear  Intermediates: Frontier Molecular Orbitals (FMOs) for H-atom Abstraction
Intermediates: Frontier Molecular Orbitals (FMOs) for H-atom Abstraction
Spectroscopic studies combined with DFT calculations can provide detailed electronic structure descriptions of Cun–O2 species (1). Electronic structure descriptions provide insight into relative chemical reactivities (18), for which the energies, molecular orbital coefficients, and overlaps of FMOs (i.e., low-energy unoccupied and high-energy occupied molecular orbitals, particularly the LUMO and HOMO) play important roles in activating specific reaction coordinates and can be obtained from DFT calculations (1). In this section, we describe the acceptor FMOs of the 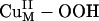 and
and  species at the CuM site in PHM, extended from spectroscopic results on well defined model complexes and consider their relative activation for H-atom abstraction.
species at the CuM site in PHM, extended from spectroscopic results on well defined model complexes and consider their relative activation for H-atom abstraction.
The 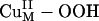 Intermediate. The spectroscopically calibrated and DFT optimized structure of the putative
Intermediate. The spectroscopically calibrated and DFT optimized structure of the putative 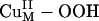 intermediate has a square pyramidal geometry with the OOH– bound in an end-on fashion (Fig. 3A) (13). The lowest energy acceptor orbital of
intermediate has a square pyramidal geometry with the OOH– bound in an end-on fashion (Fig. 3A) (13). The lowest energy acceptor orbital of 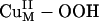 for H-atom abstraction is the spin-down LUMO, which is a Cu d-based orbital (≈61% Cu character) with low OOH–
for H-atom abstraction is the spin-down LUMO, which is a Cu d-based orbital (≈61% Cu character) with low OOH– 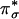 character (≈19%) because of a Cu–OOH pseudo-σ bonding interaction that is not very covalent (Fig. 3B). The OOH–
character (≈19%) because of a Cu–OOH pseudo-σ bonding interaction that is not very covalent (Fig. 3B). The OOH– 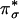 component of this spin-down LUMO is highly polarized toward the Cu with a limited coefficient (2%) on the remote oxygen atom, indicating that the spin-down LUMO is, in fact, not a good acceptor orbital for H-atom abstraction. Another possible H-atom abstraction acceptor orbital, the OOH– σ* orbital, is ≈3 eV (1 eV = 1.602 × 10–19 J) higher in energy than the spin-down LUMO and has a large coefficient on the O2 moiety (53%, Fig. 3C). However, the σ* orbital is similarly polarized toward the Cu, resulting in a much lower molecular orbital coefficient on the remote oxygen atom (13%). This polarization is mainly due to the effect of protonation and is generally observed for CuII bound OOH– (13, 14, 19), giving rise to a strengthened O–O bond.
component of this spin-down LUMO is highly polarized toward the Cu with a limited coefficient (2%) on the remote oxygen atom, indicating that the spin-down LUMO is, in fact, not a good acceptor orbital for H-atom abstraction. Another possible H-atom abstraction acceptor orbital, the OOH– σ* orbital, is ≈3 eV (1 eV = 1.602 × 10–19 J) higher in energy than the spin-down LUMO and has a large coefficient on the O2 moiety (53%, Fig. 3C). However, the σ* orbital is similarly polarized toward the Cu, resulting in a much lower molecular orbital coefficient on the remote oxygen atom (13%). This polarization is mainly due to the effect of protonation and is generally observed for CuII bound OOH– (13, 14, 19), giving rise to a strengthened O–O bond.
Fig. 3.

Acceptor FMOs of the 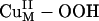 intermediate. (A) Geometry-optimized structure of
intermediate. (A) Geometry-optimized structure of 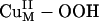 . (B) Spin-down LUMO
. (B) Spin-down LUMO 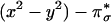 . (C) Peroxide σ* orbital. (D) rR spectra of the CuII–OOH model complex in the vO–O region excited at 568.2 nm (14).
. (C) Peroxide σ* orbital. (D) rR spectra of the CuII–OOH model complex in the vO–O region excited at 568.2 nm (14).
This increased O–O bond strength is best probed experimentally by rR spectroscopy, where the vO–O frequency and its 18O isotope shift can be used (along with the Cu–O modes) to determine the O–O force constant kO–O by means of a normal coordinate analysis. Fig. 3D gives the rR spectra of a related mononuclear model complex, L3CuII–OOH [where L3 is hydrotris(3-tert-butyl-5-isopropyl-1-pyrazolyl)borate] (14). The vO–O vibration of L3CuII–OOH occurs at 843 cm–1 and shifts to 799 cm–1 upon 18O labeling. These rR data give a kO–O for L3CuII–OOH of 3.51 mdyne/Å (1 dyne = 10 μN), which is higher than those of unprotonated 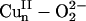 species (Table 1, row E), reflecting the strengthened O–O bond from protonation-induced polarization. The facts that the σ* orbital has a low coefficient on the remote oxygen atom because of polarization and is high in energy indicate that it is also an ineffective pathway for H-atom abstraction. Therefore, the electronic structure description of the putative
species (Table 1, row E), reflecting the strengthened O–O bond from protonation-induced polarization. The facts that the σ* orbital has a low coefficient on the remote oxygen atom because of polarization and is high in energy indicate that it is also an ineffective pathway for H-atom abstraction. Therefore, the electronic structure description of the putative 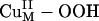 intermediate suggested that it has a strong O–O bond that is not significantly activated for H-atom abstraction.
intermediate suggested that it has a strong O–O bond that is not significantly activated for H-atom abstraction.
Table 1. Summary of vO–O frequencies and kO–O force constants of binuclear and mononuclear Cu–O2 species.
| Nature of ligand | Structure | vO–O, cm-1 | kO–O, mdyne/Å | |
|---|---|---|---|---|
| A | Peroxo | 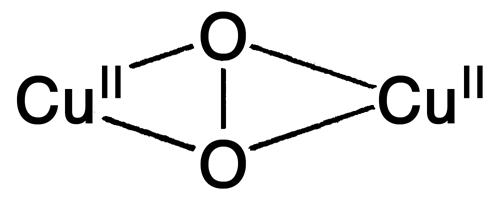 |
763 | 2.43 |
| B | Peroxo |  |
832 | 3.17 |
| C | Hydroperoxo |  |
880 | 3.52 |
| D | Peroxo | 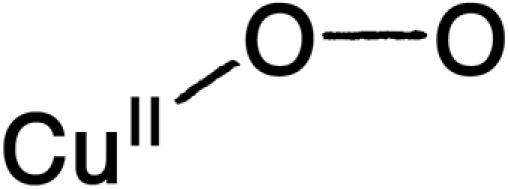 |
803 | 2.90 |
| E | Hydroperoxo |  |
843 | 3.51 |
| F | Peroxo |  |
968 | - |
| G | Oxo |  |
NA | NA |
| H | Superoxo | 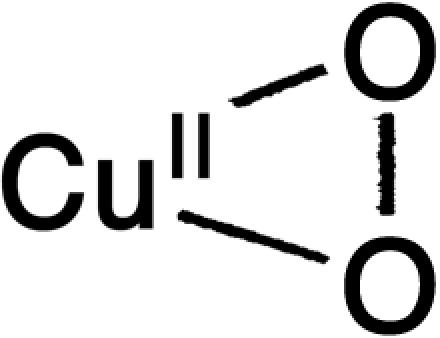 |
1,043 | 5.72 |
The Side-On  Intermediate. The optimized lowest energy structure of the CuIIM–superoxo intermediate has the superoxide ligand bound equatorially in a side-on fashion, forming a square pyramidal geometry around the Cu center with a Met as the long axial ligand (Fig. 4A) (16). This side-on geometry is very similar to the crystal structure of a well characterized side-on CuII–superoxo model complex LCuIIO2 (where L is the trispyrazolylborate ligand). The superoxide nature of this complex was experimentally determined from its vO–O frequency (1,043 cm–1) and 18O isotope shift (Δv = 59 cm–1) (Fig. 4D) (15, 20). The lowest energy acceptor orbital of the CuII –superoxo intermediate (i.e., the FMO for H-atom abstraction) is its LUMO, which is the antibonding combination of the Cu dx2–y2 and superoxide
Intermediate. The optimized lowest energy structure of the CuIIM–superoxo intermediate has the superoxide ligand bound equatorially in a side-on fashion, forming a square pyramidal geometry around the Cu center with a Met as the long axial ligand (Fig. 4A) (16). This side-on geometry is very similar to the crystal structure of a well characterized side-on CuII–superoxo model complex LCuIIO2 (where L is the trispyrazolylborate ligand). The superoxide nature of this complex was experimentally determined from its vO–O frequency (1,043 cm–1) and 18O isotope shift (Δv = 59 cm–1) (Fig. 4D) (15, 20). The lowest energy acceptor orbital of the CuII –superoxo intermediate (i.e., the FMO for H-atom abstraction) is its LUMO, which is the antibonding combination of the Cu dx2–y2 and superoxide 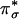 orbitals (Fig. 4B) (16). The Cu dx2–y2 and superoxide
orbitals (Fig. 4B) (16). The Cu dx2–y2 and superoxide 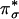 orbitals are in the CuO2 plane and have a large σ-type orbital overlap, resulting in a highly covalent singlet ground state with a large O2– coefficient (64%, Fig. 4B). This highly covalent CuII–superoxo interaction contributes to the formation of this complex as the 1-e– reduction of O2 is energetically less favorable than the 2-e– process (see below).
orbitals are in the CuO2 plane and have a large σ-type orbital overlap, resulting in a highly covalent singlet ground state with a large O2– coefficient (64%, Fig. 4B). This highly covalent CuII–superoxo interaction contributes to the formation of this complex as the 1-e– reduction of O2 is energetically less favorable than the 2-e– process (see below).
Fig. 4.

Electronic structure and spectroscopy of the 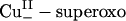 species. (A) Geometry-optimized structure of
species. (A) Geometry-optimized structure of  . (B) Acceptor FMO (LUMO) of
. (B) Acceptor FMO (LUMO) of 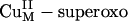 . (C) Schematic diagram of the interaction between the CuII dx2–y2 and superoxide
. (C) Schematic diagram of the interaction between the CuII dx2–y2 and superoxide  orbitals. The arrows and their widths indicate the expected CT transitions (singlets) and their relative intensities. (D) rR spectra in the vO–O region. (E) SQUID magnetic susceptibility measured effective magnetic moment μeff. B.M., Bohr magneton. Lines are the simulated curves assuming different singlet/triplet energy splittings, S/T = ES = 1 – ES = 0. (F) Absorption spectra of the CuII–superoxo model complex. (Left) UV/visible CuII d–d transitions assigned. (Right) Near-infrared CT. Vibrational overtones of mulling agent are labeled by asterisks (15).
orbitals. The arrows and their widths indicate the expected CT transitions (singlets) and their relative intensities. (D) rR spectra in the vO–O region. (E) SQUID magnetic susceptibility measured effective magnetic moment μeff. B.M., Bohr magneton. Lines are the simulated curves assuming different singlet/triplet energy splittings, S/T = ES = 1 – ES = 0. (F) Absorption spectra of the CuII–superoxo model complex. (Left) UV/visible CuII d–d transitions assigned. (Right) Near-infrared CT. Vibrational overtones of mulling agent are labeled by asterisks (15).
The CuII center (S = 1/2) and the superoxide ligand (S = 1/2) can, in principle, have either a singlet or a triplet ground state for the CuIIM–superoxo intermediate. The diamagnetic singlet ground state of the CuMM–superoxo intermediate was confirmed experimentally by SQUID (superconducting quantum interfering device) magnetic susceptibility measurements for the CuII–superoxo model complex (Fig. 4E), which showed that the effective magnetic moment (μeff) is close to zero at low temperature, as compared with μeff ≈ 2.83 Bohr magnetons for a triplet state (15). Additionally, the μeff deviates from zero at higher temperatures (Fig. 4E) because of the thermal population of an excited triplet state at ≈1,500 cm–1. This triplet state is in fact not related to the singlet ground state, which involves a spin pair in the dx2–y2/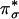 orbital in the Cu–O2 plane. This low-lying triplet derives from the interaction between the CuII dx2–y2 and the superoxide
orbital in the Cu–O2 plane. This low-lying triplet derives from the interaction between the CuII dx2–y2 and the superoxide  orbitals (Fig. 4C Upper Right). The corresponding
orbitals (Fig. 4C Upper Right). The corresponding  singlet state 1Γ(
singlet state 1Γ( ) is higher in energy than the triplet state because of the orthogonality of the two interacting orbitals and was observed experimentally as a low energy weak CT transition at ≈4,200 cm–1 in the absorption spectrum of LCuIIO2 (Fig. 4F) (15). The triplet state associated with the ground state singlet involves excitation of an electron from the
) is higher in energy than the triplet state because of the orthogonality of the two interacting orbitals and was observed experimentally as a low energy weak CT transition at ≈4,200 cm–1 in the absorption spectrum of LCuIIO2 (Fig. 4F) (15). The triplet state associated with the ground state singlet involves excitation of an electron from the 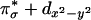 to the
to the 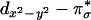 orbital, 3Γ
orbital, 3Γ , and its corresponding singlet 1Γ(
, and its corresponding singlet 1Γ( ) can be reached by means of an allowed CT transition. This CT transition should be intense in the absorption spectrum because of the large overlap between the superoxide
) can be reached by means of an allowed CT transition. This CT transition should be intense in the absorption spectrum because of the large overlap between the superoxide 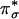 and Cu dx2–y2 orbitals (Fig. 4C). Experimentally, no intense CT transition was observed at energies up to ≈30,000 cm–1, indicating a large splitting of the bonding and antibonding combinations of the Cu dx2–y2 and superoxide
and Cu dx2–y2 orbitals (Fig. 4C). Experimentally, no intense CT transition was observed at energies up to ≈30,000 cm–1, indicating a large splitting of the bonding and antibonding combinations of the Cu dx2–y2 and superoxide 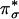 orbitals resulting from their highly covalent interaction (Fig. 4 B and C). This strong interaction leads to a covalently delocalized singlet ground state for the side-on CuII–superoxo species with no spin polarization (i.e., no net antiparallel spin localization on the CuII and the superoxide ligand), arguing against an antiferromagnetic exchange-coupled description (or biradical) previously used for the diamagnetism of CuII–superoxo species (21–23).
orbitals resulting from their highly covalent interaction (Fig. 4 B and C). This strong interaction leads to a covalently delocalized singlet ground state for the side-on CuII–superoxo species with no spin polarization (i.e., no net antiparallel spin localization on the CuII and the superoxide ligand), arguing against an antiferromagnetic exchange-coupled description (or biradical) previously used for the diamagnetism of CuII–superoxo species (21–23).
The electronic structure description of the covalently delocalized singlet ground state of the 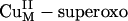 intermediate provides insight into its possible reactivity in H-atom abstraction. The LUMO of the
intermediate provides insight into its possible reactivity in H-atom abstraction. The LUMO of the 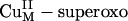 intermediate is low in energy relative to the spin-down LUMO and the σ* orbital of the
intermediate is low in energy relative to the spin-down LUMO and the σ* orbital of the  intermediate (≈4.4 eV lower than the
intermediate (≈4.4 eV lower than the  σ* orbital) and has a large orbital coefficient on the O– moiety (Fig. 4B versus Fig. 3 B and C Therefore, a
σ* orbital) and has a large orbital coefficient on the O– moiety (Fig. 4B versus Fig. 3 B and C Therefore, a 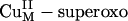 intermediate should be much more effective in H-atom abstraction than the putative
intermediate should be much more effective in H-atom abstraction than the putative 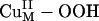 intermediate in PHM chemistry. These FMO differences should lead to differences in thermodynamics and kinetic barriers as discussed below.
intermediate in PHM chemistry. These FMO differences should lead to differences in thermodynamics and kinetic barriers as discussed below.
Correlation of Electronic Structure to Reactivity
The electronic structures derived from model studies coupled with the FMO analysis presented above provided initial insight into the relative H-atom abstraction reactivities of the  intermediates. In this section, we quantitatively compare the energetics and energy barriers of these two CuM/O2 species calculated along the reaction coordinate of H-atom abstraction.
intermediates. In this section, we quantitatively compare the energetics and energy barriers of these two CuM/O2 species calculated along the reaction coordinate of H-atom abstraction.
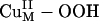 Reaction Coordinate. As predicted from the above electronic structure description, the
Reaction Coordinate. As predicted from the above electronic structure description, the 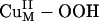 species does not appear to be activated for H-atom abstraction. The calculated potential energy surface along the H-atom transfer coordinate for the
species does not appear to be activated for H-atom abstraction. The calculated potential energy surface along the H-atom transfer coordinate for the  intermediate is shown in blue in Scheme 1, step b, by using a small substrate analogue formylglycine (FmG) (16). This reaction generates the substrate radical FmG·, a H2O product, and a CuM–oxyl species. In contrast to the L3CuIIOOH model complex, the ΔG of this reaction is thermodynamically accessible [≈6–7 kcal/mol (1 cal = 4.18 J)]. This small ΔG is due to the energy difference (≈22 kcal/mol) between the strong O–H bond of the H2O product and the activated C–H bond of the FmG reactant from resonance delocalization of the FmG· radical generated, combined with stabilization by the Met ligand, which binds in an equatorial position in the
intermediate is shown in blue in Scheme 1, step b, by using a small substrate analogue formylglycine (FmG) (16). This reaction generates the substrate radical FmG·, a H2O product, and a CuM–oxyl species. In contrast to the L3CuIIOOH model complex, the ΔG of this reaction is thermodynamically accessible [≈6–7 kcal/mol (1 cal = 4.18 J)]. This small ΔG is due to the energy difference (≈22 kcal/mol) between the strong O–H bond of the H2O product and the activated C–H bond of the FmG reactant from resonance delocalization of the FmG· radical generated, combined with stabilization by the Met ligand, which binds in an equatorial position in the 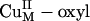 species in contrast to its axial coordination in
species in contrast to its axial coordination in 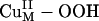 in Fig. 3A (16). However, consistent with the FMO analysis of
in Fig. 3A (16). However, consistent with the FMO analysis of 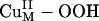 , the potential energy surface along the H-atom transfer reaction coordinate showed a large energy barrier of ≈37 kcal/mol, making this reaction kinetically highly unlikely. Therefore, H-atom abstraction by the
, the potential energy surface along the H-atom transfer reaction coordinate showed a large energy barrier of ≈37 kcal/mol, making this reaction kinetically highly unlikely. Therefore, H-atom abstraction by the  intermediate is an energetically plausible but kinetically unfavorable reaction pathway in PHM (16).
intermediate is an energetically plausible but kinetically unfavorable reaction pathway in PHM (16).
Scheme 1.

Summary of the 2-e– (left) and 1-e– (right) reaction pathways in PHM (16). For clarity, His and Met ligands are omitted in the structures. Only species that are essential to the reactions are indicated on the scheme. Free energies are referenced to the initial reactions, which are set to zero. The proton and H2O ligand in steps v and a and the H2O ligand in step iii are from the solvent. T.S., transition state.
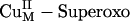 Reaction Coordinate. In contrast to the
Reaction Coordinate. In contrast to the 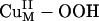 species, the calculated energetics and energy barrier indicated that CuM–superoxo is very reactive in H-atom abstraction, as predicted from the above electronic structure description. The calculated potential energy surface for the CuIIM–superoxo intermediate H-atom abstraction reaction is shown in red in Scheme 1, step ii. This reaction generates the substrate radical FmG· and an asymmetrically side-on bound CuII–hydroperoxo species that can readily convert to the end-on bound
species, the calculated energetics and energy barrier indicated that CuM–superoxo is very reactive in H-atom abstraction, as predicted from the above electronic structure description. The calculated potential energy surface for the CuIIM–superoxo intermediate H-atom abstraction reaction is shown in red in Scheme 1, step ii. This reaction generates the substrate radical FmG· and an asymmetrically side-on bound CuII–hydroperoxo species that can readily convert to the end-on bound 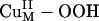 intermediate (Fig. 3A) by binding an H2O molecule (ΔG ≈ –0.3 kcal/mol) (16). The energetics of this H-atom abstraction reaction is almost thermo-neutral, ≈2 kcal/mol. More importantly, because of its highly covalent FMO, the energy barrier along the H-atom transfer coordinate is only ≈14 kcal/mol (Scheme 1, step ii), which is much lower than that for the
intermediate (Fig. 3A) by binding an H2O molecule (ΔG ≈ –0.3 kcal/mol) (16). The energetics of this H-atom abstraction reaction is almost thermo-neutral, ≈2 kcal/mol. More importantly, because of its highly covalent FMO, the energy barrier along the H-atom transfer coordinate is only ≈14 kcal/mol (Scheme 1, step ii), which is much lower than that for the  intermediate (≈37 kcal/mol) at the same active site/substrate distance (Scheme 1, step b). Therefore, the favorable reaction energetics and the low energy barrier indicate that the
intermediate (≈37 kcal/mol) at the same active site/substrate distance (Scheme 1, step b). Therefore, the favorable reaction energetics and the low energy barrier indicate that the 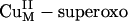 H-atom abstraction reaction is a highly favorable pathway in PHM both thermodynamically and kinetically.
H-atom abstraction reaction is a highly favorable pathway in PHM both thermodynamically and kinetically.
A reasonable reaction pathway for completion of the substrate hydroxylation was determined in ref. 16 and is summarized in Scheme 1 (green steps). After the H-atom abstraction reaction, the 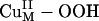 intermediate and the FmG· radical generated can undergo a direct OH group transfer to form the hydroxylated product FmG-OH and a CuIIM-oxyl species, driven by the formation of a strong product C–O bond (Scheme 1, step iv) (16). (An alternative reaction pathway, in which the
intermediate and the FmG· radical generated can undergo a direct OH group transfer to form the hydroxylated product FmG-OH and a CuIIM-oxyl species, driven by the formation of a strong product C–O bond (Scheme 1, step iv) (16). (An alternative reaction pathway, in which the 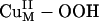 species undergoes reductive O–O bond cleavage by means of ET from CuH coupled to protonation, is ≈20 kcal/mol uphill in ΔG because of the unfavorable 1-e– reduction of the peroxide.) The CuIIM–oxyl species produced can then be reduced to the stable resting CuIIM site by the CuIH site by an intramolecular ET process and protonation from solvent (Scheme 1, step v). This long-range ET-coupled reduction process is downhill in free energy (ΔG ≈ –12 kcal/mol), which is due to the relatively high-energy nature of the CuIIM–oxyl species that provides the necessary driving force to complete the reaction.
species undergoes reductive O–O bond cleavage by means of ET from CuH coupled to protonation, is ≈20 kcal/mol uphill in ΔG because of the unfavorable 1-e– reduction of the peroxide.) The CuIIM–oxyl species produced can then be reduced to the stable resting CuIIM site by the CuIH site by an intramolecular ET process and protonation from solvent (Scheme 1, step v). This long-range ET-coupled reduction process is downhill in free energy (ΔG ≈ –12 kcal/mol), which is due to the relatively high-energy nature of the CuIIM–oxyl species that provides the necessary driving force to complete the reaction.
With an additional H2O ligand at the CuM site, geometry optimization gave an end-on CuII–superoxo species (Scheme 1, step i), which is ≈11 kcal/mol higher in free energy than the side-on  intermediate (16). The LUMO of the end-on CuII–superoxide species is also an antibonding combination of Cu dx2–y2 and superoxide
intermediate (16). The LUMO of the end-on CuII–superoxide species is also an antibonding combination of Cu dx2–y2 and superoxide  orbitals with similar molecular orbital coefficients on the oxygen atoms to those of the side-on CuIIM–superoxo intermediate. Based on its electronic structure description and FMO theory, this end-on superoxide species is predicted to be comparable to or slightly less reactive than the side-on superoxide species, not considering the steric effects of their different binding geometries (16). The structure of a Cu–O2 intermediate in PHM has recently been solved and has a four-coordinate Cu with O2 binding in an end-on mode (24). The nature of the Cu–O2 species in the structure is not known (i.e., CuI–O2, CuII–superoxide, CuII–peroxide, CuII–OOH, etc.), and further spectroscopic studies are needed to define this intermediate. In light of the reaction mechanism presented here for PHM and DβM, it might correspond to the end-on CuII–superoxide species geometry-optimized with an additional H2O ligand (without the H2O, it optimizes to the side-on structure) or the
orbitals with similar molecular orbital coefficients on the oxygen atoms to those of the side-on CuIIM–superoxo intermediate. Based on its electronic structure description and FMO theory, this end-on superoxide species is predicted to be comparable to or slightly less reactive than the side-on superoxide species, not considering the steric effects of their different binding geometries (16). The structure of a Cu–O2 intermediate in PHM has recently been solved and has a four-coordinate Cu with O2 binding in an end-on mode (24). The nature of the Cu–O2 species in the structure is not known (i.e., CuI–O2, CuII–superoxide, CuII–peroxide, CuII–OOH, etc.), and further spectroscopic studies are needed to define this intermediate. In light of the reaction mechanism presented here for PHM and DβM, it might correspond to the end-on CuII–superoxide species geometry-optimized with an additional H2O ligand (without the H2O, it optimizes to the side-on structure) or the  species generated by the H-atom abstraction (Scheme 1, step iii), both of which have a similar end-on Cu– O–O geometry. However, the O–O bond length of the Cu–O2 intermediate in the crystal structure may be too short for a peroxo species (24).
species generated by the H-atom abstraction (Scheme 1, step iii), both of which have a similar end-on Cu– O–O geometry. However, the O–O bond length of the Cu–O2 intermediate in the crystal structure may be too short for a peroxo species (24).
Structure/Function Correlations: Noncoupled Versus Exchange-Coupled Binuclear Cu Sites
Electronic structure descriptions combined with FMO analyses and reaction coordinate calculations indicate that the 1-e– reduced CuM–superoxo species is likely the reactive CuII/O2 species in H-atom abstraction by PHM, as compared with the 2-e– reduced 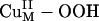 species. This CuM–superoxo mechanism is consistent with the kinetic studies by Evans et al. (17). The reaction pathway summarized in Scheme 1 (steps in red and green), from the formation of the CuM–superoxo intermediate to the H-atom abstraction reaction, and the completion of the substrate hydroxylation also provide insight into the mechanism of the inter-Cu intramolecular ET process involved in the PHM/DβM reaction and the role of the nonmagnetically coupled nature of their active sites, as compared with the coupled binuclear Cu proteins.
species. This CuM–superoxo mechanism is consistent with the kinetic studies by Evans et al. (17). The reaction pathway summarized in Scheme 1 (steps in red and green), from the formation of the CuM–superoxo intermediate to the H-atom abstraction reaction, and the completion of the substrate hydroxylation also provide insight into the mechanism of the inter-Cu intramolecular ET process involved in the PHM/DβM reaction and the role of the nonmagnetically coupled nature of their active sites, as compared with the coupled binuclear Cu proteins.
The direct bridging of the two Cu centers in the binuclear Cu proteins Hc, Tyr, and CO provides a mechanism for O2 reduction by two electrons to the side-on  species (3) and results in the strong Cu–Cu antiferromagnetic exchange coupling (–2 J ≥ 1,200 cm–1 (25). This side-on
species (3) and results in the strong Cu–Cu antiferromagnetic exchange coupling (–2 J ≥ 1,200 cm–1 (25). This side-on  peroxo species is activated for electrophilic attack at the phenyl ring of substrates (Fig. 1C). In contrast, there is no observable magnetic interaction (i.e., very small J) between the two Cu centers in the non-coupled binuclear Cu protein PHM and DβM (26), because of the large Cu–Cu distance (≈11 Å in PHM) (6, 8). Thus, a long-range intramolecular ET process is required at some stage in the mechanism for the enzymatic reactions in PHM and DβM. Marcus theory governs the ET rate constant kET (27):
peroxo species is activated for electrophilic attack at the phenyl ring of substrates (Fig. 1C). In contrast, there is no observable magnetic interaction (i.e., very small J) between the two Cu centers in the non-coupled binuclear Cu protein PHM and DβM (26), because of the large Cu–Cu distance (≈11 Å in PHM) (6, 8). Thus, a long-range intramolecular ET process is required at some stage in the mechanism for the enzymatic reactions in PHM and DβM. Marcus theory governs the ET rate constant kET (27):
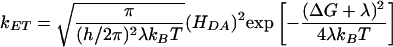 |
where HDA is the donor/acceptor electronic coupling matrix element, ΔG is the driving force, and λ is the reorganization energy, which includes the active site geometry change (λinner) and the reorientation of the solvent dipoles (λouter, λ = λinner + λouter) associated with redox. The electronic coupling matrix element HDA is related to the exchange constant J:
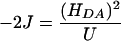 |
where U is the metal–metal CT energy (25, 28). Because J is small for the non-coupled CuM and CuH sites, the (HDA)2 between the two Cu centers also must be small. Significant geometry changes between the reduced and oxidized forms of the CuM and CuH sites have been observed experimentally (10–12, 29) and are also found in calculated structures (13, 16), which suggest a large reorganization energy (λinner) is also associated with their redox reactions (Fig. 2, A, B, C, and D, respectively). [Note that the crystal structures of PHM (6, 8) did not resolve significant differences between the oxidized and the reduced proteins, whereas EXAFS results showed significant geometry changes upon redox (10–12). EXAFS studies are more accurate in determining the metal–ligand bond lengths and differentiating Cu oxidation states.] Therefore, to have a significant kET, there must be a large driving force ΔG for the ET process from CuH to CuM. The reaction mechanism in Scheme 1 indicates that PHM could achieve this driving force through a direct OH transfer to the substrate FmG· radical after the H-atom abstraction step (Scheme 1, step iv). The reduction and protonation of the high-energy CuM–oxyl species formed could provide the necessary driving force for the intramolecular ET from the CuI site (Scheme 1, step v). This thermodynamically driven ET mechanism also suggests that superoxide channeling (7) is not a necessary event for the ET process; furthermore, neither is the substrate-mediated ET mechanism (8), because no change was observed in the EPR spectrum of resting PHM upon substrate binding, which indicates that the J value between the two Cu centers is still very small when the substrate is present (13).
The noncoupled nature of the PHM/DβM active sites is strongly correlated with their chemistry. If two Cu centers are strongly exchange-coupled, the O2 reaction with the reduced protein would lead to fast ET from both Cu sites to O2, generating a 2-e– reduced binuclear- or mononuclear-CuII–peroxide level species (O –2), depending on the distance between the two Cu atoms (Table 1, rows A–E). These CuII–peroxo/hydroperoxo complexes do not appear to be reactive in H-atom abstraction (1, 5, 30–34), nor is the mononuclear Cu–OOH species considered above (13, 16). Although 4-e– reduction of O2 by two Cu atoms could lead to a 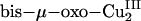 species (Table 1, row G), which is very reactive in H-atom abstraction, the existence of the CuIII oxidation state in a biological environment is not known and likely not accessible because of the inability of biological ligands (His, etc.) to stabilize the CuIII oxidation state. The inaccessible oxidation state is also the case for the mononuclear CuIII–peroxide species (Table 1, row F), which was recently synthesized with an exceptionally strong electron-donating ligand (35). Therefore, to form the 1-e– reduced superoxide level species CuII–superoxo, which from the above model would be the reactive species in H-atom abstraction and not proceed further to a thermodynamically favored 2-e– reduced peroxo species [at pH = 7, E°(O2/H2O2) = 0.28 V, E°(O2/O2-) = –0.33 V versus normal hydrogen electrode] (36), the two Cu sites have to be nonelectronically coupled. This noncoupled nature of the binuclear Cu active site provides a strategy for PHM and DβM to form a reactive CuII–superoxo species at one Cu site (CuM) for the required H-atom abstraction reactivity while maintaining the ability to provide an additional electron from another Cu site (CuH) to complete the reaction, the intramolecular ET being switched on by a high driving force at the appropriate step in the enzymatic reaction cycle.
species (Table 1, row G), which is very reactive in H-atom abstraction, the existence of the CuIII oxidation state in a biological environment is not known and likely not accessible because of the inability of biological ligands (His, etc.) to stabilize the CuIII oxidation state. The inaccessible oxidation state is also the case for the mononuclear CuIII–peroxide species (Table 1, row F), which was recently synthesized with an exceptionally strong electron-donating ligand (35). Therefore, to form the 1-e– reduced superoxide level species CuII–superoxo, which from the above model would be the reactive species in H-atom abstraction and not proceed further to a thermodynamically favored 2-e– reduced peroxo species [at pH = 7, E°(O2/H2O2) = 0.28 V, E°(O2/O2-) = –0.33 V versus normal hydrogen electrode] (36), the two Cu sites have to be nonelectronically coupled. This noncoupled nature of the binuclear Cu active site provides a strategy for PHM and DβM to form a reactive CuII–superoxo species at one Cu site (CuM) for the required H-atom abstraction reactivity while maintaining the ability to provide an additional electron from another Cu site (CuH) to complete the reaction, the intramolecular ET being switched on by a high driving force at the appropriate step in the enzymatic reaction cycle.
In summary, the mononuclear reactive CuII/O2 species in the noncoupled binuclear Cu proteins gives a different reaction mechanism in O2 activation and substrate hydroxylation from that of the coupled binuclear Cu proteins. The extent of magnetic exchange coupling between the two Cu centers in these two classes of binuclear Cu proteins plays an important role in determining the formation of the reactive CuIIn–O2 species and thus their resulting reactivities (H-atom abstraction versus electrophilic attack).
Acknowledgments
We thank Drs. Betty Eipper and Joseph Bell for collaboration. The research was supported by National Institutes of Health Grant DK-31450 (to E.I.S.). P.C. was supported by a Gerhard Casper Stanford Graduate Fellowship and a Franklin Veatch Memorial Fellowship.
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Solomon, E. I., Chen, P., Metz, M., Lee, S.-K. & Palmer, A. E. (2001) Angew. Chem. Int. Ed. 40, 4570–4590. [DOI] [PubMed] [Google Scholar]
- 2.Solomon, E. I., Sundaram, U. M. & Machonkin, T. E. (1996) Chem. Rev. 96, 2563–2606. [DOI] [PubMed] [Google Scholar]
- 3.Metz, M. & Solomon, E. I. (2001) J. Am. Chem. Soc. 123, 4938–4950. [DOI] [PubMed] [Google Scholar]
- 4.Klinman, J. P. (1996) Chem. Rev. 96, 2541–2562. [DOI] [PubMed] [Google Scholar]
- 5.Liang, H.-C., Dahan, M. & Karlin, K. D. (1999) Curr. Opin. Chem. Biol. 3, 168–175. [DOI] [PubMed] [Google Scholar]
- 6.Prigge, S. T., Kolhekar, A. S., Eipper, B. A., Mains, R. E. & Amzel, L. M. (1997) Science 278, 1300–1305. [DOI] [PubMed] [Google Scholar]
- 7.Jaron, S. & Blackburn, N. J. (1999) Biochemistry 38, 15086–15096. [DOI] [PubMed] [Google Scholar]
- 8.Prigge, S. T., Kolhekar, A. S., Eipper, B. A., Mains, R. E. & Amzel, L. M. (1999) Nat. Struct. Biol. 6, 976–983. [DOI] [PubMed] [Google Scholar]
- 9.Bell, J., Meskini, R. E., D'Amato, D., Mains, R. E. & Eipper, B. A. (2003) Biochemistry 42, 7133–7142. [DOI] [PubMed] [Google Scholar]
- 10.Blackburn, N. J., Rhames, F. C., Ralle, M. & Jaron, S. (2000) J. Biol. Inorg. Chem. 5, 341–353. [DOI] [PubMed] [Google Scholar]
- 11.Boswell, J. S., Reedy, B. J., Kulathila, R., Merkler, D. J. & Blackburn, N. J. (1996) Biochemistry 35, 12241–12250. [DOI] [PubMed] [Google Scholar]
- 12.Blackburn, N. J., Hasnain, S. S., Pettingill, T. M. & Strange, R. W. (1991) J. Biol. Chem. 266, 23120–23127. [PubMed] [Google Scholar]
- 13.Chen, P., Bell, J., Eipper, B. A. & Solomon, E. I. (2004) Biochemistry 43, 5735–5747. [DOI] [PubMed] [Google Scholar]
- 14.Chen, P., Fujisawa, K. & Solomon, E. I. (2000) J. Am. Chem. Soc. 122, 10177–10193. [Google Scholar]
- 15.Chen, P., Root, D. E., Campochiaro, C., Fujisawa, K. & Solomon, E. I. (2003) J. Am. Chem. Soc. 125, 466–474. [DOI] [PubMed] [Google Scholar]
- 16.Chen, P. & Solomon, E. I. (2004) J. Am. Chem. Soc. 126, 4991–5000. [DOI] [PubMed] [Google Scholar]
- 17.Evans, J. P., Ahn, K. & Klinman, J. P. (2003) J. Biol. Chem. 278, 49691–49698. [DOI] [PubMed] [Google Scholar]
- 18.Fleming, I. (1976) Frontier Orbitals and Organic Chemical Reactions (Wiley, New York).
- 19.Root, D. E., Mahroof-Tahir, M., Karlin, K. D. & Solomon, E. I. (1998) Inorg. Chem. 37, 4838–4848. [DOI] [PubMed] [Google Scholar]
- 20.Fujisawa, K., Tanaka, M., Moro-oka, Y. & Kitajima, N. (1994) J. Am. Chem. Soc. 116, 12079–12080. [Google Scholar]
- 21.Nappa, M., Valentine, J. S., Miksztal, A. R., Schugar, H. J. & Isied, S. S. (1979) J. Am. Chem. Soc. 101, 7744–7746. [Google Scholar]
- 22.Chaudhuri, P., Hess, M., Weyhermuller, T. & Wieghardt, K. (1999) Angew. Chem. Int. Ed. 38, 1095–1098. [DOI] [PubMed] [Google Scholar]
- 23.Cramer, C. J., Tolman, W. B., Theopold, K. H. & Rheingold, A. L. (2003) Proc. Natl. Acad. Sci. USA 100, 3635–3640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Prigge, S. T., Eipper, B. A., Mains, R. E. & Amzel, L. M. (2004) Science 304, 864–867. [DOI] [PubMed] [Google Scholar]
- 25.Tuczek, F. & Solomon, E. I. (2001) Coord. Chem. Rev. 219–221, 1075–1112. [Google Scholar]
- 26.Ljones, T. & Skotland, T. (1984) in Copper Proteins and Copper Enzymes, ed. Lontie, R. (CRC Press, Boca Raton, FL), pp. 131–157.
- 27.Marcus, R. A. & Sutin, N. (1985) Biochim. Biophys. Acta 811, 265–322. [Google Scholar]
- 28.Brunold, T. C., Gamelin, D. R. & Solomon, E. I. (2000) J. Am. Chem. Soc. 122, 8511–8523. [Google Scholar]
- 29.Scott, R. A., Sullivan, R. J., DeWolf, W. E., Jr., Dolle, R. E. & Kruse, L. I. (1988) Biochemistry 27, 5411–5417. [DOI] [PubMed] [Google Scholar]
- 30.Kitajima, N. & Moro-oka, Y. (1994) Chem. Rev. 94, 737–757. [Google Scholar]
- 31.Decker, H., Dillinger, R. & Tuczek, F. (2000) Angew. Chem. Int. Ed. 39, 1591–1595. [DOI] [PubMed] [Google Scholar]
- 32.Schindler, S. (2000) Eur. J. Inorg. Chem., 2311–2326.
- 33.Mahadevan, V., Gebbink, R. K. & Stack, T. D. (2000) Curr. Opin. Chem. Biol. 4, 228–234. [DOI] [PubMed] [Google Scholar]
- 34.Que, L., Jr., & Tolman, W. B. (2002) Angew. Chem. Int. Ed. 41, 1114–1137. [DOI] [PubMed] [Google Scholar]
- 35.Aboelella, N., Lewis, E. A., Reynolds, A. M., Brennessei, W. W., Cramer, C. J. & Tolman, W. B. (2002) J. Am. Chem. Soc. 124, 10660–10661. [DOI] [PubMed] [Google Scholar]
- 36.Sawyer, D. T. (1991) Oxygen Chemistry (Oxford Univ. Press, New York).
- 37.Pate, J. E., Cruse, R. W., Karlin, K. D. & Solomon, E. I. (1987) J. Am. Chem. Soc. 109, 2624–2630. [Google Scholar]
- 38.Baldwin, M. J., Ross, P. K., Pate, J. E., Tyeklár, Z., Karlin, K. D. & Solomon, E. I. (1991) J. Am. Chem. Soc. 113, 8671–8679. [Google Scholar]
- 39.Baldwin, M. J., Root, D. E., Pate, J. E., Fujisawa, K., Kitajima, N. & Solomon, E. I. (1992) J. Am. Chem. Soc. 114, 10421–10431. [Google Scholar]