

Abstract
Exposure of small animals to cigarette smoke is widely used as a model to study the pathogenesis of chronic obstructive pulmonary disease. However, protocols and exposure systems utilised vary substantially and it is unclear how these different systems compare.
We analysed the gene expression profile of six publically available murine datasets from different cigarette smoke-exposure systems and related the gene signatures to three clinical cohorts.
234 genes significantly regulated by cigarette smoke in at least one model were used to construct a 55-gene network containing 17 clusters. Increasing numbers of differentially regulated clusters were associated with higher total particulate matter concentrations in the different datasets. Low total particulate matter-induced genes mainly related to xenobiotic/detoxification responses, while higher total particulate matter activated immune/inflammatory processes in addition to xenobiotic/detoxification responses. To translate these observations to the clinic, we analysed the regulation of the revealed network in three human cohorts. Similar to mice, we observed marked differences in the number of regulated clusters between the cohorts. These differences were not determined by pack-year.
Although none of the experimental models exhibited a complete alignment with any of the human cohorts, some exposure systems showed higher resemblance. Thus, depending on the cohort, clinically observed changes in gene expression may be mirrored more closely by specific cigarette smoke exposure systems. This study emphasises the need for careful validation of animal models.
Short abstract
Particulate matter skews gene expression pattern in cigarette smoke-exposed mice towards an inflammatory phenotype http://ow.ly/Tpex3038fyC
Introduction
The devastating health impact of cigarette smoking is well known [1]. Despite this, over 1 billion people continue to smoke worldwide [2]. Smoking is the main risk factor of chronic obstructive pulmonary disease (COPD), an inflammatory disorder characterised by a progressive and largely irreversible airway obstruction. While emphasis should be placed on smoking prevention and cessation, a greater understanding of cellular and molecular mechanisms that contribute to the pathogenesis of COPD are equally important given the highly addictive nature and chronic persistence of cigarette smoking [1], the burden this disease places on society [3], and the fact that current pharmacologic interventions show limited effects [4].
Although animal models do not fully capture the complexity of COPD, they are valuable tools to investigate biological mechanisms that contribute to the pathogenesis and progression of COPD, and to screen novel intervention strategies. Exposure of animals to cigarette smoke is viewed as one of the most relevant experimental models for COPD, as they include the main aetiological factor associated with COPD. However, smoke exposure systems and protocols vary substantially between laboratories, which may explain the divergent and sometimes contradictory nature of results reported in the literature [5]. These discrepancies are often attributed to differences between exposure systems and experimental protocols, although our understanding of differences between models remains limited.
The purpose of this study was to compare and contrast gene expression profiles between different murine models of cigarette smoke exposure and relate the gene signatures to clinical cohorts. The analysis included six publically available mouse datasets representing different experimental systems. We found 234 genes that were significantly regulated by cigarette smoke in at least one of the experimental systems. Based on this, a functional network with 17 clusters was constructed to investigate the biological significance of differential gene expression. Experimental models with high total particulate matter (TPM) exhibited regulated genes in a higher number of clusters than models with low TPM. While low TPM mainly induced genes involved in detoxification, higher TPM concentrations activated genes involved in inflammatory processes in addition to the detoxification response. Similar to the experimental models, biological response patterns in human cohorts showed marked differences. However, unlike animal models where the response to cigarette smoke was dependent of the TPM concentrations, differences in the human cohorts were not determined by pack-year and need further investigation.
Materials and methods
Animal datasets
Lung gene expression profiles of seven publically available datasets were obtained, but only six were used in the current study. These profiles were deposited in the Gene Expression Omnibus (GEO) (accession numbers GSE55127, GSE33512, GSE18344, GSE8790, GSE17737, GSE52509 and GSE33561) [6–11] and comprised a total of 54 samples. GSE33512, GSE55127, GSE33561 and GSE52509 datasets were pre-processed as described in the corresponding source publications. GSE18344, GSE17737 and GSE8790 datasets contained samples profiled on Affymetrix Mouse Genome 430 2.0 arrays. These arrays were normalised with frozen Robust Multi-array Analysis, a procedure that allows microarrays to be pre-processed individually or in small batches and allows data to be combined into a single dataset for further analyses [12]. Since different profiling platforms contain different numbers of genes, we included 10 634 genes in the analysis that were shared among all platforms. The Distance-Weighted Discrimination (DWD) method [13] was used to remove technical variation from the datasets that were to be combined for further analyses. For a more detailed description of the analyses refer to the supplementary material.
Human dataset
Human subjects and the lung specimen collection have been described previously [14]. Non-tumour lung specimens were collected from patients undergoing lung surgery at three different sites: Institut Universitaire de Cardiologie et de Pneumologie de Québec (IUCPQ), Laval University (Quebec, Canada), University of British Columbia (Vancouver, Canada) and University of Groningen (Groningen, the Netherlands), henceforth referred to Laval, UBC and Groningen. Non-neoplastic pulmonary parenchyma was harvested and stored at −80°C. Gene expression profiling was carried out using Affymetrix arrays at the same facility using the same methods for the all the datasets, data was deposited as GSE23546.
Differential gene expression analysis
We used the “limma” package [15] to compare smoke-exposed mice from each model to control mice pooled across all models. T-statistics were followed by Benjamini–Hochberg adjustment for multiple testing [16].
Network analysis
Networks were constructed based on 234 ≥2-fold differentially expressed genes using Reactome FI plugin (Ontario Institute for Cancer Research, Toronto, ON, Canada) in Cytoscape software (Cytoscape Consortium, www.cytoscapeconsortium.org) [17, 18]. This relatively stringent ≥2-fold threshold was chosen in accordance with previous publications [19, 20]. The networks were then analysed for the presence of significant gene clusters (also termed “clusters” or “modules”) [21] and these clusters were further examined with pathway enrichment and gene ontology tools in Cytoscape. Further details of the analyses are provided in the supplementary material.
Gene set enrichment analysis
We used gene set enrichment analysis (GSEA) [22] to examine the enrichment of 17 genes previously reported to be induced by cigarette smoke in the human and mouse lung (MMP12, AHRR, SPP1, ALDH3A1, CYP1B1, GDF15, GSTA2, NQO1, PLA2G7, TREM2, CLEC5A, ACP5, ATP6V0D2, BHLHE41, NEK6, DCSTAMP and LCN2 [6]). Two out of 17 genes (DCSTAMP and BHLHE41) were unavailable on the gene expression profiling platforms; therefore, GSEA was performed using the remaining 15 genes. Analysis was performed separately for each of the models, and p-values were pooled and corrected for multiple testing using Benjamini–Hochberg multiple testing correction [16]. In the course of GSEA it is usually revealed that not all of the genes belonging to the gene set of interest exhibit modified levels of expression in the profiled sample. Therefore, it is suggested to extract the core members of high scoring gene sets that contribute to the enrichment. Such groups of core members is termed a “Leading Edge” subset and it can be interpreted as the core of a gene set that accounts for the enrichment signal.
Multivariate regression analysis
Multivariate analysis was carried out by using lm function from the R stats package (www.r-project.org). We analysed the following factors: 1) TPM, 2) sex and 3) exposure duration. In a separate analysis, we assessed absolute dose (TPM × average exposure time) and sex. For GSE33512 the TPM was provided as a range between 100 and 120 µg·L−1; for the purpose of our analysis we used the average of 110 µg·L−1. Similarly, exposure duration provided for GSE33561 was 6–7 weeks, thus we used the average of 6.5 weeks.
Results
Processing and combining independent datasets according to their exposure groups
To compare gene expression profiles in the lungs of mice exposed to cigarette smoke we acquired seven publically available, independent datasets from the GEO. We included in the analysis datasets from mice that were exposed to cigarette smoke for 6–16 weeks, regardless of strain, sex, array platform and smoke exposure system (table 1). Strong batch effects led to clustering of samples based on study, array platform and exposure (figure 1a). We next employed the DWD method for removing batch effects (see Materials and methods) resulting in sample clustering largely according to exposure (figure 1b), suggesting that most study and array platform effects were successfully removed, while the factor of interest remained. Of note, five samples from room air-exposed mice clustered within cigarette smoke-exposed samples: three samples from GSE8790, one sample from GSE55127 and one sample from GSE33561. Based on the assumption that room air samples from all studies should cluster together, we removed these five samples from the analysis. Since all room air-exposed control samples from GSE8790 were removed, we excluded this study from the analysis. Batch effect removal and filtering based on the assumption that room air-exposed samples should cluster together resulted in a collection of 46 samples. These samples formed two well-defined clusters, based on room air and cigarette smoke exposure (figure 1c). Similar results were observed in a parallel examination using principal component analysis (figure S1).
TABLE 1.
Mouse datasets downloaded from Gene Expression Omnibus (GEO)
| GEO accession [ref.] | Exposure duration weeks | Strain | Sex | Age weeks | Samples | TPM µg·L−1 | Cigarette |
| GSE8790 [9] | 6 | A/J | Male | 8 | 3 RA/3 CS | 90 | 2R4F |
| GSE33561 [11] | 6–7 | AKR/J | Male | 6–8 | 3 RA/3 CS | 90 | 2R4F |
| GSE33512 [7] | 16 | C57BL/6 | Male | 12 | 4 RA/4 CS | 100–120 | 1R3F |
| GSE52509 [10] | 16 | C57BL/6 | Female | 8–10 | 3 RA/3 CS | 500 | 3R4F |
| GSE17737 | 12 | C57BL/6 | Female | NA | 5 FA/5 CS | NA | NA |
| GSE55127 [6] | 8 | BALB/C | Female | 6–8 | 5 RA/5 CS | >600 | 3R4F# |
| GSE18344 [8] | 8 | CD-1 | Female | 13 | 4 RA/4 CS | 750 | 2R4F |
TPM: total particulate matter; RA: room air; FA: forced air; CS: cigarette smoke; NA: not applicable. #: filters removed.
FIGURE 1.

Combining datasets. a) Clustering of samples before the batch effect removal procedure (Distance-Weighted Discrimination; DWD) was performed. b) Clustering of samples after DWD. c) Clustering of samples after removing GSE8790 and samples from two control mice that clustered with the samples obtained from smoke-exposed mice. Samples were clustered using average linkage and Spearman correlation distance.
Differential gene expression analysis: 20 genes differentially expressed between cigarette smoke-exposed mice and control samples in all studies
We next performed a pair-wise differential gene expression analyses between cigarette smoke-exposed mice and the pooled control samples to examine biological processes that were significantly activated or suppressed by cigarette smoke exposure. Transcriptional changes varied markedly between studies, ranging from 62 upregulated and 124 down-regulated genes in study GSE33561 and 833 upregulated and 626 down-regulated genes in study GSE55127 (figure 2). In addition, the numbers of differentially expressed genes found in samples from GSE18344, GSE55127 and GSE17737 was significantly higher than the numbers of such genes found in the remaining three datasets (p=0.0005, Welch two sample t-test).
FIGURE 2.

Genes differentially expressed between the smoke-exposed mice in each model and the control group. Increasing fold changes are highlighted with the increasing level of shading. In each category the genes are sorted by the fold-change values obtained from GSE18344. The colour coding is different for the all fold changes and fold change ≥2.
To examine similarities between the six studies we focused on gene transcripts with ≥2-fold change between the pooled control samples and cigarette smoke-exposed mice. A full list of 234 genes that changed ≥2-fold in at least one of the studies is shown in table S1. The majority of these genes were upregulated in the smoke-exposed mice (figure S2). Of the 234 genes, 20 were differentially expressed between the cigarette smoke-exposed mice and the controls in all six studies (figure 2). This included a number of genes that have previously been implicated in cigarette smoke-induced inflammatory processes, as well as xenobiotic and anti-oxidant responses. The remaining 214 genes were differentially expressed between the cigarette smoke-exposed mice and the controls in decreasing number of studies with GSE18344, GSE55127 and GSE17737 showing greater overall change in gene expression compared to the other three studies. These findings suggest that despite similarities, there are marked differences in transcriptional changes between studies.
Analysis of pathways and clusters: 17 clusters regulated by cigarette smoke in mouse lung in all studies
Next, we built a functional interaction network based on the 234 genes differentially expressed between cigarette smoke-exposed mice and controls. Of the 234 genes, 55 constructed a complex network containing 17 clusters (figure 3a). Each cluster was then analysed to discover pathways and gene ontology processes represented by genes belonging to the cluster. Table 2 presents the genes involved in the clusters and their descriptions based on the results of this analysis (table S2).
FIGURE 3.

Functional interaction network based on the 234 differentially expressed genes. a) Functional network with annotated gene clusters. Each cluster is contained within a separate NTA: grey oval, with the ID superimposed on it. b) Expression of the gene clusters in each of the mouse models. Clusters containing at least one upregulated gene are marked in pink; clusters containing at least one downregulated gene are marked in green; clusters containing both up- and downregulated genes are marked in grey. Clusters not containing any differentially expressed genes are marked by black contour. Models are sorted based on the total particulate matter used in the model: from lowest (lower left corner) to highest (upper right corner).
TABLE 2.
Functional annotation of the gene clusters
| Cluster | Genes in cluster |
Function derived from Pathway Enrichment and Gene Ontology analyses |
Gene list |
| 0 | 6 | TLR signalling: unfolded protein response | DNAJB1, DNAJC5B, HSPA1A, HSPB1, SAA1, TLR2 |
| 10 | 2 | TLR signalling | CD86, CXCL9 |
| 1 | 6 | Chemokines: signalling | CCL17, CCL22, CCL4, CCR1, CXCL13, CXCL5 |
| 7 | 3 | Chemokines: activity | CCL2, CCL3, CCL7 |
| 11 | 2 | Chemokines: NOD-like receptor signalling | CXCL1, CXCL2 |
| 2 | 5 | Muscle contraction | ACTN2, MYH6, MYL1, TCAP, TNNI3 |
| 3 | 5 | Integrins | CD14, ITGAM, ITGAX, ITGB2, SPON2 |
| 4 | 4 | Circadian clock | ARNTL, DBP, NR1D1, PER1 |
| 5 | 3 | Osteoclast differentiation | CLEC5A, TREM2, TYROBP |
| 6 | 3 | Interleukins: FC-epsilon receptor I signalling | FCER1G, FCGR2B, IL12B |
| 14 | 2 | Interleukins | IL1R2, IL1RN |
| 8 | 3 | p53 pathway | IGF1, IGFBP3, IGFBP6 |
| 9 | 3 | – | AHRR, ARNT2, MAFB |
| 12 | 2 | Class I MHC mediated antigen processing and presentation | CYBA, NCF4 |
| 13 | 2 | Immune response (complement pathway) | C1QA, C1QB |
| 15 | 2 | Glutathione metabolism | GCLC, GCLM |
| 16 | 2 | Heterotrimeric G-protein signalling pathway-Gq alpha and Go alpha mediated pathway | RGS1, RGS16 |
There were no pathways or processes significantly represented by cluster 9. TLR: Toll-like receptor; NOD: nucleotide-binding oligomerisation domain; MHC: major histocompatibility complex.
To examine the differences in the regulation of clusters between the models, we defined the direction of regulation of each of the clusters (see Materials and methods), and highlighted the clusters accordingly (figure 3b). A clustergram showed marked differences in how clusters were regulated between studies (figure 4a). GSE33561 and GSE33512 showed relatively few clusters that were regulated by smoke exposure. Studies GSE52509, GSE17737 and GSE55127 showed regulation of the majority of clusters, while all clusters were regulated in GSE18344. Cluster 1 was upregulated in all studies, while clusters 7 and 11 were upregulated in five out of six studies. These clusters related to chemokine signalling and activity, and NOD-like receptor signalling.
FIGURE 4.

Clusters from the functional network. a) Models clustered by the numbers of differentially expressed genes in each of the clusters. Numbers of the differentially expressed genes are indicated on the heatmap. Branches of the vertical dendrogram are named using the cluster IDs (0–16). Each cluster is functionally annotated based on the Pathway Enrichment and Gene Ontology analyses (see table 3). Cluster 9 is marked by “–” since it did not show any significant representation of any of the known processes. #: cluster contained both up- and downregulated genes. b) Correlation between total particulate matter (TPM) and number of differentially regulated clusters across mouse models. *: indicates mouse models. TLR: Toll-like receptor; MHC: major histocompatibility complex; NOD: nucleotide-binding oligomerisation domain. r2 =0.9348, p-value = 0.0072.
The number of regulated clusters correlates with total particulate matter
Next, we examined factors that correlate with regulated clusters across the six studies. We examined TPM, sex and duration of the exposure. Study GSE17737 was omitted from the analysis, as TPM concentration was not publically available. TPM was the only factor that correlated with the number of affected clusters (p=0.0368), while neither sex nor exposure duration reached statistical significance. Based on this, we plotted the number of regulated clusters as a function of total particulate matter across the five datasets, for which TPM information was available (figure 4b). Fitted linear regression analysis revealed a significant correlation between total particulate matter and the number of regulated clusters (p=0.0072, 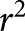 =0.9348). Additionally, we performed multivariate regression analysis to examine the correlation between regulated clusters, the absolute dose (TPM × average exposure time) and sex; however, the results did not reach statistical significance (p=0.2389).
=0.9348). Additionally, we performed multivariate regression analysis to examine the correlation between regulated clusters, the absolute dose (TPM × average exposure time) and sex; however, the results did not reach statistical significance (p=0.2389).
Enrichment for genes regulated by cigarette smoke in mice and humans lungs
Previous reports revealed 17 genes that are significantly regulated by cigarette smoke in both mice and humans [6]. Based on these findings, we examined whether the samples obtained from cigarette smoke-exposed mice in the 6 studies are enriched in these 17 genes using GSEA [22]. All models showed that 15 out of 17 genes (MMP12, AHRR, SPP1, ALDH3A1, CYP1B1, GDF15, GSTA2, NQO1, PLA2G7, TREM2, CLEC5A, ACP5, ATP6V0D2, NEK6 and LCN2) were significantly induced, as shown by the significant enrichment in the list of these genes (p<0.05) (see Materials and methods). Figure S3 shows the enrichment of the signature in each of the studies. Based on the enrichment plots, the majority of the examined genes contributed to these results. This observation is supported by the findings that the majority of the genes belong to the Leading Edge (table S3). These genes are of interest, as they are shared among models and probably point towards fundamental mechanisms involved in the response to cigarette smoke.
Mapping results obtained from mouse gene clusters on human data
As we observed marked differences in numbers of clusters regulated between cigarette smoke-exposure studies, we next sought to investigate whether similar differences exist in the regulation of these clusters between human cohorts. We used three human cohorts obtained from three different centres. Both current smokers and never-smokers were represented in each cohort (table 3). We analysed the expression of the 55 genes used to construct the network in the mouse studies. As described in the Materials and methods section, genes showing a significant differential expression were used to establish the regulation of the clusters. Similar to mice, we observed differences between the different cohorts (figure 5a). While relatively few clusters were regulated in the UBC cohort, the Groningen and LAVAL cohorts showed regulation in most clusters. Clusters 5 and 9 (reflecting Osteoclast differentiation and without functional annotation, respectively) were consistently upregulated across all cohorts, but no consistent pattern emerged with any of the other clusters. For example, clusters 1, 7 and 11 (reflecting chemokine clusters (signalling, activity and NOD-like receptor signalling)) were found to be either upregulated or not regulated, while cluster 10 (reflecting Toll-like receptor signalling) exhibited all possible modes of regulation: up-, down- and bi-direction regulation. The clustergram of the Laval and Groningen cohorts was more similar to the mouse GSE52509 and GSE55127 datasets, while the UBC cohort displayed greater similarities to GSE33561 and GSE33512 (figure 5b). Figure 5c shows a plot depicting the number of regulated clusters in mouse models as a function of TPM (same as in figure 4b) with the number of regulated clusters in human cohorts superimposed as horizontal lines. Similar to mice, there are marked differences in the number of regulated clusters among cohorts and, none of the experimental models exhibit a complete alignment with any of the human cohorts.
TABLE 3.
Clinical characteristics of subjects
| Variables | Groningen | LAVAL | UBC | |||
| Never-smokers | Current smokers | Never-smokers | Current smokers | Never-smokers | Current smokers | |
| Subjects | 12 | 45 | 27 | 82 | 17 | 88 |
| Male/female | 5 (42%)/7 | 23 (51%)/22 | 6 (22%)/21 | 37 (45%)/45 | 8 (47%)/9 | 54 (61%)/34 |
| Age years | 48.3±15.4 | 57.5±8.7 | 55.8±11.6 | 63±9.2 | 59.6±14.6 | 62±9.4 |
| Smoking pack-years | 0 | 37.9±17.4 | 0 | 53±21.8 | 0 | 53.7±27.1 |
| FEV1 % pred | 98.6±9.38 (7) | 73.3±19.8 (17) | 94.1±13.2 (4) | 75.1±14.4 (1) | 105.4±33.1 (7) | 76.4±18.2 (7) |
| FVC % pred | 98.8±7.3 (7) | 87.1±16.3 (18) | 96.5±12.5 (6) | 87. 1±13.5 (5) | 99.1±31.2 (6) | 86.7±15.7 (7) |
Data are presented as mean±sd, unless otherwise stated. The number of missing values is shown in parentheses. LAVAL: Laval University; UBC: University of British Columbia; FEV1: forced expiratory volume in 1 s; FVC: forced vital capacity.
FIGURE 5.

Regulation of clusters in human datasets. a) Expression of the gene clusters in each human cohort. Clusters containing at least one upregulated gene are marked in pink; clusters containing at least one downregulated gene are marked in green; clusters containing both up- and downregulated genes are marked in grey. Clusters not containing any differentially expressed genes are marked by black contour. b) Models clustered by the numbers of differentially expressed genes in each of the clusters across human data and mouse models. Numbers of the differentially expressed genes are indicated on the heatmap. Branches of the vertical dendrogram are named using the cluster IDs (0–16). Each cluster is functionally annotated based on the Pathway Enrichment and Gene Ontology analyses (see table 3). Cluster 9 is marked by “–” since it did not show any significant representation of any of the known processes. #: cluster contained both up- and downregulated genes. c) Correlation between total particulate matter (TPM) and number of differentially regulated clusters across mouse models and human datasets. Human datasets are marked by horizontal lines indicating the number of regulated clusters in each dataset. *: indicates mouse models. Laval: Laval University; GRNG: Groningen; UBC: University of British Columbia; TLR: Toll-like receptor; MHC: major histocompatibility complex; NOD: nucleotide-binding oligomerisation domain.
Discussion
Animal models are widely used to study the pathogenesis of smoking-related diseases such as COPD. There are, however, marked differences between cigarette smoke exposure systems and protocols used by different laboratories. Consequently, data generated are often difficult to compare and may even appear conflicting. In the present study, we compared gene expression profiles from different smoke exposure systems to assess similarities and differences between systems/protocols and to determine what approach most accurately reflects observations in clinical cohorts.
To address this we analysed seven publically available datasets in which mice were exposed to cigarette smoke for 6–16 weeks regardless of strain and sex. We purposely used broad inclusion criteria to increase the number of datasets included in the analysis. In contrast to previous studies that used specific exposure protocols to identify genes that contribute to cigarette smoke-induced pathology, the analytical approach in this study aimed at establishing similarities and differences in the gene expression profile between different experimental models of cigarette smoke exposure. We used batch effect removal procedures and quality assurance filtering procedures with the assumption that datasets from room air-exposed mice should cluster together, with the effects of smoke being stronger than the effects of the strain or the sex. No assumptions were made regarding the clustering of datasets from cigarette smoke-exposed mice as it is conceivable that, in some of the studies, the effects of cigarette smoke are subtle, leading to clustering of these samples within control groups rather than samples from smoke-exposed mice.
We previously identified 17 smoking-induced genes conserved between a large-scale transcriptomic human dataset and one of the mouse datasets used in the current study [6]. Of note, this gene list was enriched in all murine datasets, indicating that all models mimic the molecular behaviour of these conserved genes.
In our study, 234 genes were differentially expressed by ≥2-fold in at least one of the datasets. Of these, only 20 genes were significantly regulated in all datasets, highlighting the marked difference between models. Most of the upregulated genes were related to immune inflammatory processes (CCL6 [23], CD84, CXCL5 [24], GPNMB [25], LGALS3 [26], MMP12 [27], MS4A7, PTGIR and TREM2 [28]) or detoxification responses (AKR1B8 [29], CD68 [30], CYP1B1 [31], PLA2G7 and SLC7A11). Of note, CTSK [32], CXCL5 [33], CYP1B1, LGALS3 [34], MMP12 [35], PLA2G7, PTGIR [36], SLC7A11 [37] and TREM2 [38] have been described previously, while the remaining genes (AKR1B8, CCL6, CD68, CD84, GPNMB, IGFBP6, LHFPL2, MS4A7, MYO5A, SLC39A4 and ZRANB3), to the best of our knowledge, have not been reported previously in association with smoking-related diseases. A description of the full gene names and a summary of their known functions are provided in table S4. Despite marked differences between models, these latter genes are of particular interest to further study as they are shared among all animal models and human cohorts and probably point towards fundamental mechanisms involved in the response to cigarette smoke. However, not all smokers develop COPD and as many as one out of four Americans with COPD have never smoked cigarettes [39]; thus, while these genes are shared and conserved among mice and humans, they may not be related to the development of COPD. In addition, many processes exist between transcription and translation, and observed gene regulation does not necessarily imply protein-related changed. Hence, further analyses should validate these differentially expressed genes at the protein level.
Of the 234 genes with >2-fold regulation, 55 genes formed a network organised into 17 clusters. Most clusters were associated with functions within inflammatory and immune responses (Toll-like receptor signalling, chemokines, integrins, interleukins, class 1 major histocompatibility complex-mediated antigen processing and presentation, immune response and G-protein signalling). Additional clusters related to muscle contraction, circadian clock, the p53 signalling pathway and osteoclast differentiation. Finally, we observed a cluster related to glutathione metabolism. Glutathione is an important reducing agent and has been shown to render protection against reactive oxygen species, as well as regulation of intracellular redox status.
The regulation of the clusters varied markedly among smoke exposure systems. In some of the datasets, we observed predominant activation of genes involved in detoxification, with little or no regulation of genes associated with immune inflammatory clusters. In contrast, other systems showed regulation of almost all clusters. Analysis of the influence of sex, strain, duration of the exposure and TPM concentration revealed that only TPM showed a significant correlation with the number of affected clusters. While the samples do show a tendency to cluster based on sex, the effect of sex is not strong enough to reach significance. The lack of sex effect is consistent with the controversial information reported for COPD patients [40, 41]. However, we are not excluding the possibility of sex-related differences in our groups. Similarly, due to the limitation in number of mouse models available for different strains, we were unable to examine the effect of the strain and are, therefore, not excluding a possibility of strain differences.
Based on our data, we suggest that two main processes are involved in the response to cigarette smoke. At low TPM concentration, cigarette smoke exposure triggers a xenobiotic and detoxification response. Of note, the activation of detoxification genes was conserved among all datasets regardless of TPM concentration. At higher TPM concentrations, however, we also observed effects on clusters associated with immune inflammatory processes. Our data are in agreement with reports by March et al. [42] and Hodge-Bell et al. [43], documenting that cigarette smoke-induced inflammation is a function of TPM. However, the association between TPM levels and changes in gene expression patterns has not been reported previously. It is plausible that: 1) these immune inflammatory processes render protection if the xenobiotic and detoxification responses are overwhelmed; or 2) that the damage associated with higher TPM concentrations drives immune inflammatory processes. Ultimately, our data suggest that TPM concentration acts as a rheostat regulating the intensity and nature of the response to cigarette smoke.
To evaluate the relevance of these observations in humans, we compared all the mouse models to three distinct human cohorts. The goal of this comparison was to determine if functional interacting networks derived from mice exposed to cigarette smoke were also regulated in humans and, ultimately, find the smoke exposure system that most accurately reflects what is observed clinically. Similar to mice, we observed marked differences in the regulation of clusters between the clinical cohorts. Four clusters were regulated in the UBC cohort, whereas Groningen and Laval cohorts showed regulation of almost all clusters. The UBC cohort clustered with the mice exposed to the lowest TPM, while Laval and Groningen cohorts clustered with mice exposed to higher levels of TPM. No significant correlation was found between number of regulated clusters and pack-years. Factors that contribute to the marked differences between clinical cohorts are currently not well understood and may include factors such as socioeconomic status, environmental exposure, age, sex and body mass index to mention a few. What these findings ultimately show is that there is no ideal experimental smoke exposure protocol and that different models may relate more closely to certain cohorts. Clearly, further research is required to validate animal models against the clinical conditions.
There are limitations to the present study that should be noted. The relatively low number of available models can limit our ability to detect true associations that have small effect sizes. For example, C57BL/6 was the only strain that was used in multiple datasets (n=3). Unfortunately, one of these datasets did not provide information about TPM and was excluded from the full analysis. In future, it will be important to incorporate more models with repeated characteristics such as strain and sex. The comparison with human data was restricted to clusters derived from mice data containing 55 genes. The differences observed in the regulation of clusters across cohorts do not reflect global gene expression changes induced by smoking in the human lung. Our previous gene expression study revealed 599 transcripts consistently altered by smoking in these three human cohorts [14]. Functional interaction network analysis based on smoking-induced genes in humans is warranted.
In the present study, we found that higher TPM concentrations skewed the gene expression response towards an inflammatory phenotype and positively affected the intensity of the response in mice. Our data further suggest that mouse models mimic the molecular behaviour of conserved genes affected by cigarette smoking in human tissues. However, there are marked differences between gene expression profiles among animal models and human cohorts. Hence, animal models may be used to study biological mechanisms that contribute to cigarette smoke-induced inflammation and damage/repair responses, but the translation of these findings to human disease requires validation that these pathways are activated in the corresponding patient cohort.
Acknowledgements
We are thankful to the members of the Merck-Laval-UBC-Groningen Lung eQTL consortium. We would like to thank the staff at the Respiratory Health Network Tissue Bank of the FRQS (Laval University, Quebec City, Canada) for their valuable assistance with the lung eQTL dataset.
Footnotes
This article has supplementary material available from openres.ersjournals.com
Support statement: This work was supported by the Canadian Institutes of Health Research (CIHR) (MOP-64390 and MOP-142353). A. Dvorkin-Gheva is supported by a postdoctoral fellowship from the Canadian Breast Cancer Foundation (CBCF), Ontario chapter. G. Vanderstocken is supported by the Canadian Respiratory Research Network (CRRN). Y. Bossé holds a Canada Research Chair in Genomics of Heart and Lung Diseases. The lung eQTL study at Laval University was supported by the Fondation de l'Institut universitaire de cardiologie et de pneumologie de Québec, the Respiratory Health Network of the FRQS, the Canadian Institutes of Health Research (MOP-123369), and the Cancer Research Society and Read for the Cure. Funding information of this article has been deposited with the Open Funder Registry.
Conflict of interest: None declared.
References
- 1.Centers for Disease Control and Prevention, National Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health. How Tobacco Smoke Causes Disease: The Biology and Behavioral Basis for Smoking-Attributable Disease: A Report of the Surgeon General Atlanta, Centers for Disease Control and Prevention, 2010. [Google Scholar]
- 2.Ng M, Freeman MK, Fleming TD, et al. . Smoking prevalence and cigarette consumption in 187 countries, 1980–2012. JAMA 2014; 311: 183–192. [DOI] [PubMed] [Google Scholar]
- 3.Toy EL, Gallagher KF, Stanley EL, et al. . The economic impact of exacerbations of chronic obstructive pulmonary disease and exacerbation definition: a review. COPD 2010; 7: 214–228. [DOI] [PubMed] [Google Scholar]
- 4.Yasothan U, Kar S. Therapies for COPD. Nat Rev Drug Discov 2008; 7: 285–286. [DOI] [PubMed] [Google Scholar]
- 5.Stämpfli MR, Anderson GP. How cigarette smoke skews immune responses to promote infection, lung disease and cancer. Nat Rev Immunol 2009; 9: 377–384. [DOI] [PubMed] [Google Scholar]
- 6.Morissette MC, Lamontagne M, Bérubé J-C, et al. . Impact of cigarette smoke on the human and mouse lungs: a gene-expression comparison study. PLoS One 2014; 9: e92498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shi Y, Cao J, Gao J, et al. . Retinoic acid-related orphan receptor-α is induced in the setting of DNA damage and promotes pulmonary emphysema. Am J Respir Crit Care Med 2012; 186: 412–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gebel S, Diehl S, Pype J, et al. . The transcriptome of Nrf2-/- mice provides evidence for impaired cell cycle progression in the development of cigarette smoke-induced emphysematous changes. Toxicol Sci 2010; 115: 238–252. [DOI] [PubMed] [Google Scholar]
- 9.Rangasamy T, Misra V, Zhen L, et al. . Cigarette smoke-induced emphysema in A/J mice is associated with pulmonary oxidative stress, apoptosis of lung cells, and global alterations in gene expression. Am J Physiol Lung Cell Mol Physiol 2009; 296: L888–L900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.John-Schuster G, Hager K, Conlon TM, et al. . Cigarette smoke-induced iBALT mediates macrophage activation in a B cell-dependent manner in COPD. AJP Lung Cell Mol Physiol 2014; 307: L692–L706. [DOI] [PubMed] [Google Scholar]
- 11.Podowski M, Calvi C, Metzger S, et al. . Angiotensin receptor blockade attenuates cigarette smoke-induced lung injury and rescues lung architecture in mice. J Clin Invest 2012; 122: 229–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McCall MN, Bolstad BM, Irizarry RA. Frozen robust multiarray analysis (fRMA). Biostatistics 2010; 11: 242–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Benito M, Parker J, Du Q, et al. . Adjustment of systematic microarray data biases. Bioinformatics 2004; 20: 105–114. [DOI] [PubMed] [Google Scholar]
- 14.Bossé Y, Postma DS, Sin DD, et al. . Molecular signature of smoking in human lung tissues. Cancer Res 2012; 72: 3753–3763. [DOI] [PubMed] [Google Scholar]
- 15.Wettenhall JM, Smyth GK. limmaGUI: a graphical user interface for linear modeling of microarray data. Bioinformatics 2004; 20: 3705–3706. [DOI] [PubMed] [Google Scholar]
- 16.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B 1995; 57: 289–300. [Google Scholar]
- 17.Shannon P, Markiel A, Ozier O, et al. . Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 2003; 13: 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu G, Stein L. A network module-based method for identifying cancer prognostic signatures. Genome Biol 2012; 13: R112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Heintzman ND, Hon GC, Hawkins RD, et al. . Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 2009; 459: 108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van't Veer LJ, Dai H, van de Vijver MJ, et al. . Gene expression profiling predicts clinical outcome of breast cancer. Nature 2002; 415: 530–536. [DOI] [PubMed] [Google Scholar]
- 21.Newman ME. Modularity and community structure in networks. Proc Natl Acad Sci USA 2006; 103: 8577–8582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Subramanian A, Tamayo P, Mootha VK, et al. . Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA 2005; 102: 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ma B, Zhu Z, Homer RJ, et al. . The C10/CCL6 chemokine and CCR1 play critical roles in the pathogenesis of IL-13-induced inflammation and remodeling. J Immunol 2004; 172: 1872–1881. [DOI] [PubMed] [Google Scholar]
- 24.Gear ARL, Camerini D. Platelet chemokines and chemokine receptors: linking hemostasis, inflammation, and host defense. Microcirculation 2010; 10: 335–350. [DOI] [PubMed] [Google Scholar]
- 25.Ripoll VM, Irvine KM, Ravasi T, et al. . Gpnmb is induced in macrophages by IFN-gamma and lipopolysaccharide and acts as a feedback regulator of proinflammatory responses. J Immunol 2007; 178: 6557–6566. [DOI] [PubMed] [Google Scholar]
- 26.Gao P, Simpson JL, Zhang J, et al. . Galectin-3: its role in asthma and potential as an anti-inflammatory target. Respir Res 2013; 14: 136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lagente V, Le Quement C, Boichot E. Macrophage metalloelastase (MMP-12) as a target for inflammatory respiratory diseases. Expert Opin Ther Targets 2009; 13: 287–295. [DOI] [PubMed] [Google Scholar]
- 28.Colonna M. TREMs in the immune system and beyond. Nat Rev Immunol 2003; 3: 445–453. [DOI] [PubMed] [Google Scholar]
- 29.Shen Y, Ma J, Yan R, et al. . Impaired self-renewal and increased colitis and dysplastic lesions in colonic mucosa of AKR1B8-deficient mice. Clin Cancer Res 2015; 21: 1466–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gottfried E, Kunz-Schughart LA, Weber A, et al. . Expression of CD68 in non-myeloid cell types. Scand J Immunol 2008; 67: 453–463. [DOI] [PubMed] [Google Scholar]
- 31.Nebert DW, Dalton TP, Okey AB, et al. . Role of aryl hydrocarbon receptor-mediated induction of the CYP1 enzymes in environmental toxicity and cancer. J Biol Chem 2004; 279: 23847–23850. [DOI] [PubMed] [Google Scholar]
- 32.Golovatch P, Mercer BA, Lemaître V, et al. . Role for cathepsin K in emphysema in smoke-exposed guinea pigs. Exp Lung Res 2015; 35: 631–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nikota JK, Shen P, Morissette MC, et al. . Cigarette smoke primes the pulmonary environment to IL-1/CXCR-2-dependent nontypeable Haemophilus influenzae-exacerbated neutrophilia in mice. J Immunol 2014; 193: 3134–3145. [DOI] [PubMed] [Google Scholar]
- 34.Pilette C, Colinet B, Kiss R, et al. . Increased galectin-3 expression and intra-epithelial neutrophils in small airways in severe COPD. Eur Respir J 2007; 29: 914–922. [DOI] [PubMed] [Google Scholar]
- 35.Mocchegiani E, Giacconi R, Costarelli L. Metalloproteases/anti-metalloproteases imbalance in chronic obstructive pulmonary disease: genetic factors and treatment implications. Curr Opin Pulm Med 2011; 17: Suppl 1, S11–S19. [DOI] [PubMed] [Google Scholar]
- 36.Wilson SM, Shen P, Rider CF, et al. . Selective prostacyclin receptor agonism augments glucocorticoid-induced gene expression in human bronchial epithelial cells. J Immunol 2009; 183: 6788–6799. [DOI] [PubMed] [Google Scholar]
- 37.Knackstedt LA, LaRowe S, Mardikian P, et al. . The role of cystine-glutamate exchange in nicotine dependence in rats and humans. Biol Psychiatry 2009; 65: 841–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Koth LL, Cambier CJ, Ellwanger A, et al. . DAP12 is required for macrophage recruitment to the lung in response to cigarette smoke and chemotaxis toward CCL2. J Immunol 2010; 184: 6522–6528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tan WC, Sin DD, Bourbeau J, et al. . Characteristics of COPD in never-smokers and ever-smokers in the general population: results from the CanCOLD study. Thorax 2015; 70: 822–829. [DOI] [PubMed] [Google Scholar]
- 40.Sørheim I-C, Johannessen A, Gulsvik A, et al. . Gender differences in COPD: are women more susceptible to smoking effects than men? Thorax 2010; 65: 480–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kirkpatrick DP, Dransfield MT. Racial and sex differences in chronic obstructive pulmonary disease susceptibility, diagnosis, and treatment. Curr Opin Pulm Med 2009; 15: 100–104. [DOI] [PubMed] [Google Scholar]
- 42.March TH, Wilder JA, Esparza DC, et al. . Modulators of cigarette smoke-induced pulmonary emphysema in A/J mice. Toxicol Sci 2006; 92: 545–559. [DOI] [PubMed] [Google Scholar]
- 43.Hodge-Bell KC, Lee KM, Renne RA, et al. . Pulmonary inflammation in mice exposed to mainstream cigarette smoke. Inhal Toxicol 2007; 19: 361–376. [DOI] [PubMed] [Google Scholar]