

Abstract
Defining processes that are synthetic lethal with p53 mutations in cancer cells may reveal possible therapeutic strategies. In this study, we report the development of a signal-oriented computational framework for cancer pathway discovery in this context. We applied our bipartite-graph-based functional module discovery algorithm to identify transcriptomic modules abnormally expressed in multiple tumors, such that the genes in a module were likely regulated by a common, perturbed signal. For each transcriptomic module, we applied our weighted k-path merge algorithm to search for a set of somatic genome alterations (SGA) that likely perturbed the signal, i.e., the candidate members of the pathway that regulate the transcriptomic module. computational evaluations indicated that our methods identified pathways perturbed by SGA. In particular, our analyses revealed that SGA affecting TP53, PTK2, YWHAZ and MED1 perturbed a set of signals that promote cell proliferation, anchor-free colony formation and epithelial-mesenchymal transition (EMT). These proteins formed a signaling complex that mediate these oncogenic processes in a coordinated fashion. Disruption of this signaling complex by knocking down PTK2, YWHAZ, or MED1 attenuated and reversed oncogenic phenotypes caused by mutant p53 in a synthetic lethal manner. This signal-oriented framework for searching pathways and therapeutic targets is applicable to all cancer types, thus potentially impacting precision medicine in cancer.
Introduction
Cancer is driven by aberrations in cellular signaling pathways, commonly caused by SGAs, such as somatic mutations and copy number alterations, and epigenetic alterations that affect signaling proteins (1,2). Large-scale cancer genomic studies, such as The Cancer Genome Atlas (TCGA) provide an unprecedented opportunity to comprehensively investigate cancer pathways perturbed by SGAs.
Discovering cancer drivers and pathways is an active research area. Different algorithms have been designed to search for cancer driver genes using cancer genomic data availed by large-scale projects such as The Cancer Genome Atlas (TCGA) and the International Cancer Genome Consortium (ICGC) (3–5) (see Djosta Nono et al (6) for a comprehensive review). However, a majority of the current methods concentrates on finding genes that are mutated above a baseline frequency according to a certain statistical model, without information regarding the functional impacts of mutation events, such as the biological processes perturbed by the mutation events. As such, it is difficult to organize candidate drivers observed in different tumors into pathways. To address this challenge, researchers have designed a family of algorithms that searches for a set of mutated genes that exhibit mutually exclusive patterns in tumors and identifies them as members of a candidate pathway, based on the observation that SGA events affecting a pathway seldom co-occur in a tumor (7–12). This framework has limitations in that mutual exclusivity is neither a necessary nor a sufficient condition for genes involved in a pathway. That is, not all genes in a pathway show mutual exclusivity, and not all mutually exclusive genes are in a common pathway. Other researchers resort to the knowledge of biological networks, e.g., protein-protein interaction (PPI) networks, to search for genes that are closely located within a biological network and are commonly perturbed by SGAs (13–16). The limitation of this approach is that PPI data can be noisy and that member proteins of a pathway do not always physically interact. Importantly, the aforementioned driver-identification or pathway-discovery algorithms do not take into account the functional impacts of SGA events, which is critical in order to determine if an SGA-perturbed gene is a driver and whether a set of SGAs perturb a common cellular signal.
In this study, we developed a novel signal-oriented computational framework, which utilizes the intrinsic relation between the perturbation of a signaling pathway and the expression changes of down-stream genes regulated by the pathway, to search for driver SGAs and reconstruct signaling pathways. To this end, we first applied a bipartite-graph-based functional module discovery (BFMD) algorithm to identify transcriptomic modules (17,18) as signatures of cellular signals that are perturbed in tumors. We then applied a weighted k-path merge (WKM) algorithm to identify a set of SGAs that perturb a common signal (12,19) with respect to a transcriptomic module as a means to discover the members of a cancer pathway.
Applying our framework to breast cancer tumors from TCGA, we defined the most commonly perturbed transcriptomic modules and investigated the candidate pathways driving their aberrant expression. We discovered a p53-centered signaling complex involving TP53, PTK2, YWHAZ, and MED1, which drives multiple oncogenic processes in breast cancer and is associated with worse clinical outcome. We show that disrupting the complex, by knocking down PTK2, YWHAZ and MED1 proteins, specifically attenuates or reverses the oncogenic phenotypes induced by mutant p53. Given that p53 is the most frequently mutated gene (>40% of all types of tumors) and is associated with worse outcome (1,20), the effective targeting of p53-mutation-mediated signals could have a significant impact on a large number of cancer patients.
The signal-oriented framework is a general approach that can be used to find other cancer pathways and signaling complexes, which could potentially have a broad impact on precision medicine in cancer.
Materials and Methods
Identification of response modules
An overview of our study is shown in the supplementary Fig S1a – S1f. In each tumor, differentially expressed genes annotated with closely related GO terms in the Biological Process domain were grouped into non-disjoint sets, with each set summarized by a GO term using a method previously reported (18,21) (Algorithm S1). Genes summarized by a common GO term and tumors were organized in a tumor-vs-gene bipartite graph (Fig 1a). Search for a densely connected sub-graph was performed using a previously reported algorithm (18) (Algorithm S2–1, S2–2). Since the differentially expressed genes in such a transcriptomic module were repeatedly co-differentially expressed in multiple tumors, we hypothesized that they were likely regulated by a common cellular signaling pathway that was perturbed in these tumors, and thus we refer to them as a response module.
Figure 1. Identification of response modules predictive of clinical outcomes.
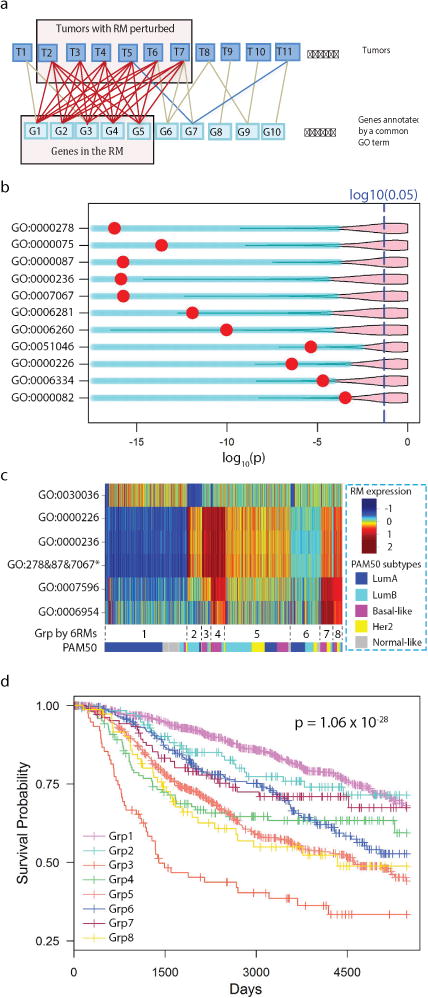
a) Diagram illustrating the search for a response module by finding a densely connected bipartite graph. Tumors and DEGs annotated by a common GO term are represented as the nodes in the graph. An edge indicates that a gene is differentially expressed in a tumor. A response module consists of a set of co-differentially expressed genes in multiple tumors satisfying a specified connectivity. b) A volcano plot illustrating response modules that are predictive of patient outcomes. A row corresponds to a response module, and a red dot indicates the Kaplan-Meier p-value of METABRIC patients dichotomized according to the state of the response module. The pink area indicates the upper 95% of p-values that can be obtained when dividing patients conditioning on randomly drawn gene sets of the same size as the corresponding response module. c) A heat map illustrating the averaged within-module expression values of response modules (response modules annotated with GO:0000278, GO:0000087, and GO:0007067 are merged and indicated by an asterisk). The pseudo-color represents the relative expression value of a response module in tumors by the number of standard deviations from the mean. The tumors were clustered into subgroups with group IDs indicated below the heat map; the proportion of tumors belonging to PAM50 subtypes within a cluster are shown as colors. d) Kaplan-Meier survival curves of the 8 patient groups identified by clustering analysis.
Searching for members of a candidate pathway
To search for the SGAs in tumors perturbing a pathway that regulates the expression of a response module, we designed the WKM algorithm (Supplementary Fig S1e, Algorithm S3–1, S3–2, S4) with the following steps: 1) Apply Fisher’s exact test to assess the strength of statistical association between each SGA and the expression state of the response module. 2) Instantiate a human protein-protein interaction (PPI) network in which nodes represent proteins and edges reflect physical interactions between them. In the network, the proteins that were perturbed by SGAs in TCGA data are assigned a positive weight (the negative log of the p-value from Step 1), and the unaffected proteins are assigned a weight of 0. Thus, the weight of a protein reflects the amount of information a gene/protein carries with respect to the expression state of the response module (the smaller the p-value, the bigger the weight). 3) Iteratively search for k-paths (22) consisting of a set of k proteins (including SGA-affected proteins and the nodes connecting them) with maximal total weight. 4) Iteratively merge intersecting k-paths (by order of descending weight) to construct a subnetwork, such that the total amount of information carried by the sub-network with respect to the response module is maximal. After merging each k-path into the sub-network, we reassessed the strength of the collective association of the SGAs in the sub-network with respect to the response module, and we stopped the procedure when subsequent merges resulted in a less than 10% increase of the negative log p-value. Detailed information for computational algorithms and cell biology experiments are provided in the Supplementary Method section.
Cell experimental procedures
Breast cancer cell lines were obtained from the Integrative Cancer Biology Program (ICBP) 45 breast cancer cell line kit (ICBP45) of the National Cancer Institute. The cell lines were obtained by the ICBP program in 2010 and were thawed two weeks before experiments (2013–2014). There were less than 10 passages between thawing of the cells, and the experiments described in this study. In general, mycoplasma testing is performed periodically on all cell lines used in the laboratory (approx every 3 months), but in this case this was not necessary since all lines were passaged less then 10 times since obtaining from NCI-ATCC ICBP program. Detailed experimental procedures for cell biology experiments are provided in the Supplementary Methods section.
Results
Transcriptomic signatures reflect the cellular states of tumors
In order to identify aberrant pathways as potential therapeutic targets, we first searched for transcriptomic modules that reflect aberrations in cellular signaling, i.e., expression signatures of aberrant pathways. Our approach is based on the assumption that if a module of genes is involved in a common biological process and exhibits coordinated changes in multiple tumors, this transcriptomic module is likely regulated by a common signaling pathway (17,18,23,24) that is aberrant in these tumors. We have developed the BFMD algorithm (see Methods and supplementary Fig S1a, S1b, Algorithm S2–1, S2–2) to discover such transcriptomic modules.
We obtained the genomic (somatic mutations and copy number alterations) and transcriptomic data of 533 breast cancer tumors in TCGA (25). For each tumor, we first identified differentially expressed genes by contrasting the expression value of a gene against the distribution of the control breast samples profiled by TCGA (see Methods). We then grouped differentially expressed genes into subsets, with each set consisting of genes involved in a common biological process summarized by a Gene Ontology (GO) term (26), using the methods developed in our previous studies (18,21,27) (see Supplementary Methods). To search for a module of genes that was co-differentially expressed in multiple tumors, we organized the genes annotated by a common GO term and all tumors as a bipartite graph (Fig 1b) and searched for a dense subgraph (18) enclosing a set of tumors and a module of genes, i.e., a response transcriptomic module. To avoid finding trivial modules that contained few genes or were differentially expressed in few tumors, we discarded the subgraphs that enclosed less than 10 genes or less than 30 tumors (< 5% of tumors). Since each response module is annotated with a unique GO term, we used the GO term to represent the response module.
We identified 88 putative response modules (Table S1) among the TCGA breast cancers. Many response modules contained genes that are involved in biological processes intrinsic to cancer cells (28), such as cell cycle checkpoint (GO:000075), cell migration (GO:0016477), and programmed cell death (GO:0012501), whereas other response modules reflected biological processes related to the changed environment of tumors, such as wound healing (GO:0042060) and inflammatory response (GO:0006954).
Signal-oriented response modules are informative features for cancer subtyping and clinical outcome prediction
The expression state of these response modules provided a more concise representation of the state of signaling pathways in a tumor than did individual genes (17,29). To investigate whether this pathway-oriented representation is informative of patient outcome, we examined the expression state of these 88 putative response modules in another large breast cancer cohort, METABRIC (30), which had been followed for a longer period than the TCGA cohort and was thus more suitable for survival analysis. We trained a classifier for each of the 88 response modules to detect the expression statuses of these modules in METABRIC samples, and clustering analysis indicates that the expression statuses of the modules exhibit similar patterns among the two cohorts (Fig S2a – S2h). There is no significance in the distribution of patient age and race across the clusters.
We then set out to test the hypothesis that, because they reflect the state of cellular signals, the expression states of response modules can be used as informative features for predicting patient survival. Conditioning on the expression state of each response module, we iteratively divided the METABRIC cohort into two groups and conducted a survival analysis to assess whether each modular feature was informative of outcome. After correcting for potential false discoveries (31) due to multiple testing and random discovery of informative modules, we identified 11 response modules that are significantly informative of patient outcome (Fig 1b, Fig S3).
Since some of the remaining 88 response modules, though not predictive when evaluated alone, could have enhanced predictive power when analyzed in combination with others, we examined all combinations of 4 (from the full set of 88 response modules) as features and used these to build decision trees (Fig S4a, S4b, supplementary Methods) to divide the patients into subgroups, followed by a survival analysis. We retained the top 10 combinations that yielded significant survival differences, which were derived from 8 response modules (Table S2). Interestingly, this procedure included response modules annotated as inflammation response (GO:0006954) and blood coagulation (GO:0007596, which is a subordinate concepts of the biological process wound healing GO:0042060), both of which were deemed non-informative when evaluated alone. These 8 response modules can be organized into 3 main axes important to cancer (17,28,32): cell proliferation, cytoskeleton organization/cellular mobility, and inflammation/wound healing. Among the 8 modules, 3 are related to different stages of cell cycle and their states are highly correlated; therefore we merged them into a combined module, which results in 6 informative response modules.
Using the above 6 response modules as features, we performed consensus clustering analysis (33) to search for breast cancer subtypes. Unlike the 4 breast cancer subtypes reported by TCGA, our analysis divided breast cancer into 8 new subgroups (Fig 1c, 1d, S5a, S5b) that exhibited significant differences in survival. The results indicate that the response modules provide a signal-oriented perspective for identifying novel subtypes of tumors. Compared with conventional breast cancer subtyping based on individual genes, our approach not only identified response modules highly predictive of patient outcome, but it also enabled us to search for a candidate pathway underlying the aberrant expression of each response module as a potential therapeutic target.
Combining genomic alteration data and response modules enables pathway discovery
Availability of TCGA genomic data enabled us to search for SGAs that likely perturbed signaling pathways regulating each response module. We hypothesize that if a set of SGAs is consistently associated with the aberrant expression of a response module, and if the proteins affected by these SGA events are closely located within a PPI network, then the SGAs likely perturb a signaling pathway that regulates the response module of interest.
We applied the WKM algorithm to search for the perturbation module regulating each response module (n=88), the results of which are available at a supplementary website (http://breastcancersignalingsignatures.dbmi.pitt.edu/). The relationship between genes in response and perturbation modules are biologically sensible. For example, for a response module containing genes annotated by GO:0016477 (cell migration), we examined the genes in its corresponding perturbation module by performing an extensive literature search, and we found that 30 of the 40 genes in the perturbation module are related to cell migration or metastasis (Table S3), including AKT1, CREB1, GAB2, and CCT2. In another example, for a response module containing 23 genes annotated by GO:0012501 (programmed cell death), we found that 35 of the 46 proteins (genes) in its corresponding perturbation module are reported to regulate cell death, apoptosis, or autophagy (Table S4).
In depth analysis of a pathway influencing breast cancer outcome
To validate that our approach is capable of identifying biologically meaningful pathways, we further investigated the candidate pathway that drove the aberrant expression of the mitosis response module (GO:0007067), which was the pathway most highly predictive of patient survival (P < 1.87−16). We performed detailed computational analyses of the relationships between the members of this candidate pathway and further experimentally tested the hypothesis that members of this candidate pathway (revealed by our methods) truly regulate common signals.
Fig 2a shows the members of the perturbation module and their genomic alteration status in TCGA breast cancer tumors. We organized tumors along the x-axis and SGAs along the y-axis, such that a block indicates a subset of tumors in which a gene was perturbed by certain types (color-coded) of SGAs. In the figure, the vertical line between Groups D and E dichotomizes tumors into those that aberrantly expressed the response module (to the left) and those that did not (to the right). Fig 2a shows that mutations of TP53 and copy number amplifications of YWHAZ, FADD, PTK2, and MED1 were significantly associated with the expression of the mitosis response module. In 134 tumors (Groups B & C), SGAs affecting any of these genes (i.e., mutation of TP53 or amplification of any of YWHAZ, FADD, PTK2 and MED1) were sufficient to drive the aberrant expression of the mitosis response module, and these SGA events exhibited a mutually exclusive pattern in these tumors, which strongly suggests that they perturb a common signal (7,9). On the other hand, 76 tumors (Group A) harbored a TP53 mutation in combination with copy number amplification of one or more of YWHAZ, FADD, PTK2, and MED1, and the frequency of their being co-perturbed was significantly greater than by chance (Table 1). The high prevalence of combinatorial perturbations indicates that genetic interactions (34) among these genes provides an oncogenic advantage in these tumors. In the PPI network, p53 is a hub directly interacting with PTK2, YWHAZ, and MED1 proteins (Fig 2b) (35,36), providing a molecular basis for the genetic interactions at the protein level. Since the PPI interactions among these proteins provide a strong indication that they may function together, we concentrate on studying the relationships among these four proteins in the following subsections.
Figure 2. TPYM encodes a common set of signals.
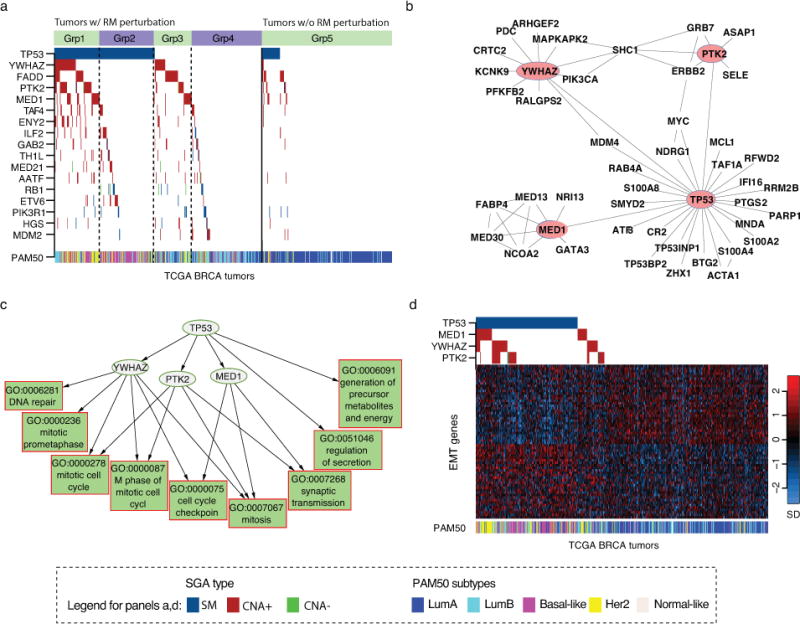
a) The distribution patterns of SGAs significantly associated with the expression state of the mitosis response module (GO:0007067). Tumors are on the x-axis, and SGA-perturbed genes are along the y-axis. A color-coded box indicates the tumors in which a gene is altered. The solid line separates the tumors with the response module aberrantly expressed (left) from those with normal expression (right). Dotted lines separate tumors into subsets with different combinations of TPYM alterations. b) PPI interaction between TPYM and their close neighbors. c) The relationships between TPYM alterations and their target response modules are organized as a directed acyclic graph. d) The expression status of EMT signature genes and the SGA status of TPYM. TCGA breast cancers are arranged along the x-axis, and EMT signature genes are arranged along the y-axis. The expression value of a gene is normalized to have a mean of 0 and unit standard deviation, and the pseudo-color is mapped to the range covering 90% of the tumors. PAM50 classifications of the tumors are shown as a color bar below.
Table 1.
Statistical significance of joint SGA events
| Gene | TP53 | YWHAZ | PTK2 | FADD | MED1 |
|---|---|---|---|---|---|
| TP53 | — | < 10−5 | < 10−5 | 0.51068 | 9×10−5 |
| YWHAZ | — | < 10−5 | 0.17147 | 2.22×10−4 | |
| PTK2 | — | 0.15215 | 0.02283 | ||
| FADD | — | 0.02298 | |||
| MED1 | — |
Results from the cBioPortal are calculated using all provisional breast cancer samples from TCGA using cBioPortal web (accessed on May 7th, 2014).
SGAs affecting TP53, PTK2, YWHAZ, and MED1 perturb a common set of signals
Based on the discovered genetic interactions and known physical interactions among TP53, PTK2, YWHAZ, and MED1 (hereafter abbreviated as TPYM), we hypothesized that their corresponding proteins form a signaling complex that cooperatively encodes a common set of signals. To test this hypothesis, we identified all the response modules regulated by the TPYM SGAs, such that each SGA was associated with a collection of response modules reflecting the distinct cellular signals perturbed by the SGA. We then organized the SGAs and the response modules regulated by them in a directed acyclic graph to reveal the nested-effects relationships among the target response modules regulated by these SGAs (18,37) (Fig 2c). In this graph, a directed edge (or a path) from an SGA to a response module indicates that the SGA regulates the expression of the response module, and an edge between two SGAs indicates that the response modules regulated by the child SGA are subsumed by those regulated by the parent SGA. Fig 2c shows that perturbing TP53 led to the aberrant expression of a broad range of response modules, and the response modules affected by PTK2, YWHAZ, and MED1 were largely overlapping and subsumed by those affected by TP53. The results indicate that the SGAs affecting TPYM perturb a common set of signals manifested by the common response modules.
Because it is reported that the individual genes in TPYM are involved in epithelial-mesenchymal transition (EMT) (38–40), we examined the relationships of the SGAs perturbing these genes and the expression profiles of EMT-signature genes (41) in TCGA breast cancer tumors, and indeed the SGAs affecting TPYM were associated with the differential expression of EMT signature genes (Fig 2d, Table S5). In sum, our results demonstrate that SGAs perturbing TPYM affect cell cycle progression, cell proliferation, and EMT.
TPYM form a protein complex
Based on the observed genetic interaction (frequent joint alteration of TPYM) and the known protein interactions from PPI databases, we hypothesized that the 4 proteins form a physical complex that mediates the aforementioned set of signals. To test this hypothesis, we first examined whether the 4 proteins form a protein complex in breast cancer cell lines (Table 2). Fig 3a shows the baseline expression of each of the 4 proteins in different cell lines. Co-immunoprecipitation results show that the 4 proteins form a complex in the MDA-MB231 cell line (Fig 3b) and in other cell lines (Fig S6). When we used siRNA to knock down each of the proteins individually followed by co-immunoprecipitation (Fig 3c – 3e, Fig S7), the complex was disrupted.
Table 2.
Genomic status of TPYM in the studied cell lines
| TP53 | PTK2 | YWHAZ | MED1 | |
|---|---|---|---|---|
| MDA-MB-231 | p53(R280K)/p53(null) | wt | wt | wt |
| HCC38 | p53(R273L)/p53(wt) | wt | wt | wt |
| MCF-7 | wt | wt | copy number amplified | wt |
| ZR75.1 | wt | wt | wt | wt |
Figure 3. P53, PTK2, YWHAZ, and MED1 form a protein complex.
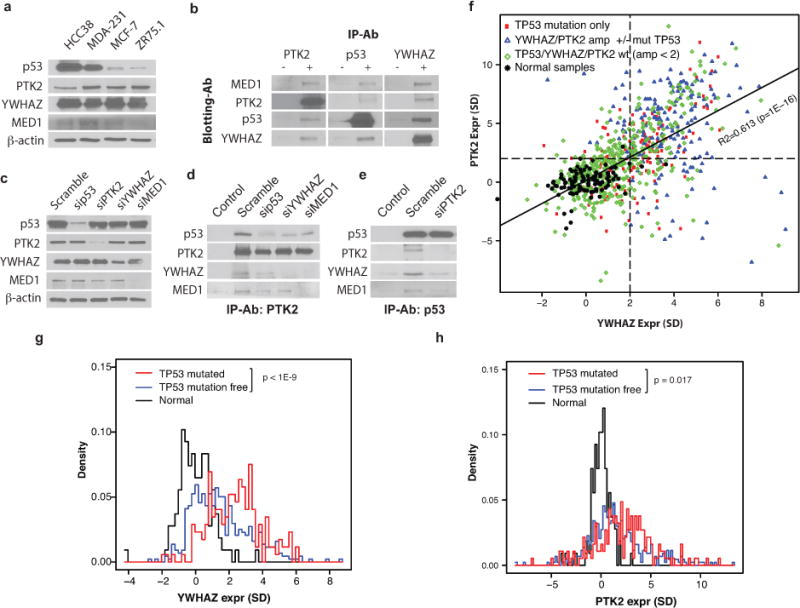
a) Baseline protein expression of TPYM proteins in different cell lines. b) Co-IP of the four proteins. Results are organized according to antibody for Co-IP (“−” denotes control IgG, “+” indicates specific antibody) and antibody for Western blot (rows). c) Western blot showing the impact of siRNA treatments on the expression of the four proteins. d – e) The impact of depleting member proteins of the tetramer complex. f) Scatter plot of PTK2 (y-axis) and YWHAZ (x-axis) expression in tumors. Each point represents a tumor, and its genomic status of TP53, PTK2, and YWHAZ are indicated by color and shape. Samples are normalized according to the mean and standard deviation (SD) of normal breast samples; the unit of axis represents the number of SDs from the mean of the normal samples for the corresponding genes. The dashed lines indicate 2 SDs above the mean of the normal samples for PTK2 and YWHAZ respectively. The solid line is the regression line with R-squared and p-values shown. g – h) Histograms of normal samples (black), tumor samples without SGAs affecting TP53, PTK2, or YWHAZ (blue), and tumor samples with TP53 mutation but normal status for PTK2 and YWHAZ (red). The P-values for the contrasts between TP53 mutant tumors and those from the tumors without SGAs are shown.
Since we observed that a single SGA affecting one of TPYM proteins was often sufficient to mediate the signals of the entire complex in some tumors (Fig 2a), we hypothesized that an SGA affecting one member of the complex was sufficient to activate a feed-forward mechanism to induce the formation of the signaling complex. To test this hypothesis, we examined whether the transcription of PTK2, YWHAZ, and MED1 were coordinated, and we also investigated the impact of TP53 mutations on their expression. Fig 3f shows that expression of PTK2 and YWHAZ are overexpressed and strongly correlated (R = 0.613, P ≤ 1.0E-16) in tumors with different combinations of mutation or copy number alterations in TP53, PTK2, and YWHAZ. Interestingly, copy number amplification of either PTK2 or YWHAZ (with or without TP53 mutation) tended to be accompanied by over-expression of the other (Fig 3f). Mutations in TP53 alone were sufficient to induce the expression of both PTK2 and YWHAZ (Fig 3f, 3g, 3h) in tumors without copy number alterations of the latter genes. We found that transcriptomic expression of MED1 was not correlated with the other members.
In summary, the results indicate that a transcriptomic mechanism likely coordinates the expression between PTK2 and YWHAZ and with respect to TP53 status, whereas MED1 is likely regulated by unknown post-transcriptional mechanisms.
Disrupting TPYM complex attenuates oncogenic behaviors
Because TPYM form a protein complex and the SGAs perturbing TPYM affect a common set of signals, we hypothesized that formation of the TPYM complex mediates the oncogenic signals that are commonly associated with the SGAs perturbing its members (e.g., mutations of p53 or amplification of PTK2) and that disrupting this signaling complex could block the pathologic aberrant signals.
To test this hypothesis, we performed a series of knockdown experiments to disrupt the complex in breast cancer cell lines (Fig 3c – 3e). Based on our computational analyses (results from Fig 2c and 2d), we predicted that disrupting the complex would affect cell cycle progression, cell proliferation, colony formation, and cell migration (the latter two phenotypes are related to EMT), and we examined the impact of knockdown on these phenotypes. We found (Fig 4a – 4d, S8) that knocking down any member of the complex significantly attenuated the above oncogenic phenotypes in cell lines hosting p53 mutations (HCC38 and MDA-MB231), whereas the impact of knock-downs was moderate in cell lines with wild type TP53 (MCF7 and ZR75.1).
Figure 4. Knocking down TPYM proteins attenuates oncogenic cellular phenotypes.
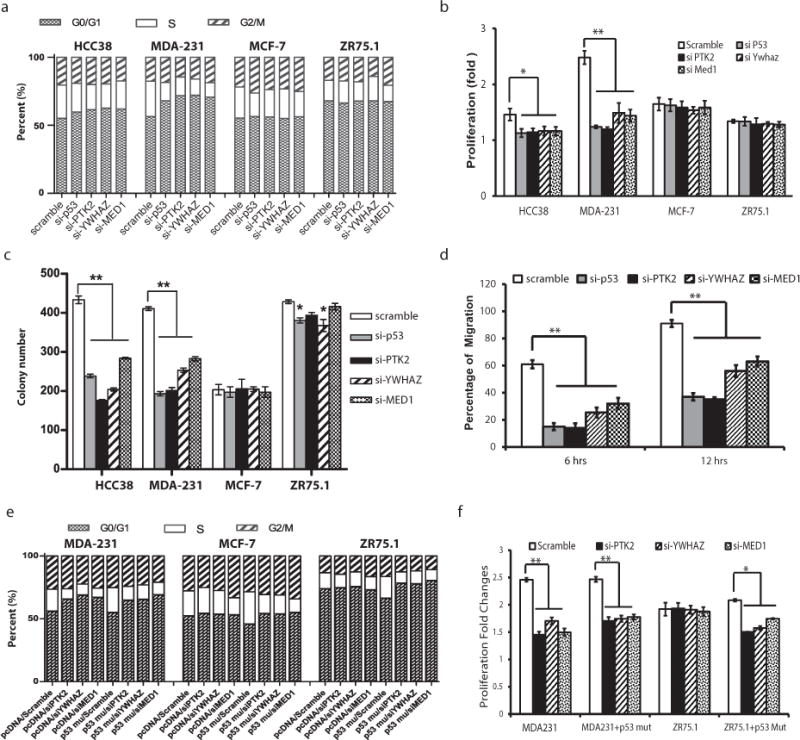
The impact of knocking down TPYM on a) cell cycle, b) cell proliferation, c) colony formation, and d) cell migration. e) Impact of introducing mutant p53 (R231K) on cell cycle and impact of co-transfection of mutant p53 and siRNA against PYM. f) Impact of introducing mutant p53 (R231K) on cell proliferation and impact of co-transfection of mutant p53 and siRNA against PYM.
To examine whether knocking down PTK2, YWHAZ, and MED1 specifically blocks the oncogenic signal originated by p53 mutations, we introduced a mutant p53 (R231K) cDNA into cell lines with wild type TP53 (ZR75.1 and MCF-7) and a cell line with mutant TP53 (MDA-MB231) to examine the impact of p53 mutation on oncogenic behavior. Our results (Fig 4e – 4f) showed a significant increase in the proportion of cells in S and G2/M phase and in cell proliferation induced by mutant p53. As expected, the introduction of mutant p53 into MDA-MB231 did not change cellular phenotype. Importantly, our results (Fig 4e – 4f) demonstrate that the impact of mutant p53 in ZR75.1 and MCF-7 was reversed by depletion of PTK2, YWHAZ, or MED1. Taken together, our results indicate that the oncogenic effect of mutant p53 requires the presence of the other members of TPYM and that the oncogenic effect of mutant p53 can be blocked by knocking down PTK2, YWHAZ, and MED1, presumably through disrupting the TPYM complex.
Discussion
We designed and applied a signal-oriented computational framework, in which we first characterize perturbed cellular signals in the form of response modules, and then we identify the SGAs (and presumably associated signaling pathways) regulating these response modules. This framework enables us to discover the relationships between SGAs and the transcriptomic modules by teasing out specific information buried in highly convoluted transcriptomic and genomic alteration data. While this study mainly concentrates on breast cancer, the framework is generally applicable to study cancer pathways for all cancer types. We anticipate this framework will be applied broadly to different types of cancers for discovering novel cancer pathways and therapeutic targets per the example shown in this paper.
Our analyses highlight a previously unappreciated signaling complex that drives multiple oncogenic processes and is associated with poor outcome in breast cancer. Although the oncogenic effects of SGAs affecting individual genes in TPYM are well known (38–40,42,43) and their relationships have been studied separately (36,44), the interplay among these genes in cancer, in particular their coordinated “mutual activation” to form a signaling complex, had not been well appreciated. Identification of this pathway reflects the strength of signal-oriented multi-omic analyses in discovering novel pathways, which reveals connections that are difficult to discern when studying different types of omics data separately.
Our results provide a novel hypothesis for the “gain of function” (45–47) of p53 mutations. It is well appreciated that tumors with mutations in TP53 are more aggressive than tumors with homozygous loss of TP53. That is, mutant p53 not only loses the tumor suppressor function but also gains oncogenic properties (45–47). However, the exact mechanism of such gain of function remains unclear. Our results demonstrate that mutations in p53 are associated with formation of the TPYM signaling complex, which in turn mediates multiple oncogenic processes such as EMT. Thus, the “gain of function” of mutant p53 is likely mediated by the formation of the complex.
Our results show that mutations in TP53 often result in the accumulation of mutant p53 proteins, due to the loss of a feedback regulation of p53 protein (2,48); our results also show that mutation of TP53 is often associated with coordinated transcription induction of PTK2 and YWHAZ, thus leading to an optimal stoichiometry that would facilitate the formation of the TPYM complex. The higher than expected co-occurrence of mutation in TP53 and copy number amplification of PTK2 or WYHAZ also indicates that such joint alterations may provide oncogenic advantages. As for the mechanism by which mutation in TP53 induces expression of PTK2, it is known that wild type p53 binds to the promoter of PTK2 (44), potentially suppressing the gene. Thus loss of the transcription factor function of mutant p53 would release such a suppression and lead to the induction of PTK2. It can be hypothesized that regulation of YWHAZ is controlled in a similar fashion. We hypothesize that wild type p53 may also be able to form the TPYM complex. However, the p53 protein is usually tightly controlled in TP53-wt cells, thus the rate and the quantity of the TPYM complex may be limited. This may explain why the p53-mutant cell lines are more sensitive to disrupting the TPYM complex than the cell lines with wild type p53 are, because the former may be more addicted to the oncogenic propertiesy of the complex.
Identification of the TPYM signaling complex could translate into a therapeutic strategy for treating tumors with aberrations in the TPYM pathway by disrupting the complex, which is supported by our proof-of-concept experiments. Although the knockdown of each individual member of TPYM could uniquely affect each oncogenic phenotype (e.g., cell cycle proliferation, cell proliferation, colony formation, cell migration) via a distinct mechanism (independent of other complex members), it is a more concise and plausible hypothesis that the loss of oncogenic phenotypes associated with each knockdown results from the disruption of the complex. As such, all four proteins could be targeted to disrupt the signaling complex to block its signal, thus expanding treatment options. For example, for tumors hosting p53 mutations, one can target any of the other three members, even if their genomic statuses are wild type. Furthermore, targeting the complex creates a “synthetic lethal” effect (49) with respect to p53 mutations. Our results show that targeting the 3 members of TPYM other than p53 has a more prominent impact on cell lines with p53 mutations, indicating that the oncogenic properties of these cell lines are more dependent on the complex.
Although our results are derived from breast cancer, the TPYM pathway is perturbed in multiple cancers (collectively covering over 90% of ovarian cancers, uterine carcinosarcoma, and lung squamous cancers, and over 70% of small cell lung cancers, head and neck squamous carcinomas, and esophageal adenocarcinoma in TCGA) (50). Therefore, any successful therapeutic strategy targeting the members of the TPYM complex would have a broad impact. Furthermore, our signal-oriented computational framework will facilitate the discovery of additional novel pathways capable of being exploited by targeted therapeutic strategies.
One limitation of the method reported in this study is the requirement of GO annotations in order to find transcriptomic modules related to a common function, which may miss genes that are not well annotated. Another potential limitation is that the k-path algorithm relies on a PPI network to search for candidate driver SGAs, which subjects the method to the incompleteness and noisiness of our knowledge in PPIs. Nonetheless, we show that the method in general can identify important response and perturbation modules that are informative of patient survival. Future studies may include using the statuses of response or perturbation modules to predict the efficacy of anticancer drugs.
Supplementary Material
Acknowledgments
The authors would like to thank Drs. Andrew Stern, Steffi Osterreich, Robert Edwards, Xin Huang for discussions and Michelle Kienholz for assistance editing the manuscript, and Dr Nancy Davidson’s generous contribution of the breast cancer cell lines from the NCI-ATCC ICBP program. The research was partially supported by following NIH grants: R01LM 010144, R01LM 011155, R01LM012011, U54HG008540, R01CA154695, R01CA094118, K99LM011673, and P01CA97132.
Footnotes
The authors declare no conflict of interest pertaining to this manuscript.
References
- 1.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546–58. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weinberg RA. The biology of cancer. New York, NY: Garland Science, Taylor & Francis Group; 2013. [Google Scholar]
- 3.Joly Y, Dove ES, Knoppers BM, Bobrow M, Chalmers D. Data sharing in the post-genomic world: the experience of the International Cancer Genome Consortium (ICGC) Data Access Compliance Office (DACO) PLoS Comput Biol. 2012;8:e1002549. doi: 10.1371/journal.pcbi.1002549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dees ND, Zhang Q, Kandoth C, Wendl MC, Schierding W, Koboldt DC, et al. MuSiC: identifying mutational significance in cancer genomes. Genome research. 2012;22:1589–98. doi: 10.1101/gr.134635.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214–8. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Djotsa Nono ABD, Chen K, Liu X. Comutational prediction of genetic drivers in cancer. eLS. 2016 [Google Scholar]
- 7.Ciriello G, Cerami E, Sander C, Schultz N. Mutual exclusivity analysis identifies oncogenic network modules. Genome research. 2012;22:398–406. doi: 10.1101/gr.125567.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vandin F, Upfal E, Raphael BJ. Algorithms for detecting significantly mutated pathways in cancer. J Comput Biol. 2011;18:507–22. doi: 10.1089/cmb.2010.0265. [DOI] [PubMed] [Google Scholar]
- 9.Vandin F, Upfal E, Raphael BJ. De novo discovery of mutated driver pathways in cancer. Genome Res. 2012;22:375–85. doi: 10.1101/gr.120477.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Raphael BJ, Dobson JR, Oesper L, Vandin F. Identifying driver mutations in sequenced cancer genomes: computational approaches to enable precision medicine. Genome medicine. 2014;6:5. doi: 10.1186/gm524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim YA, Cho DY, Dao P, Przytycka TM. MEMCover: integrated analysis of mutual exclusivity and functional network reveals dysregulated pathways across multiple cancer types. Bioinformatics. 2015;31:i284–92. doi: 10.1093/bioinformatics/btv247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lu S, Lu KN, Cheng SY, Hu B, Ma X, Nystrom N, et al. Identifying Driver Genomic Alterations in Cancers by Searching Minimum-Weight, Mutually Exclusive Sets. PLoS computational biology. 2015;11:e1004257. doi: 10.1371/journal.pcbi.1004257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim YA, Salari R, Wuchty S, Przytycka TM. Module cover – a new approach to genotype-phenotype studies. Pacific Symposium on Biocomputing Pacific Symposium on Biocomputing. :2013135–46. [PMC free article] [PubMed] [Google Scholar]
- 14.Kim YA, Przytycki JH, Wuchty S, Przytycka TM. Modeling information flow in biological networks. Phys Biol. 2011;8:035012. doi: 10.1088/1478-3975/8/3/035012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tuncbag N, McCallum S, Huang SS, Fraenkel E. SteinerNet: a web server for integrating ‘omic’ data to discover hidden components of response pathways. Nucleic Acids Res. 2012;40:W505–9. doi: 10.1093/nar/gks445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hofree M, Shen JP, Carter H, Gross A, Ideker T. Network-based stratification of tumor mutations. Nature methods. 2013;10:1108–15. doi: 10.1038/nmeth.2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Segal E, Friedman N, Koller D, Regev A. A module map showing conditional activity of expression modules in cancer. Nat Genet. 2004;36:1090–8. doi: 10.1038/ng1434. [DOI] [PubMed] [Google Scholar]
- 18.Lu S, Jin B, Cowart LA, Lu X. From data towards knowledge: Revealing the architecture of signaling systems by unifying knowledge mining and data mining of systematic perturbation data. PLoS ONE. 2013 doi: 10.1371/journal.pone.0061134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lu S, Lu X. Integrating genome and functional genomics data to reveal perturbed signaling pathways in ovarian cancers. AMIA Summits on Translational Science proceedings AMIA Summit on Translational Science. 2012;2012:72–8. [PMC free article] [PubMed] [Google Scholar]
- 20.Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333–9. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheng WY, Ou Yang TH, Anastassiou D. Development of a prognostic model for breast cancer survival in an open challenge environment. Science translational medicine. 2013;5:181ra50. doi: 10.1126/scitranslmed.3005974. [DOI] [PubMed] [Google Scholar]
- 22.Lu S, Zhang F, Chen J, Sze S-h. Finding pathway structures in protein interaction networks. Algorithmica. 2007;48:363–74. [Google Scholar]
- 23.Segal E, Kim SK. The modular era of functional genomics. Genome Biol. 2003;4:317. doi: 10.1186/gb-2003-4-5-317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wong DJ, Nuyten DS, Regev A, Lin M, Adler AS, Segal E, et al. Revealing targeted therapy for human cancer by gene module maps. Cancer Res. 2008;68:369–78. doi: 10.1158/0008-5472.CAN-07-0382. [DOI] [PubMed] [Google Scholar]
- 25.Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nature genetics. 2000;25:25–9. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jin B, Lu X. Identifying informative subsets of the Gene Ontology with information bottleneck methods. Bioinformatics. 2010;26:2445–51. doi: 10.1093/bioinformatics/btq449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 29.Lu S, Cai C, Osmanbeyoglu HU, Chen L, Day R, Cooper G, et al. Identify Informative Modular Features for Predicting Cancer Clinical Outcomes. 2012 [Google Scholar]
- 30.Curtis C, Shah SP, Chin S-F, Turashvili G, Rueda OM, Dunning MJ, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012:1–7. doi: 10.1038/nature10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Venet D, Dumont JE, Detours V. Most random gene expression signatures are significantly associated with breast cancer outcome. PLoS Comput Biol. 2011;7:e1002240. doi: 10.1371/journal.pcbi.1002240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–7. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Monti S, Tamayo P, Mesirov J, Golub T. Consensus clustering: A resampling-based method for class discovery and visualization of gene expression microarray data. Machine Learning. 2003;52:91–118. [Google Scholar]
- 34.Mani R, St Onge RP, Hartman JLt, Giaever G, Roth FP. Defining genetic interaction. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:3461–6. doi: 10.1073/pnas.0712255105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chatr-Aryamontri A, Breitkreutz BJ, Heinicke S, Boucher L, Winter A, Stark C, et al. The BioGRID interaction database: 2013 update. Nucleic Acids Res. 2013;41:D816–23. doi: 10.1093/nar/gks1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Golubovskaya VM, Cance WG. FAK and p53 protein interactions. Anti-cancer agents in medicinal chemistry. 2011;11:617–9. doi: 10.2174/187152011796817619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Markowetz F, Kostka D, Troyanskaya OG, Spang R. Nested effects models for high-dimensional phenotyping screens. Bioinformatics. 2007;23:i305–12. doi: 10.1093/bioinformatics/btm178. [DOI] [PubMed] [Google Scholar]
- 38.Chang CJ, Chao CH, Xia W, Yang JY, Xiong Y, Li CW, et al. p53 regulates epithelial-mesenchymal transition and stem cell properties through modulating miRNAs. Nature cell biology. 2011;13:317–23. doi: 10.1038/ncb2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lu J, Guo H, Treekitkarnmongkol W, Li P, Zhang J, Shi B, et al. 14–3–3 zeta Cooperates with ErbB2 to promote ductal carcinoma in situ progression to invasive breast cancer by inducing epithelial-mesenchymal transition. Cancer cell. 2009;16:195–207. doi: 10.1016/j.ccr.2009.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Danes CG, Wyszomierski SL, Lu J, Neal CL, Yang W, Yu D. 14–3–3 zeta down-regulates p53 in mammary epithelial cells and confers luminal filling. Cancer Res. 2008;68:1760–7. doi: 10.1158/0008-5472.CAN-07-3177. [DOI] [PubMed] [Google Scholar]
- 41.Kim JE, Leung E, Baguley BC, Finlay GJ. Heterogeneity of expression of epithelial-mesenchymal transition markers in melanocytes and melanoma cell lines. Frontiers in genetics. 2013;4:97. doi: 10.3389/fgene.2013.00097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Menon R, Deng M, Ruenauver K, Queisser A, Pfeifer M, Offermann A, et al. Somatic copy number alterations by whole-exome sequencing implicates YWHAZ and PTK2 in castration-resistant prostate cancer. The Journal of pathology. 2013;231:505–16. doi: 10.1002/path.4274. [DOI] [PubMed] [Google Scholar]
- 43.Menon R, Deng M, Ruenauver K, Queisser A, Peifer M, Offermann A, et al. Somatic copy number alterations by whole-exome sequencing implicates YWHAZ and PTK2 in castration-resistant prostate cancer. The Journal of pathology. 2013;231:505–16. doi: 10.1002/path.4274. [DOI] [PubMed] [Google Scholar]
- 44.Wei CL, Wu Q, Vega VB, Chiu KP, Ng P, Zhang T, et al. A global map of p53 transcription-factor binding sites in the human genome. Cell. 2006;124:207–19. doi: 10.1016/j.cell.2005.10.043. [DOI] [PubMed] [Google Scholar]
- 45.Cadwell C, Zambetti GP. The effects of wild-type p53 tumor suppressor activity and mutant p53 gain-of-function on cell growth. Gene. 2001;277:15–30. doi: 10.1016/s0378-1119(01)00696-5. [DOI] [PubMed] [Google Scholar]
- 46.Muller PA, Vousden KH. Mutant p53 in cancer: new functions and therapeutic opportunities. Cancer cell. 2014;25:304–17. doi: 10.1016/j.ccr.2014.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zheng T, Wang J, Zhao Y, Zhang C, Lin M, Wang X, et al. Spliced MDM2 isoforms promote mutant p53 accumulation and gain-of-function in tumorigenesis. Nat Commun. 2013;4:2996. doi: 10.1038/ncomms3996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387:296–9. doi: 10.1038/387296a0. [DOI] [PubMed] [Google Scholar]
- 49.Kaelin WG., Jr Synthetic lethality: a framework for the development of wiser cancer therapeutics. Genome Med. 2009;1:99. doi: 10.1186/gm99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Science signaling. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.