

Abstract
Most proteases are synthesized as inactive precursors which are processed by proteolytic cleavage into a mature active form, allowing regulation of their proteolytic activity. The activation of the glutamic-acid-specific extracellular metalloprotease (Mpr) of Bacillus subtilis has been examined. Analysis of Mpr processing in defined protease-deficient mutants by activity assay and Western blotting revealed that the extracellular protease Bpr is required for Mpr processing. pro-Mpr remained a precursor form in bpr-deficient strains, and glutamic-acid-specific proteolytic activity conferred by Mpr was not activated in bpr-deficient strains. Further, purified pro-Mpr was processed to an active form by purified Bpr protease in vitro. We conclude that Mpr is activated by Bpr in vivo, and that heteroprocessing, rather than autoprocessing, is the major mechanism of Mpr processing in vivo. Exchange of glutamic acid for serine in the cleavage site of Mpr (S93E) allowed processing of Mpr into its mature form, regardless of the presence of other extracellular proteases, including Bpr. Thus, a single amino acid change is sufficient to convert the Mpr processing mechanism from heteroprocessing to autoprocessing.
The gram-positive, spore-forming bacterium Bacillus subtilis secretes at least eight extracellular proteases at the end of the exponential phase of growth (21). Two major extracellular proteases are produced during the onset of sporulation, the alkaline serine protease subtilisin (AprE) (18, 38) and a neutral protease (NprE) (40). The minor extracellular proteases include Epr (2, 27), bacillopeptidase F (Bpr) (30, 31, 39), metalloprotease (Mpr) (20, 23, 28), neutral protease B (NprB) (35), wall-associated extracellular protease (WprA) (17), and Vpr (29). Expression of these proteolytic enzymes seems to be tightly regulated (33), and their major function is thought to be supplying amino acids for growth via degradation of extracellular proteins. However, some physiological roles have been discussed. For example, it has been suggested that WprA is involved in proteolysis of misfolded proteins during membrane translocation (32).
Many bacterial proteolytic enzymes are synthesized as inactive precursors, or zymogens, to prevent unwanted protein degradation and to enable spatial and temporal regulation of proteolytic activity (12). Biochemical studies of the activation mechanism of individual proteases have provided insights into their physiological functions. Every extracellular protease in B. subtilis is synthesized as a preproenzyme in the cytoplasm and is processed to a mature enzyme in the extracellular milieu. The activation mechanism of B. subtilis subtilisin has been well studied. The zymogen form of subtilisin is converted to an active form via intramolecular autoprocessing (19) in the extracellular space (9). The propeptide has been shown to guide the proper folding of subtilisin in vivo and in vitro (34). Most extracellular proteases in B. subtilis seem to be activated by autoprocessing. However, the Mpr protease might be an exception in that it is not able to activate itself. Reportedly, Mpr has a high substrate specificity, recognizing a glutamic acid residue as a P1 cleavage site (20). However, a glutamic acid residue is not present in the Mpr propeptide cleavage site, suggesting that it is not autoprocessed. Many glutamic-acid-specific proteases such as V8 protease (5) and SPase (43) have been found in Staphylococcus aureus and some nonpathogenic bacteria (10, 13, 42); however, the processing mechanisms of glutamyl endopeptidases have not been clearly defined.
Here, we report that Mpr can be specifically activated by other extracellular proteases and that Mpr can be converted to an autoproteolytic protease by placing a glutamic acid residue in the propeptide cleavage site. The molecular evolution of protease activation mechanisms in B. subtilis is also discussed.
MATERIALS AND METHODS
Bacterial strains, plasmids, media, and growth conditions.
The bacterial strains and plasmids used in this study are listed in Table 1. Cultivation was carried out in 200 ml of Luria-Bertani medium in a 2-liter flask at 37°C with shaking (200 rpm). Culture supernatants for examining protease expression and activity, as well as purification, were collected after 12 to 36 h of growth as described above. Where necessary, media were supplemented with 100 μg of ampicillin (Amp; purchased from DUCHEFA)/ml for Escherichia coli and with 50 μg of kanamycin (Kan; purchased from DUCHEFA)/ml and 15 μg of tetracycline (Tc; purchased from Sigma)/ml for B. subtilis.
TABLE 1.
Strains and plasmids used in this study
| Strain or plasmid | Genotype and genetic markersa | Comment(s) | Reference or source |
|---|---|---|---|
| Strains | |||
| B. subtilis 168 | trpC2 | Wild type | 36 |
| B. subtilis PB100 | bpr | bpr-deficient B. subtilis 168 | This study |
| B. subtilis DB104 | nprE aprE | nprE- and aprE-deficient B. subtilis 168 | 4 |
| B. subtilis SB300 | nprE aprE epr | epr-deficient DB104 | This study |
| B. subtilis PB300 | nprE aprE bpr | bpr-deficient DB104 | This study |
| B. subtilis DB428 | nprE aprE epr bpr | Four-exoprotease gene-deficient B. subtilis 168 | 4 |
| B. subtilis LB700 | nprE aprE epr bpr mpr nprB wprA F−araD139 Δ(ara-leu)7696 galE15 | Seven-exoprotease gene-deficient B. subtilis 168 | 15 |
| E. coli MC1061 | galK16 ΔlacX74 rpsL (Strr) hsdR2 (rK− mK+) mcrA mcrB1 | Host for DNA manipulation | 25 |
| Plasmids | |||
| pSM704 | Kanr; Bacillus ori | Shuttle expression vector with subtilisin promoter and signal sequence | 15 |
| pSP704 | Kanr Ampr; Bacillus ori, E. coli ori | Shuttle expression vector with subtilisin promoter | This study |
| pMpr | Kanr Ampr; Bacillus ori, E. coli ori | Mpr secretion plasmid | This study |
| pMprH6 | Kanr Ampr; Bacillus ori, E. coli ori | C-terminally His6-tagged Mpr secretion plasmid | This study |
| pMprH6(S93E) | Kanr Ampr; Bacillus ori, E. coli ori | pMprH6 plasmid harboring S93E mutation | This study |
| pBpr | Kanr Ampr; Bacillus ori, E. coli ori | Bpr secretion plasmid | This study |
| pUC19 | Ampr; E. coli ori | Cloning vector | 41 |
| pUCTV2 | Kanr Ampr; Bacillus ts-ori, E. coli ori | Temperature sensitive suicide vector | 37 |
| pDEPR | Tcr Ampr; Bacillus ts-ori, E. coli ori | Gene replacement vector with epr::tet | This study |
| pDBPR | Tcr Ampr; Bacillus ts-ori, E. coli ori | Gene replacement vector with bpr::tet | This study |
ori, origin of replication; ts-ori, temperature-sensitive ori.
DNA manipulations and PCR.
All DNA manipulations were carried out by standard protocols (25). DNA restriction digestions were performed as recommended by the supplier (New England Biolabs, Inc.). E. coli plasmids were isolated with the Qiaprep spin miniprep kit (QIAGEN). Genomic DNA from B. subtilis was prepared with the Genomic DNA purification kit (Promega). Competent cells of B. subtilis and E. coli were prepared and transformed by standard protocols (8, 24). PCR was carried out following standard procedures with LA-Taq DNA polymerase (TaKaRa).
Construction of mpr and bpr expression vectors.
The subtilisin promoter from pSM704 (15) was PCR amplified with two primers, SP-F (5′-GCCGTCTGTACGTTCCTTAAG) and SP-R (5′-TCTGAGCTCTCTCCTTTTAAAAAA). The EcoRI- and SacI-digested PCR product was subcloned into EcoRI-SacI-digested pSM704 for the removal of the signal sequences. The resulting plasmid was termed pSP704. The mpr gene was amplified from genomic DNA of B. subtilis 168 with primers Mpr-F (5′-AGGGAGCTCACAAAATGAAAT) and Mpr-R (5′-ATGGATCCTTATTGCCCAATATTGAATAT). The mpr PCR product (1.0 kb) and pSP704, both digested with SacI and BamHI, were ligated to create pMpr, where expression of the mpr gene is controlled by the subtilisin promoter. To facilitate detection of the Mpr protein, we introduced a C-terminal His tag in the mpr gene product as follows: the mpr gene was amplified with Mpr-F and MprH6-R (5′-ATGGATCCTTAATGATGATGATGATGATGACTACTTCCTTGATTTGCCCAATATTGAAT), and the SacI- and BamHI-digested PCR product was inserted into SacI- and BamHI-digested pSP704 to make pMprH6.
The bpr gene was amplified from genomic DNA of B. subtilis 168 with primers Bpr-F (5′-GGGGAGCTCAATGAGGAAAAAAACGAA AAACAGACTCAT)and Bpr-R (5′-TTGGATCCACTTAATTTTCTGTGTTCATATTAAGTTTT). The bpr PCR product (1.0 kb) and pSP704, both digested with SacI and BamHI, were ligated to make pBpr.
Construction of gene disruption vectors.
bpr (4.5 kb) was amplified from chromosomal DNA of B. subtilis 168 with primers BPR-EF (5′-GGAATTCAAAAAAACGAAAAACAGACTCATCAG) and BPR-ER (5′-GGAATTCCGCCCGGATCAGGGAACTCTGTATCCC). epr (1.9 kb) was amplified with primers EPR-EF (5′-AGAGAATTCAATCATGAAAAACAT) and EPR-ER (5′-GGTGAATTCTAGAAGGTTTTTGGT). The PCR products were digested with EcoRI and cloned in EcoRI-digested pUC19 to make pUC19-Epr and pUC19-Bpr, respectively. PvuII-digested pUC19-Epr and Bst1107I- and EcoRV-digested pUC19-Bpr were each ligated with XhoI linker to make pUC19-EprX and pUC19-BprX. The tetracycline resistance gene (tet; 1.7kb) excised from pUCTV2 (37) with XhoI was inserted into XhoI-digested pUC19-EprX and pUC19-BprX to make pUC19-EprTet and pUC19-BprTet, respectively. DNA fragments obtained from EcoRI-digested pUC19-EprTet and pUC19-BprTet were cloned in the EcoRI site of tet gene-deleted pUCTV2, to construct gene disruption plasmids pDBPR and pDEPR, respectively.
Inactivation of bpr and epr in B. subtilis DB104.
Strain DB104 was transformed with plasmid pDBPR or pDEPR, and tetracycline-resistant clones were selected at 30°C and then grown for 36 h at 42°C (15). Five tetracycline-resistant colonies were randomly selected for each construct, and PCR was used to confirm that the strains carried the inactivated bpr or epr gene (data not shown). Primers BprP-F (5′-TAACAAACAGATAATTAGACCCATTTA) and Tet-R (5′-TAAAATTTGGTTGTGTCGTAAATTCGA) plus BprP-R (5′-CTTGACGTCTTTCAGATTGTCATCGGA) and Tet-F (5′-TGGCTGGTTACCTTGATGTATATAAA) were used to confirm the inactivation of the bpr gene. Primers BprP-F and BprP-R are located outside the cloned sequence in the pDBPR vector, and primers Tet-R and Tet-F are located in the tet gene. Inactivation of the epr gene with the tet gene was confirmed with primer pairs EprP-F (5′-ATCCGAGCTTATCGGCCCACTCGTTCC) and Tet-R and EprP-R (5′-CTGCAGGTCGACGACGGAGCATGAGAA) and Tet-F. epr- and bpr-inactivated DB104 strains were designated SB300 and PB300, respectively.
Site-directed mutagenesis.
To introduce the point mutation S93E at the propeptide cleavage site in pro-Mpr, PCR mutagenesis was carried out with pMprH6 as a template and primers S93E-F (5′-CAAACCTTACAGCCTGAGAGCATTATCGGAACT) and S93E-R (5′-AGTTCCGATAATGCTCTCAGGCTGTAAGGTTTG). The resulting PCR product was digested with DpnI to remove parental pMprH6 and subsequently transformed into E. coli XL-1 Blue. The resulting plasmid was named pMprH6(S93E), and the presence of the S93E point mutation was confirmed by DNA sequencing.
SDS-PAGE and immunoblot analysis.
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was carried out by Laemmli's method (14). Western blot analysis was performed by standard protocols (25). A His probe (Santa Cruz Biotechnology) and anti-rabbit immunolgobulin G-peroxidase conjugate (Bio-Rad) were used as primary and secondary antibodies, to detect His6-tagged Mpr. The light-emitting nonradioactive ECL kit (Amersham) was used for signal detection.
Plate assay for determination of proteolytic activity.
The culture supernatants from 12- to 36-h spent-culture broths of B. subtilis were placed on Luria-Bertani agar plates containing 3% (wt/vol) skim milk and Kan (25 μg/ml). Proteolytic activity was determined by observing the clear zone around colonies after 12 h of incubation at 37°C.
Determination of glutamic-acid-specific proteolytic activity.
Glutamic-acid-specific protease activity was determined by a slightly modified chromogenic assay (20). Ten microliters of culture supernatant was added to 90 μl of reaction solution (50 mM Tris-Cl [pH 7.5], 2 mM CaCl2, 1 mM acetyl-Glu-p-nitroanilide [Bachem], 2% dimethylformamide). After incubation of the culture supernatant for 8 h at 37°C, the absorbance was measured at 410 nm (Bio-Rad microwell plate reader, model 550).
Protein purification.
A 36-h broth culture of B. subtilis LB700 carrying either pBpr or pMpr was centrifuged (6,000 × g; 30 min), and the supernatant was obtained. Ammonium sulfate was added to 80% saturation. After 4 h of being stirred on ice, the precipitate was collected by centrifugation (8,000 × g; 30 min at 4°C), redissolved in 20 mM Tris-HCl (pH 8.0), and dialyzed overnight against the same buffer at 4°C.
For the purification of Bpr, the dialysate was concentrated with an YM-3 membrane (Amicon Corp.) and applied to a DEAE Sepharose CL-6B (Pharmacia) column equilibrated with 20 mM Tris-HCl prior to Mono-Q (Pharmacia) chromatography. Bpr was eluted from both columns by an NaCl gradient.
Purification of pro-Mpr was carried out as follows. The dialysate was applied to a DEAE Sepharose CL-6B (Pharmacia) column equilibrated with 20 mM Tris-HCl (pH 8.0). pro-Mpr was eluted from the column by an NaCl gradient. The active fractions were dialyzed against 20 mM Tris-HCl (pH 8.0), concentrated with an YM-3 membrane, loaded onto a Sephacryl S-100 (Pharmacia) column, and fractionated.
The purification of His6-tagged pro-Mpr(S93E) was performed as follows. A 36-h broth culture of B. subtilis LB700 carrying pMprH6(S93E) was centrifuged (6,000 × g; 30 min), and the supernatant was applied to a Ni-nitrilotriacetic acid superflow (QIAGEN) column. pro-Mpr(S93E) was eluted with imidazole buffer. The protein concentration was measured by Lowry's method (16).
In vitro processing of pro-Mpr by Bpr.
Purified Bpr and pro-Mpr were mixed at different ratios with 20 mM Tris-Cl (pH 7.5) in a total volume of 100 μl. After incubation at 37°C for 30 min, the processing of pro-Mpr was analyzed by SDS-PAGE, and Mpr activities of mixtures were measured as above described.
In vitro processing of pro-Mpr(S93E).
A mixture of purified pro-Mpr(S93E) with 20 mM Tris-Cl (pH 7.5) in a total volume of 100 μl was incubated for 4 to 12 h at 37°C. The reaction was stopped by the addition of 10 μl of trichloroacetic acid. After centrifugation, the precipitant was analyzed by immunoblotting.
Protein sequencing.
The protein on a SDS gel was electroblotted onto a polyvinylidene difluoride membrane. The protein band was excised from the Coomassie brilliant blue-strained polyvinylidene difluoride membrane and subjected to N-terminal amino acid sequence analysis.
RESULTS
Propeptide cleavage of metalloprotease (Mpr) by heteroprocessing in B. subtilis.
The signal peptide and propeptide sequences of Mpr are reported to be composed of 34 and 59 amino acids, respectively, (28), and a previous study showed that the propeptide cleavage site should be between Ser93-Ser94 in pro-Mpr (see Fig. 6). It was reported that proteolytic activity of Mpr is highly specific to glutamic acid (20). Therefore, we thought that Mpr was unlikely to be processed by a self-cleaving mechanism like that of subtilisin. To investigate Mpr processing, we overexpressed Mpr in multiple-protease-deficient strains of B. subtilis and examined the secreted proteins in supernatants of 36-h cultures by SDS-PAGE for patterns of Mpr processing. We noted a remarkable difference in the molecular masses of Mpr proteins (overexpressed from pMpr) between strain DB104, which lacks the NprE and AprE extracellular proteases, and strain DB428, which lacks the Epr and Bpr extracellular proteases as well as NprE and AprE. While a 33-kDa protein was overexpressed in DB428, a 28-kDa protein was overexpressed in DB104 (see Fig. 2A). N-terminal amino acid sequences of the 33- and 28-kDa proteins indicated (see Fig. 6) were determined to be AENPQ and SIIGT, respectively. The molecular masses of these proteins correspond well to those of pro-Mpr and mature Mpr, suggesting that Bpr and/or Epr is a strong candidate(s) in the processing of pro-Mpr and further, that pro-Mpr was activated not by itself (autoprocessing), but by other proteolytic activities (heteroprocessing).
FIG. 6.

The deduced amino acid sequence of Mpr (A) and heteroprocessing and autoprocessing of Mpr in the extracellular milieu of B. subtilis (B). (A) Asterisks indicate the experimentally determined first amino acid residues of pro-Mpr and mature Mpr, respectively. Glutamic acid residues are underlined. (B) The initial translation product of the mpr transcript consists of a signal sequence (Pre; amino acids 1 to 34), a propeptide region (Pro; amino acids 35 to 93), and the mature enzyme (Mature; amino acids 94 to 313). Lined and dashed arrows indicate heteroprocessing by other extracellular proteases, and autoprocessing, respectively.
FIG. 2.

Characterization of processing and activation of Mpr in different B. subtilis strains. SDS-PAGE analysis (A), Western blotting (B), and artificial substrate colorimetric assay (C) of the culture supernatants of different B. subtilis strains carrying pMprH6. Lanes 1, B. subtilis 168; lanes 2, DB104; lanes 3, SB300; lanes 4, PB300; lanes 5, DB428; lanes 6, LB700. p, precursor; m, mature; Abs410nm, absorbance at 410 nm.
Activation and processing of pro-Mpr in vivo by Bpr and not by Epr.
To investigate whether Epr, Bpr, or both was required for the processing of pro-Mpr, we expressed Mpr and MprH6 in different multiple-protease-deficient B. subtilis strains including DB104 (aprE nprE), SB300 (aprE nprE epr), PB300 (aprE nprE bpr), and DB428 (aprE nprE epr bpr) and detected the proteolytic activity on skim milk-agar plates. We observed a prominent clear zone around colonies of DB104 and epr-deficient SB300 strains but not around colonies of bpr-deficient PB300, DB428, and LB700 (aprE nprE epr bpr mpr nprB wprA) strains (Fig. 1) at 36 h of growth. We examined pro-Mpr secretion in DB104 and SB300 strains by SDS-PAGE (Fig. 2 A) and immunoblot (Fig. 2B) analyses and found that pro-Mpr was completely processed into the mature form. However, in bpr-deficient strains pro-Mpr protein remained throughout the time course.
FIG. 1.

Plate assay of the activation of Mpr and MprH6(S93E) in different multiple extracellular protease gene-deficient B. subtilis strains. The activation of pro-Mpr was monitored by clear zones around B. subtilis colonies oversecreting Mpr for the indicated time. Lane 1, B. subtilis strain carrying pSP704 (negative control); lane 2, pMpr; lane 3, pMprH6; lane 4, pMprH6(S93E).
To confirm that Mpr was processed into an active form, we tested whether the culture supernatants of different strains were able to hydrolyze a synthetic chromogenic substrate of Mpr, acetyl-Glu-p-nitroanilide. The glutamic acid-specific proteolytic activity of Mpr was observed in the culture supernatants of SB300 and DB104 strains harboring pMpr (Fig. 2C). However, in the culture supernatants of bpr-deficient PB300, DB428, and LB700 strains carrying pMpr, no glutamic acid-specific proteolytic activity was observed (Fig. 2C).
Activation of pro-Mpr by Bpr in vitro.
We purified both Bpr and pro-Mpr from culture supernatants of overexpressing strains to examine pro-Mpr processing in vitro. We observed that purified Bpr activated purified pro-Mpr in vitro as detected by the production of the mature form of Mpr by SDS-PAGE analysis (Fig. 3A) and by the activation of Mpr in the colorimetric assay for glutamic-acid-specific proteolytic activity (Fig. 3B). From these results, we conclude that Bpr specifically converts inactive pro-Mpr to active Mpr through proteolytic processing of the Mpr propeptide.
FIG. 3.

In vitro processing of pro-Mpr by Bpr. SDS-PAGE analysis (A) and artificial substrate colorimetric assay (B) of purified pro-Mpr processed by purified Bpr in vitro. Purified pro-Mpr (3.3 μg) was added to each mixture (100 μl) in the results shown in lanes 2 to 5. Lane 1, 4.5 μg of purified Bpr; lane 2, 4.5 μg of Bpr; lane 3, 1.5 μg of Bpr; lane 4, 0.75 μg of Bpr; lane 5, pro-Mpr. b, Bpr; p, pro-Mpr; m, mature Mpr; Abs410nm, absorbance at 410 nm..
Activation of pro-Mpr(S93E) mutant in vivo by autoprocessing.
Because Mpr was reported to have glutamic acid-specific endopeptidase activity (20), the propeptide cleavage site of Mpr was mutated to include a glutamic acid residue (mutation S93E) to determine whether Mpr was capable of autoprocessing. Prominent clear zones around colonies of all strains expressing this mutant protein were observed in the skim milk-agar plate assay even after only 12 h of growth (Fig. 1). SDS-PAGE (Fig. 4A) and immunoblot (Fig. 4B) analyses showed that the mutant pro-Mpr was largely processed into an active form from 12 h of growth, even in bpr-deficient strains. The Mpr(S93E) mutant displayed proteolytic activity similar to or greater than that of wild-type Mpr in the colorimetric assay, suggesting that S93E mutant may be activated more efficiently by an autoprocessing mechanism (Fig. 4C).
FIG. 4.

Characterization of processing and activation of Mpr(S93E) in different B. subtilis strains. SDS-PAGE analysis (A), Western blotting (B), and artificial substrate colorimetric assay (C) of the culture supernatants of different B. subtilis strains carrying pMprH6(S93E). Lane 1, B. subtilis 168; lane 2, DB104; lane 3, SB300; lane 4, PB300; lane 5, DB428; lane 6, LB700. p, precursor; m, mature.
Activation of pro-Mpr(S93E) mutant in vitro by autoprocessing.
We tried to purify pro-Mpr(S93E) from culture supernatants of overexpressing strains to examine pro-Mpr(S93E) processing in vitro. We could obtain a complex consisting of pro-Mpr(S93E) and mature Mpr(S93E) because secreted pro-Mpr(S93E) is immediately converted into the mature form during the purification process. However, we could observe that purified pro-Mpr (S93E) was activated by autoprocessing in vitro throughout the time course as detected by the increase of the mature form of Mpr(S93E) in immunoblot analysis (Fig. 5). Thus, we conclude that the S93E mutation allows efficient autoprocessing of Mpr.
FIG. 5.
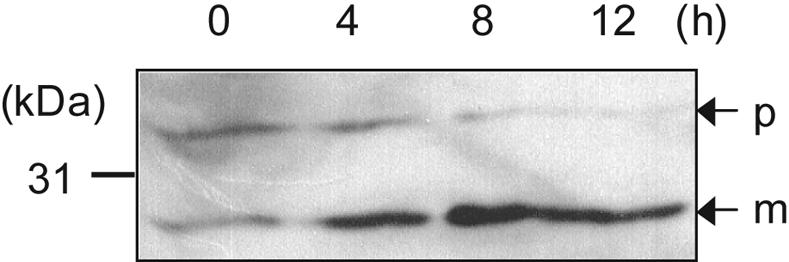
In vitro autoprocessing of Mpr(S93E). Time course processing of purified pro-Mpr(S93E) was analyzed by immunoblotting. The numbers (0, 4, 8, and 12) indicate the incubation time in hours. p, precursor; m, mature.
DISCUSSION
The activation mechanism of many extracellular proteases in B. subtilis has been investigated. Subtilisin is well characterized and known to be activated by autoprocessing (34). Most extracellular proteases in B. subtilis like subtilisin, have been thought to be autoprocessed. It was reported that Bpr might be susceptible to autolysis (22). We observed that Epr and NprB were each fully processed when expressed from a plasmid in the seven-protease-deficient LB700 strain, indicating that they might be activated by autoprocessing (data not shown). In the case of Mpr, however, the recent report that Mpr is a glutamic-acid-specific endopeptidase (20) and that glutamic acid is absent in the propeptide cleavage site of pro-Mpr led us to consider whether Mpr was activated by another protease.
Proteolytic activity was observed on skim milk-agar plates in an aprE nprE mutant carrying pMpr but not in an aprE nprE epr bpr mutant carrying pMpr, suggesting that Mpr might require Epr or Bpr for activation rather than being autoprocessed. To identify which was required for Mpr activation, we investigated pro-Mpr processing in bpr-deficient and epr-deficient derivatives of DB104 (PB300 and SB300, respectively) (Table 1). The skim milk plate assay indicated that bpr, but not epr, was required for Mpr processing (Fig. 1).
This finding was confirmed by SDS-PAGE analysis with Coomassie blue staining, demonstrating that pro-Mpr (33 kDa) was accumulated and not converted into the mature form (28 kDa) in culture supernatants of bpr-deficient strains carrying pMprH6 (Fig. 2A). However, a 28-kDa form could be detected in bpr-deficient strains PB300 and DB428 by immunoblotting (Fig. 2B). To determine whether pro-Mpr was properly processed in the bpr-deficient strains or nonspecifically processed by other proteolytic activities to an apparently mature size, we examined the glutamic-acid-specific protease activity in various B. subtilis strains carrying pMprH6. As shown in Fig. 2C, no glutamyl proteolytic activity was observed in the culture supernatants of bpr-deficient strains carrying pMprH6. This suggests that the 28-kDa protein detected by Western blotting in the bpr-deficient strains is an inactive form of MprH6, although it is processed at the N terminus because the C-terminal His6 tag is being detected.
These in vivo findings are supported by in vitro studies demonstrating that purified Bpr processes purified pro-Mpr to a proteolytically active form of the expected size in vitro (Fig. 3). We conclude from these in vivo and in vitro results that Bpr specifically processes pro-Mpr to mature Mpr in vivo. This heteroprocessing activity represents a novel function of bacillopeptidase F in B. subtilis.
We cannot exclude the possibility that subtilisin (AprE) and neutral protease (NprE) also play roles in Mpr processing. We could not determine whether Mpr was activated by AprE and NprE by the skim milk plate assay, because strains that are aprE+ nprE+ (strains 168 and PB100) form clear zones regardless of whether they are overexpressing Mpr. However, we found significant glutamyl proteolytic activities and fully processed (28-kDa) Mpr bands in the culture supernatants of wild-type strain 168 and also in bpr-deficient PB100, suggesting that Apr and/or Npr could be involved in Mpr processing (data not shown). Multiple proteases may allow pro-Mpr to be more rapidly and fully converted into an active form in the extracellular milieu. Epr had little effect on Mpr processing (Fig. 2A and B). However, we observed that Mpr had slightly lower levels of glutamyl proteolytic activity in the absence of Epr than in the presence of Epr (Fig. 2C). This result implies that Epr contributes to Mpr processing.
In pathogenic bacteria, extracellular proteases can enhance bacterial virulence through degradation of critical host proteins and by mimicking the activity of host regulatory proteases that control important zymogen systems (6). In the staphylococcal extracellular proteolytic system, zymogen activation by other extracellular proteases was recently reported (26). In nonpathogenic bacteria such as B. subtilis, extracellular proteases may have other functions. As mentioned, all extracellular proteases of B. subtilis are known to have no effect on either exponential growth in complex medium or sporulation (29). However, production and secretion of extracellular proteases increase during postexponential growth (7). During that period, protein degradation by extracellular proteases supplies nutrients for the cells. Moreover, the extracellular proteases play a role in the activation of other proteins. For example, WprA, Apr, and Vpr are reported to activate an antimicrobial subtilin (3). The present study shows a novel function of extracellular proteases, which is to activate other extracellular proteases in B. subtilis.
Because Mpr is a glutamic-acid-specific endopeptidase that specifically forms a peptide bond after the P1 glutamic acid site (20) but does not have a P1 glutamic acid at its own processing site, we constructed the His6-tagged Mpr(S93E) mutant to determine its effect on processing. Interestingly, the S93E mutant was fully processed into an active form in all the B. subtilis strains tested, regardless of protease deficiencies (Fig. 4), indicating that pro-Mpr(S93E) was activated by autoprocessing. The in vitro studies demonstrated that purified pro-Mpr(S93E) was processed to a proteolytically active form of the expected size in vitro by the time course (Fig. 5). From these in vivo and in vitro results, we conclude that the S93E mutant allows autoprocessing. Moreover, we confirmed that processing of Mpr(S93E) occurs at the proper site between amino acids 93 and 94 (Fig. 6) by N-terminal sequence analysis of the mature form from LB700. As in the results shown in Fig. 4C, there was an appoximately twofold decrease in activity in the LB700 strain that lacks seven exopeptidases. The decrease in Mpr activity in LB700 carrying pMpr(S93E) may be due to the absence of the wprA gene product (LB700 is the only strain examined here which lacks wprA) which is associated with the protein secretion apparatus in B. subtilis (1). Absence of WprA might influence the secretion of Mpr and so affect Mpr processing. Alternatively, the decrease might be due to effects of the multiple mutations on growth rate or other factors that affect the colorimetric assay. Regardless, the processing of pro-Mpr(S93E) in the absence of Bpr and other exoproteases suggests strongly that S93E allows autoprocessing.
With regards to the autoprocessing mechanism, the alteration of serine into glutamic acid residue in pro-Mpr might help its propeptide fit properly into the P1 pocket for processing by self cleavage. This indicates that the propeptide sequence determines whether Mpr undergoes heteroprocessing or autoprocessing. This alteration of the processing mechanism could be explained in the viewpoint of the molecular evolution of proteases. It could be deduced that Mpr was originally activated by autoprocessing like other extracellular proteases and might have evolved to the present form so that it could be activated by other proteases for posttranslocational regulation. Moreover, the role of Mpr propeptide remains to be resolved in future work, since propeptides are known to be involved in guiding the correct folding or inhibition of protease activity (11).
In summary, we report that glutamic-acid-specific Mpr could be specifically activated by a heteroprocessing mechanism in B. subtilis by Bpr protease and also that only a single amino acid exchange at the Mpr processing site switches heteroprocessing to autoprocessing.
Acknowledgments
This work was supported by a research grant from the Korean Agency for Defense Department.
We are grateful to S. L. Wong for providing B. subtilis DB428. We thank F. Meinhardt for supplying plasmid pUCTV2.
REFERENCES
- 1.Bolhuis, A., H. Tjalsma, K. Stephenson, C. R. Harwood, G. Venema, S. Bron, and J. M. van Dijl. 1999. Different mechanisms for thermal inactivation of Bacillus subtilis signal peptidase mutants. J. Biol. Chem. 274:15865-15868. [DOI] [PubMed] [Google Scholar]
- 2.Bruckner, R., O. Shoseyov, and R. H. Doi. 1990. Multiple active forms of a novel serine protease from Bacillus subtilis. Mol. Gen. Genet. 221:486-490. [DOI] [PubMed] [Google Scholar]
- 3.Corvey, C., T. Stein, S. Dusterhus, M. Karas, and K. Entian. 2003. Activation of subtilin precursors by Bacillus subtilis extracellular serine proteases subtilisin (AprE), WprA, and Vpr. Biochem. Biophys. Res. Commun. 304:48-54. [DOI] [PubMed] [Google Scholar]
- 4.Doi, R. H. 1983. Isolation of Bacillus subtilis chromosomal DNA, p. 162-163. In R. L. Rodrirez and R. C. Tait (ed.), Recombinant DNA techniques. Addison-Wesley Publishing Co., Inc., Reading, Mass.
- 5.Drapeau, G. R., Y. Boily, and J. Houmard. 1972. Purification and properties of an extracellular protease of Staphylococcus aureus. J. Biol. Chem. 247:6720-6726. [PubMed] [Google Scholar]
- 6.Gillaspy, A. F., C. Y. Lee, S. Sau, A. L. Cheung, and M. S. Smeltzer. 1998. Factors affecting the collagen binding capacity of Staphylococcus aureus. Infect. Immun. 66:3170-3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gonzalez-Pastor, J. E., E. C. Hobbs, and R. Losick. 2003. Cannibalism by sporulating bacteria. Science 301:510-513. [DOI] [PubMed] [Google Scholar]
- 8.Hanahan, D. 1983. Studies on transformation of Escherichia coli with plasmids. J. Mol. Biol. 166:557-580. [DOI] [PubMed] [Google Scholar]
- 9.Inouye, M. 1991. Intramolecular chaperone: the role of the pro-peptide in protein folding. Enzyme 45:314-321. [DOI] [PubMed] [Google Scholar]
- 10.Kakudo, S., N. Kikuchi, K. Kitadokoro, T. Fujiwara, E. Nakamura, H. Okamoto, M. Shin, M. Tamaki, H. Teraoka, H. Tsuzuki, and N. Yoshida. 1992. Purification, characterization, cloning, and expression of glutamic acid-specific protease from Bacillus licheniformis ATCC 14580. J. Biol. Chem. 267:23782-23788. [PubMed] [Google Scholar]
- 11.Kessler, E., and M. Safrin. 1994. The propeptide of Pseudomoas aeruginosa elastase acts an elastase inhibitor. J. Biol. Chem. 269:22726-22731. [PubMed] [Google Scholar]
- 12.Khan, A. R., and M. N. James. 1998. Molecular mechanisms for the conversion of zymogens to active proteolytic enzymes. Protein Sci. 7:815-836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kitadokoro, K., E. Nakamura, M. Tamaki, T. Horii, H. Okamoto, M. Shin, T. Sato, T. Fujiwara, H. Tsuzuki, N. Yoshida, and H. Teraoka. 1993. Purification, characterization and molecular cloning of acidic amino acid-specific proteinase from Streptomyces fradiae ATCC 14544. Biochem. Biophys. Acta 1163:149-157. [DOI] [PubMed] [Google Scholar]
- 14.Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680-685. [DOI] [PubMed] [Google Scholar]
- 15.Lee, S. J., D. M. Kim, K. H. Bae, S. M. Byun, and J. H. Chung. 2000. Enhancement of secretion and extracellular stability of staphylokinase in Bacillus subtilis by wprA gene disruption. Appl. Environ. Microbiol. 66:476-480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lowry, O. H., N. J. Rosebrough, A. L. Farr, and R. J. Randall. 1951. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 193:265-275. [PubMed] [Google Scholar]
- 17.Margot, P., and D. Karamata. 1996. The wprA gene of Bacillus subtilis 168, expressed during exponential growth, encodes a cell-wall-associated protease. Microbiology 142:3437-3444. [DOI] [PubMed] [Google Scholar]
- 18.Millet, J. 1970. Characterization of proteinases excreted by Bacillus subtilis Marburg strain during sporulation. J. Appl. Bacteriol. 33:207-219. [DOI] [PubMed] [Google Scholar]
- 19.Ohta, Y., and M. Inouye. 1990. Pro-subtilisin E: purification and characterization of its autoprocessing to active subtilisin E in vitro. Mol. Microbiol. 4:295-304. [DOI] [PubMed] [Google Scholar]
- 20.Okamoto, H., T. Fujiwara, E. Nakamura, T. Katoh, H. Iwamoto, and H. Tsuzuki. 1997. Purification and characterization of a glutamic-acid-specific endopeptidase from Bacillus subtilis ATCC 6051; application to the recovery of bioactive peptides from fusion proteins by sequence-specific digestion. Appl. Microbiol. Biotechnol. 48:27-33. [DOI] [PubMed] [Google Scholar]
- 21.Priest, F. G. 1977. Extracellular enzyme synthesis in the genus Bacillus. Bacteriol. Rev. 41:711-753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roitsch, C. A., and J. H. Hageman. 1983. Bacillopeptidase F: two forms of a glycoprotein serine protease from Bacillus subtilis 168. J. Bacteriol. 155:145-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rufo, G. A., B. J. Sullivan, A. Sloma, and J. Pero. 1990. Isolation and characterization of a novel extracellular protease from Bacillus subtilis. J. Bacteriol. 172:1019-1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sadaie, Y., and T. Kada. 1983. Formation of competent Bacillus subtilis cells. J. Bacteriol. 153:813-821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 26.Shaw, L., E. Golonka, J. Potempa, and S. J. Foster. 2004. The role and regulation of the extracellular proteases of Staphylococcus aureus. Microbiology 150:217-228. [DOI] [PubMed] [Google Scholar]
- 27.Sloma, A., A. Ally, D. Ally, and J. Pero. 1988. Gene encoding a minor extracellular protease in Bacillus subtilis. J. Bacteriol. 170:5557-5563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sloma, A., C. F. Rudolph, G. A. Rufo, Jr., B. J. Sullivan, K. A. Theriault, D. Ally, and J. Pero. 1990. Gene encoding a novel extracellular metalloprotease in Bacillus subtilis. J. Bacteriol. 172:1024-1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sloma, A., A. G. Rufo, Jr., K. A. Theriault, M. Dwyer, S. W. Wilson, and J. Pero. 1991. Cloning and characterization of the gene for an additional extracellular serine protease of Bacillus subtilis. J. Bacteriol. 173:6889-6985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sloma, A., G. A. Rufo, Jr., C. F. Rudolph, B. J. Sullivan, K. A. Theriault, and J. Pero. 1990. Bacillopeptidase F of Bacillus subtilis: purification of the protein and cloning of the gene. J. Bacteriol. 172:1470-1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sloma, A., G. A. Rufo, Jr., C. F. Rudolph, B. J. Sullivan, K. A. Theriault, and J. Pero. 1990. Bacillopeptidase F of Bacillus subtilis: purification of the protein and cloning of the gene. J. Bacteriol. 172:5520-5521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stephenson, K., and R. C. Harwood. 1998. Influence of a cell-wall-associated protease on production of α-amylase by Bacillus subtilis. Appl. Environ. Microbiol. 64:2875-2881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Strauch, M. A., and J. A. Hoch. 1993. Transition-state regulators: sentinels of Bacillus subtilis post-exponential gene expression. Mol. Microbiol. 7:337-342. [DOI] [PubMed] [Google Scholar]
- 34.Strausberg, S., P. Alexander, L. Wang, F. Schwarz, and P. Bryan. 1993. Catalysis of a protein folding reaction: thermodynamic and kinetic analysis of subtilsin BPN interactions with its propeptide fragment. Biochemistry 32:8112-8119. [DOI] [PubMed] [Google Scholar]
- 35.Tran, L., X. C. Wu, and S. L. Wong. 1991. Cloning and expression of a novel protease gene encoding an extracellular neutral protease from Bacillus subtilis. J. Bacteriol. 173:6364-6372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Trowsdale, J., and C. Anagnostopoulos. 1976. Differences in the genetic structure of Bacillus subitilis strains carrying the trpE26 mutation and strain 168. J. Bacteriol. 126:609-618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wittchen, K. D., and F. Meinhardt. 1995. Inactivation of the major extracellular protease from Bacillus megaterium DSM319 by gene replacement. Appl. Microbiol. Biotechnol. 42:871-877. [DOI] [PubMed] [Google Scholar]
- 38.Wong, S. L., C. W. Price, D. S. Goldfarb, and R. H. Doi. 1984. The subtilisin E gene of Bacillus subtilis is transcribed from a σ 37 promoter in vivo. Proc. Natl. Acad. Sci. USA 81:1184-1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu, X. C., S. Nathoo, A. S. Pang, T. Carne, and S. L. Wong. 1990. Cloning, genetic organization, and characterization of a structural gene encoding bacillopeptidase F from Bacillus subtilis. J. Biol. Chem. 265:6845-6850. [PubMed] [Google Scholar]
- 40.Yang, M. Y., E. Ferrari, and D. J. Henner. 1984. Cloning of the neutral protease gene of Bacillus subtilis and the use of the cloned gene to create an in vitro-derived deletion mutation. J. Bacteriol. 160:15-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yanisch-Perron, C., J. Vieira, and J. Messing. 1985. Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene 33:103-119. [DOI] [PubMed] [Google Scholar]
- 42.Yoshida, N., S. Tsuruyama, K. Nagata, K. Hirayama, K. Noda, and S. Makisumi. 1988. Purification and characterization of acidic amino acid specific endopeptidase of Streptomyces griseus obtained from a commercial preparation (pronase). J. Biochem. (Tokyo) 104:451-456. [DOI] [PubMed] [Google Scholar]
- 43.Yoshikawa, K., H. Tsuzuki, T. Fujiwara, E. Nakamura, H. Iwamoto, K. Matsumoto, M. Shin, N. Yoshida, and H. Teraoka. 1992. Purification, characterization and gene cloning of a novel glutamic acid-specific endopeptidase from Staphylococcus aureus ATCC 12600. Biochem. Biophys. Acta 1121:221-228. [DOI] [PubMed] [Google Scholar]