

Abstract
We describe a simple theoretical framework for identifying orthologous sets of genes that deviate from a clock-like model of evolution. The approach used is based on comparing the evolutionary distances within a set of orthologs to a standard intergenomic distance, which was defined as the median of the distribution of the distances between all one-to-one orthologs. Under the clock-like model, the points on a plot of intergenic distances versus intergenomic distances are expected to fit a straight line. A statistical technique to identify significant deviations from the clock-like behavior is described. For several hundred analyzed orthologous sets representing three well-defined bacterial lineages, the α-Proteobacteria, the γ-Proteobacteria, and the Bacillus-Clostridium group, the clock-like null hypothesis could not be rejected for ∼70% of the sets, whereas the rest showed substantial anomalies. Subsequent detailed phylogenetic analysis of the genes with the strongest deviations indicated that over one-half of these genes probably underwent a distinct form of horizontal gene transfer, xenologous gene displacement, in which a gene is displaced by an ortholog from a different lineage. The remaining deviations from the clock-like model could be explained by lineage-specific acceleration of evolution. The results indicate that although xenologous gene displacement is a major force in bacterial evolution, a significant majority of orthologous gene sets in three major bacterial lineages evolved in accordance with the clock-like model. The approach described here allows rapid detection of deviations from this mode of evolution on the genome scale.
The classical molecular clock concept holds that the sequence of a given gene (protein) evolves at a constant rate as long as its biological function remains unchanged (5, 26, 56, 57). The actual evolutionary rates for the entire set of genes in a genome show a broad distribution, with the rates for the slowest genes differing from the rates for the fastest genes by at least 2 orders of magnitude (18, 21, 26, 28). However, the molecular clock hypothesis holds that, within the same set of orthologs (i.e., homologous genes related via vertical inheritance) (16, 44), the rate does not change, within some inevitable dispersion. The theoretical foundation of the molecular clock concept is the neutral theory of molecular evolution, which holds that, at least when no functional changes occur, the great majority of fixed substitutions in nucleotide and protein sequences are neutral, their rate being determined by the fixed, gene-specific functional constraints (13, 26, 39). The modern reincarnation of neutralism, the near-neutral theory, predicts a greater dispersion of the molecular clock because the probability of fixation of slightly deleterious mutations critically depends on the effective population size, which is prone to major fluctuations (13, 39). Numerous tests of the molecular clock revealed both a general pattern conforming to the theory and numerous violations. It has been repeatedly shown that the molecular clock is overdispersed; i.e., the variation of evolutionary rates within a set of orthologs is greater than that predicted by a null model based on the Poisson distribution (8, 18, 19).
Deviations from the molecular clock are thought to result from lineage-specific acceleration of evolution, which could be due either to functional changes entailing relaxation of purifying selection or positive selection or to increased mutational pressure caused, at least in part, by effective population size effects (5). These phenomena cause overdispersion of the molecular clock, which is manifested in unequal lengths of tree branches coming out of the same node, under the assumption that the topology of the phylogenetic tree for a given set of orthologs is known. Typically, the same species tree topology is assumed for all genes. This approach is likely to be valid for the multicellular eukaryotes, which, historically, have been the objects of the analyses that led to the molecular clock concept. However, recent comparative genomic studies strongly suggest that in addition to the regular pattern of vertical inheritance, evolution of prokaryotic genomes is dramatically affected by horizontal gene transfer (HGT) (6, 11, 12, 27, 30, 32, 38, 55). On many occasions, HGT seems to occur between evolutionarily distant organisms, although it has been argued that there could be a decreasing gradient of the HGT rate from closely related species to distantly related species; the existence of such a gradient could be one of the reasons why a species tree can be constructed at all, in spite of extensive HGT (20).
There seem to be certain connections between the amount of HGT and the biology of prokaryotic genes. In particular, the so-called complexity hypothesis holds that HGT is much less common among genes that encode subunits of macromolecular complexes, such as those involved in translation, transcription, and replication, than in genes coding for metabolic enzymes (25). While this prediction might hold statistically, subsequent studies have shown that there are very few, if any, genes that are completely refractory to HGT. In particular, evidence of HGT has been obtained for several ribosomal proteins, translation factors, and the major RNA polymerase subunits (3, 4, 24, 34).
From a comparative genomic perspective, HGT events have been classified into three categories: (i) acquisition of genes that are novel to a given phylogenetic lineage; (ii) acquisition of paralogs of genes preexisting in the given lineage; and (iii) xenologous gene displacement (XGD), in which the original gene from a given set of orthologs is displaced by a member of the same set of orthologs from a different lineage (30).
Obviously, if an HGT event, particularly XGD, goes unnoticed in the course of phylogenetic analysis, an apparent gross violation of this molecular clock will be seen when evolutionary rates are measured in the affected set of orthologous genes on the basis of the assumed species tree topology. HGT is detected through anomalies in the topology of phylogenetic trees of individual sets of orthologs or by so-called surrogate approaches, which, in the case of HGT between distant species, are based primarily on the phyletic distribution of the homologs of a given gene (7, 30, 41). Simply put, unexpected phyletic patterns (e.g., the presence of orthologs of a given gene in all or nearly all sequenced bacterial genomes but in only one archaeon) suggest that there has been HGT (in this case from a bacterium to the archaeon) (27, 30). These patterns can be expressed either in terms of presence-absence only or, more quantitatively, by comparing the significance levels of taxon-specific best hits. The general validity of this approach seems to be supported by the biological plausibility of some of the trends in the apparent horizontal gene fluxes that were detected by phyletic pattern analysis. Thus, hyperthemophilic bacteria showed a clear preponderance of genes of possible archael origin compared to mesophiles (2, 29, 36), and probable gene transfer from eukaryotic hosts to some bacterial pathogens also has been inferred (17, 40). In principle, phylogenetic tree analysis is supposed to be a more precise indicator of probable HGT events than similarity-based surrogate methods because of inaccuracies in the latter resulting from the lack of exact correspondence between sequence similarity and phylogenetic affinity (31). A genome-wide phylogenetic analysis, aimed specifically at detection of horizontally transferred genes, has been described (43). However, it is well known that phylogenetic analysis is fraught with its own slew of artifacts, such as long branch attraction, particularly when fast methods, such as minimal evolution or neighbor joining, are employed (14). In addition, phylogenetic analysis can be prohibitively expensive computationally when it is attempted on the genome scale, especially with powerful methods, such as complete maximum-likelihood analysis, and large sets of species. Therefore, surrogate, similarity-based methods have proved to be extremely useful, at least as a rapid, first-tier strategy that allows workers to delineate a set of HGT candidates.
We were interested in investigating a surrogate approach to genome-wide study of prokaryotic evolution, which combines a test of the validity and an analysis of the rate distribution of the molecular clock with detection of potential HGT events and lineage-specific acceleration of evolution. Using the Clusters of Orthologous Groups (COGs) database for proteins (46, 47), we analyzed the molecular clock behavior of COGs from three major bacterial lineages, the α-Proteobacteria, the γ-Proteobacteria, and the low-G+C-content gram-positive bacteria. We found that clock-like evolution was dominant in all three groups, but we also detected many anomalies, some of which are best explained by XGD.
MATERIALS AND METHODS
Sequences.
Three unequivocally identified and well-characterized bacterial lineages, the γ-Proteobacteria, the α-Proteobacteria, and the Bacillus-Clostridium group of gram-positive bacteria, were selected for the present study. The γ-proteobacterial set included six species: Escherichia coli K-12, Haemophilus influenzae, Pasteurella multocida, Salmonella enterica serovar Typhimurium LT2, Vibrio cholerae, and Yersinia pestis. The α-proteobacterial set included seven species: Agrobacterium tumefaciens C58 Cereon, Brucella melitensis, Caulobacter crescentus CB15, Mesorhizobium loti, Rickettsia conorii, Rickettsia prowazekii, and Sinorhizobium meliloti. The Bacillus-Clostridium set included eight species: Bacillus halodurans, Bacillus subtilis, Clostridium acetobutylicum, Listeria innocua, Lactococcus lactis, Staphylococcus aureus N315, Streptococcus pneumoniae TIGR4, and Streptococcus pyogenes M1 GAS. For each of these species sets, we identified a set of COGs (46, 48) in which each of the relevant species was represented by exactly one protein (i.e., all species present, no paralogs). Additionally, the constituent proteins were required to have a sufficient alignable length (at least 60 amino acids in conserved blocks [see below]). This search resulted in 563 COGs for the γ-proteobacterial set, 274 COGs for the α-proteobacterial set, and 234 COGs for the Bacillus-Clostridium set; the overlap for the three sets comprised 114 COGs.
Alignments.
Multiple alignments of sequence families within the bacterial groups were produced by using the MAP program (23). Sequence families involving wider taxonomic sampling of proteins were aligned by using the T-Coffee program (37). Multiple alignments were aggressively filtered for potential incorrectly aligned positions; only conserved blocks with no gaps containing 10 or more positions were retained for further analysis (53).
Evolutionary distances between genes and genomes and phylogenetic trees.
Maximum-likelihood distances between individual protein sequences were computed for each of the COGs analyzed by using the PAML package, with the JTT substitution model corrected for observed amino acid frequencies and the α parameter of γ-distribution of intraprotein evolutionary rate variability set to 1.0; this estimate includes a correction for possible multiple substitutions in the same site (54). Additionally, multiple alignments consisting of 21 sequences each were produced for the 114 COGs in which all three groups were represented, and pairwise distances were similarly computed from this alignment. Phylogenetic trees for individual COGs were constructed by using the maximum-likelihood method implemented in the Tree-Puzzle package, with the expected likelihood weight determined for the tree topology involving split versus topologies in which the respective lineage remained monophyletic (42).
To calculate intergenomic evolutionary distances, all intergenic distances obtained from the same pair of genomes in the set of 114 conserved COGs were pooled, and the median of the distribution of these distances was taken to represent the intergenomic distance (21, 52). Neighbor-joining and least-squares trees were reconstructed from the pairwise genome distance matrices by using the programs NEIGHBOR and FITCH of the PHYLIP package, respectively (15).
Limited tree analysis.
A comprehensive phylogenetic analysis of a protein family, even if it is limited to a set of orthologs from completely sequenced genomes, is rarely feasible. There are several reasons that usually preclude this type of analysis, as follows: in many cases, the sequences of orthologs from the most distant lineages, such as bacteria and archaea, are only weakly similar, which complicates the construction of an alignment suitable for building a phylogenetic tree; the large number of sequences hampers the use of advanced methods for reconstruction of the phylogeny; and the presence of in-paralogs of various ages hinders the interpretation of results. Therefore, we elected to perform a limited tree analysis for the cases where the split distance analysis indicated a likely split of a set of orthologs from a particular bacterial lineage into two subsets with distinct evolutionary histories; these groups are referred to below as left ({L}) and right ({R}) sets. For each sequence from the given COG, global pairwise alignment scores (calculated by using the ALIGN program with default parameters) (35) for alignments with the other COG members were obtained (sequences from closely related species were excluded). Members of the COG not belonging to either {L} or {R} (i.e., the sequences of orthologs outside the lineage analyzed) were arranged into two ordered lists, <HL> and <HR>, according to their similarity scores against {L} and {R}, respectively. Multiple alignments that included {L}, {R}, and the top five sequences from <HL> and <HR> were constructed by using the T-Coffee program (37). Maximum-likelihood trees were constructed by using the ProtML program of the MOLPHY package (1). For all internal branches separating the {L} and {R} subsets (see Fig. 4), the RELL bootstrap values were determined. The highest bootstrap value observed on such a branch indicated the level of support for the separation hypothesis.
FIG. 4.

Limited tree procedure. Formally, let us consider an internal branch B, which partitions all nodes into four subsets, {S1 } to {S4}. If the partition satisfies the criteria (i) {S1} contains all of {L} and none of {R}, (ii) {S2} contains all of {R} and none of {L}, and (iii) {S3} and {S4} contain neither {L} nor {R}, the branch provides the evolutionary separation between {L} and {R} regardless of the true position of the tree root.
RESULTS AND DISCUSSION
Theory. (i) Molecular clock and deviations from clock-like evolution.
The approach developed here is based on comparing the evolutionary distances between genes (proteins) in a given COG to the distances between the corresponding genomes. There is no single correct way to calculate the intergenomic distances. In principle, a reference gene believed to be a good clock, such as rRNA, or a set of genes believed to evolve under the same model, such as genes for ribosomal proteins (3, 22, 51), can be employed for this calculation. We chose one of the genome-wide approaches to evolutionary distance determination, in which the median of the distribution of the distances between orthologs from a given pair of genomes is used as a proxy for the intergenomic distance (51, 52) (see Materials and Methods).
Under a perfect molecular clock and strict vertical inheritance, the evolutionary rates of all genes differ from each other only by proportionality constants, so the distance between any pair of genes (dAB) is proportional to the distance between genomes A and B (DAB):
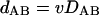 |
(1) |
where v is the relative rate of evolution for the given gene (COG) (v = 1 for COGs that evolve at the median rate). Obviously, in this case, a scatter plot of dAB versus DAB for all pairs of genomes is a straight line with the intercept at 0 and the slope equal to v (Fig. 1A).
FIG. 1.

Clock-like model of evolution: predictions and detection of deviations. (A) Clock-like evolution with vertical inheritance. (B) Clock-like evolution with one HGT from an outside source. The red branches in the trees indicate the genes from species B and C that are inferred to have been transferred from the unknown, distant species, X, and the red points on the plot at the bottom right indicate the evolutionary distances that are taken as evidence of this HGT event.
Various events that could occur during evolution could distort this idealized picture. Accelerated evolution on one terminal branch of the tree would increase all distances involving the corresponding species; as a result, the points corresponding to this species on the scatter plot are located above the main line. Similar acceleration on an internal branch of the tree would result in an upward shift of the points corresponding to two or more species. We were particularly interested in cases of HGT of a gene encoding a protein of a given COG from an outside source, resulting in displacement of the original gene characteristic of the given lineage (i.e., XGD). In these cases, the evolutionary distance between the transferred and nontransferred sequences is determined by the distance between the transfer source and the recipient lineage (D*), rather than by the intragroup distances. The scatter plot is expected to consist of two distinct sets of points: (i) points corresponding to nontransferred genes that fit a line with the intercept at 0 and the slope v, like those obtained with the clock model, and (ii) points corresponding to the transferred gene (i.e., the distances between this gene and all other members of the COG; these points fit a horizontal line with the intercept corresponding to the distance to the transfer source) (Fig. 1B).
(ii) Statistical model.
The accumulation of substitutions in protein sequences was treated as a Poisson stochastic process. The number of substitutions in a given gene, accumulated over a given time interval, is a Poisson-distributed variable with the variance proportional to the expected value:
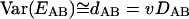 |
(2a) |
If the fact that the observed distance between proteins encoded by genomes A and B has a random error, EAB (which includes Poisson-distributed sampling error, error in deriving evolutionary distances from observed differences, and branch-to-branch variation of evolutionary rates), is taken into account, equation 1 becomes:
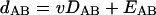 |
(2b) |
By using equation 2a, equation 2b can be rewritten as
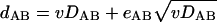 |
(3a) |
where eAB is a random variable with the expectation of 0 and the variance independent of DAB and v. Similarly, if one of the two orthologs was acquired via HGT from a distant source (Fig. 1B):
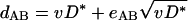 |
(3b) |
(iii) Statistical analysis: vertical inheritance.
Let us consider a set of N genomes {G}, each with a single ortholog in a given COG. One can measure the distance between orthologs in the given COG (dIJ) and the distance between the genomes (DIJ) for all N′ = N(N − 1)/2 pairs ([I,J]) from {G}. Minimizing the square error over all such pairs,
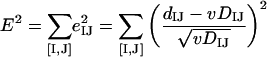 |
we get the optimal value of v:
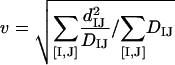 |
(4a) |
and the fit error (s2) and residual variance (u2) of eIJ:
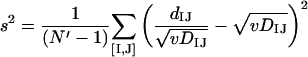 |
(4b) |
 |
(iv) Statistical analysis: HGT.
If genes in one of the lineages in a COG were acquired via HGT from a distant source, all intergenic distances in this COG fall into two groups: those that reflect vertical inheritance (DAD and DBC in Fig. 1B) and those that reflect the transfer distance (DAB, DAC, DBD, and DCD in Fig. 1B). Considering all pairs that correspond to vertical inheritance ([I,J]) and all pairs that correspond to HGT ([K,L]), the square error of the approximation is:
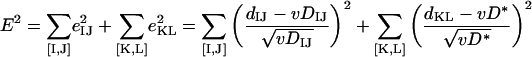 |
Minimization of E2 over v and D* yields:
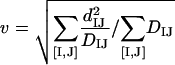 |
(5a) |
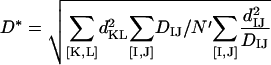 |
(5b) |
and
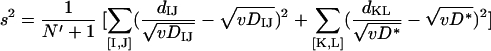 |
(5c) |
 |
(v) Statistical analysis: baseline noise.
If the pattern of a gene's inheritance is completely disjointed from the pattern of intergenomic relationships, neither equation 3a nor equation 3b adequately describes the relationships between the intergenomic and intergenic distances. In the absence of a clear dependence between these variables, the scatter plot for dAB versus DAB represents random scatter of points, and the following simple equation applies:
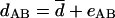 |
(6a) |
where d̄ is simply the mean intergenic distance over all pairs. The baseline variance of eIJ is:
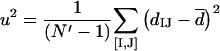 |
(6b) |
(vi) Statistical analysis of COG evolution.
Each COG was analyzed with the three models described above.
(a) Noisy data, no clock-like evolution (equation 6a).
The baseline variance of intergenic evolutionary distances (uN2) was calculated by using equation 6b.
(b) Simple molecular clock (equation 3a).
The residual variance of the straight line fit (uC2) and the fit error (sC2) were calculated by using equation 4b. The relative evolutionary rate (v) was calculated by using equation 4a.
(c) Single significant deviation from molecular clock (equations 3a and 3b).
The genomes were partitioned into two sets by breaking each branch of the species tree. For each of the possible splits, the residual variance (uT2) and the fit error (sT2) were calculated by using equation 5c. The split with the minimal sT2 was accepted. The relative evolutionary rate (v) was calculated by using equation 5a, and the distance to the transfer source was calculated by using equation 5b. Additionally, to detect the transfers originating from outside the group, the relative transfer distance (DT) was calculated as D*/max(DKL), with the maximum taken over all cross-group pairs.
Two statistical tests were performed for each COG. The first test aimed at discriminating between H0 (the data do not follow either of the two clock-like models) and H1 (the data fit either the simple-clock or single-transfer model). The ratio FC = u2N/min(u2C, u2T) was subjected to Fisher's test with (N′ − 1, N′ − 3) degrees of freedom. If the value of FC exceeded the critical level at the 0.05 level of significance (1.94 to 2.64, depending on the group of species analyzed), H0 was rejected, and the data were considered to conform to the molecular clock model.
The second test discriminated between H0 (the data fit the simple-clock model) and H1 (the data fit the single-transfer model). The ratio FT = sC2/sT2 was subjected to Fisher's test with (N′ − 2, N′ − 3) degrees of freedom. If the value of FT exceeded the critical level at the 0.05 level of significance (1.95 to 2.66 depending on the group of species analyzed), H0 was rejected, and the data were considered to conform significantly better to the single-transfer model. In this case, the value of v calculated by using equation 5a (rather than that calculated by using equation 4a) was used to describe the relative evolutionary rate for the COG in question; a DT value of >1 was considered to be an indication of HGT from an outside source.
Empirical results. (i) Molecular clock and deviations from clock-like evolution in bacteria.
We applied the theory described above to an analysis of the evolution of three major bacterial lineages, the α-Proteobacteria, the γ-Proteobacteria, and low-G+C-content gram-positive bacteria. For each of these groups, the COGs that contained a single representative from each species were selected for analysis (Table 1). Examples of relationships between the intergenomic and intergenic distances are shown in Fig. 2. Figure 2B shows clear evidence of the evolutionary heterogeneity of the COG in question. Table 1 shows a breakdown of the COGs analyzed according to their fit to one of the three models of evolution described above. For the great majority of the COGs (90 to 99%), the data could be reconciled with either the clock-like model or the model that involved one significant deviation from the clock (Table 1). For ∼70% of the COGs in each lineage, the clock-like model was found to be compatible with the data, whereas the evolution of the rest of the COGs was better explained when the group was split into two subsets with different histories. For 13 to 22% of the COGs in the three lineages, the apparent acceleration of evolution in one of the clades (DT > 1) was significant enough to suspect HGT from an outside source.
TABLE 1.
Alternative evolutionary models for COGs
| Lineage | No. (%) of COGsa
|
Total no. | |||
|---|---|---|---|---|---|
| Random scatter | Fit to the models
|
||||
| Clock-like, no anomalies | Split into two subsets
|
||||
| DT ≤ 1 | DT > 1 | ||||
| γ-Proteobacteria | 8 (1.4) | 387 (68.7) | 42 (7.5) | 126 (22.4) | 563 |
| α-Proteobacteria | 4 (1.5) | 193 (70.4) | 37 (13.5) | 40 (14.5) | 274 |
| Gram positive | 21 (9.0) | 173 (73.9) | 9 (3.8) | 31 (13.2) | 234 |
Number of COGs (percentage of the total number of COGs analyzed) for the given lineage.
FIG. 2.

Three representative examples of the relationships between intergenic and intergenomic distances. (A) Clock-like evolution. (B) Clock-like evolution with one strong deviation probably due to HGT from an outside source. (C) Random scatter of points: uncertain evolutionary scenario. The magenta lines in panels B and C show the horizontal trend lines with the best fit to the data; the evolutionary distances thought to reflect XGD in panel B are indicated by magenta diamonds.
(ii) Relative evolutionary rates in the three bacterial lineages.
All three bacterial lineages analyzed displayed a wide range of relative evolutionary rates (i.e., evolutionary rates normalized by using the intergenomic rate). The difference between the COGs that evolved fastest and the COGs that evolved slowest reached almost 2 orders of magnitude for the γ-Proteobacteria and almost 1 order of magnitude within the set of 108 conserved COGs that fit one of the two evolutionary models (Table 2). The distributions of relative evolution rates were close to log normal in all three lineages (Fig. 3). Because the median intergenomic distance was calculated only for the 114 COGs that are conserved in all three bacterial groups (see Materials and Methods for details), the medians of the rate distributions within each of the groups were not equal to one. Of the three groups, the γ-Proteobacteria showed the fastest median rate and the broadest variation in the relative evolutionary rates (Fig. 3A). This is probably due to the fact that the selected set of γ-Proteobacteria species forms a tighter (more recently diverged) cluster than the sets of α-Proteobacteria and Bacillus-Clostridium group bacteria analyzed, as indicated by the comparison of the calculated intergenomic distances (data not shown). Accordingly, the set of γ-proteobacterial COGs is more functionally diverse and includes faster-evolving genes than the COGs from the other two lineages. This conclusion is supported by the comparison of the rate distributions for the full set of 555 γ-proteobacterial COGs and the 108-COG subset that is conserved in all three lineages; not unexpectedly, the distribution in the latter set is considerably narrower and is shifted toward lower rates (Fig. 3B). Within the conserved set of 108 conserved COGs, the relative evolutionary rates are strongly correlated among the three lineages (Table 3). However, statistically significant differences between the lineages were detected; in the same COG, the rate among γ-Proteobacteria tended to be the lowest, and the rate among the α-Proteobacteria tended to be the highest (Table 3).
TABLE 2.
Relative evolution rates
| Group | Relative rate
|
Ratio of fastest to slowest
|
||||
|---|---|---|---|---|---|---|
| Minimum | Median | Maximum | All | Conserveda | 90%b | |
| γ-Proteobacteria | 0.10 | 1.40 | 9.32 | 97.6 | 35.9 | 12.0 |
| α-Proteobacteria | 0.21 | 1.08 | 4.17 | 19.4 | 18.4 | 5.6 |
| Gram positive | 0.19 | 0.97 | 4.00 | 13.0 | 9.4 | 6.9 |
Only the proteins from the 108 conserved COGs that fit one of the two evolutionary models were included.
Ratio after removal of 5% of the fastest-evolving proteins and 5% of the slowest-evolving proteins.
FIG. 3.

Distributions of the relative evolutionary rates in the three bacterial lineages analyzed and in the conserved set of COGs represented in all lineages. (A) Distributions of the relative evolutionary rates in the three lineages. gamma, γ-Proteobacteria; alpha, α-Proteobacteria; bacil, Bacillus-Clostridium group. (B) Distributions of the relative evolutionary rates in the full set of γ-Proteobacteria (gamma-555) and in the subset of COGs represented in all lineages (gamma-108). The distributions for the three bacterial lineages and the corresponding log-normal approximations (dashed lines) are color coded. The scale for the horizontal axis is logarithmic.
TABLE 3.
Evolution of the same COG in different groups
| Group | Correlation coefficient or difference between evolutionary ratesa
|
||
|---|---|---|---|
| γ-Proteobacteria | α-Proteobacteria | Gram-positive bacteria | |
| γ-Proteobacteria | 0.74 | 0.82 | |
| α-Proteobacteria | γ-Proteobacteria < α-Proteobacteria (7 × 10−4) | 0.85 | |
| Gram-positive bacteria | γ-Proteobacteria < gram-positive bacteria (2 × 10−2) | α-Proteobacteria > gram-positive bacteria (1 × 10−2) | |
The values above the diagonal are linear correlation coefficients for the relative evolution rates of the three bacterial lineages in the same COG (the correlation between the logarithms of the rates for 108 conserved COG was calculated). The data below the diagonal indicate the sign and significance level for the difference between the evolutionary rates for the three lineages as determined by the two-tailed paired t-test.
(iii) Apparent violations of molecular clock and anomalies in gene phylogenies.
All COGs that fit the two-mode model better than the simple clock-like model were analyzed further by using the limited tree procedure (see Materials and Methods for details).
Briefly, the purpose of this procedure was to investigate whether the two sets of species separated by the statistical analysis described above were also separated by a strongly supported internal branch in the phylogenetic tree for the given COG (Fig. 4). Not surprisingly, the COGs that had the longest split distance (DT) also showed a consistent tendency to have anomalies in the reconstructed phylogenies (Table 4).
TABLE 4.
COGs with most pronounced deviations from the clock-like model
| COG | Function (gene) | Proposed split | FCa | FTb | DT | v | Tree-Puzzle ELWc | Bootstrap support (%)d | Conclusions |
|---|---|---|---|---|---|---|---|---|---|
| COG2171 | Tetrahydrodipicolinate N-succinyltransferase (dapD) | (V. cholerae), (γ-Proteobacteria) | 1,050.2 | 263.1 | 29.72 | 0.287 | 1.0000 | 88 | XGD in Vibrio and Pseudomonas; probable multiple HGT |
| COG0137 | Argininosuccinate synthase (argG) | (V. cholerae), (γ-Proteobacteria) | 3,835.1 | 578.8 | 11.81 | 0.505 | 1.0000 | 100 | Uncertain; extremely long branch for a subset of γ-Proteobacteria; the remaining γ-Proteobacteria either cluster with α-Proteobacteria (P. aeruginosa) or are scattered around the tree base (Xylella fastidiosa, V. cholerae, Buchnera sp.); possible combination of acceleration with HGT |
| COG0221 | Inorganic pyrophosphatase (ppa) | (H. influenzae), (γ-Proteobacteria) | 317.8 | 96.3 | 10.00 | 0.480 | 1.0000 | 97 | Uncertain; long branch of H. influenzae and Neisseria at the base of the Proteobacteria are separated from βγ-Proteobacteria and attached to the bacterial root; acceleration in one of these and HGT between them? |
| COG0207 | Thymidylate synthase (thyA) | (E. coli, S. enterica serovar Typhimurium, Y. pestis), (γ-Proteobacteria) | 466.1 | 125.4 | 7.75 | 0.602 | 0.9685 | 98 | XGD in H. influenzae, P. multocida, and V. cholerae from gram-positive, bacteria |
| COG0783 | DNA-binding ferritin-like protein, oxidative damage protectant (dps) | (E. coli, S. enterica serovar Typhimurium, Y. pestis), (γ-Proteobacteria) | 112.9 | 76.0 | 6.26 | 1.642 | 1.0000 | 96 | XGD, probably in more thae lineage, as a result of multiple HGT |
| COG0554 | Glycerol kinase (glpK) | (Y. pestis), (γ-Proteobacteria) | 1,529.6 | 624.9 | 6.07 | 0.676 | 1.0000 | 100 | HGT of a paralog to Yersinia; Yersinia groups with paralogs of glpK (kinases with still unknown specificity) from Pseudomonas and Agrobacterium |
| COG0157 | Nicotinate-nucleotide pyrophosphorylase (nadC) | (H. influenzae, P. multocida), (γ-Proteobacteria) | 66.2 | 21.5 | 4.33 | 1.853 | 1.0000 | 100 | HGT acquisition of paralog; the proteins of H. influenzae and P. multocida group with ModD of E. coli O157:H7, not with NadC, the only COG member in E. coli K-12; loss of nadC and acquisition of modD via HGT from archaea in H. influenzae, P. multocida, and E. coli O157:H7 |
| COG1526 | Uncharacterized protein required for formate dehydrogenase activity (fdhD) | (V. cholerae), (γ-Proteobacteria) | 137.0 | 90.7 | 4.09 | 1.949 | 1.0000 | 100 | XGD in Vibrio and β-Proteobacteria, probable HGT from gram-positive bacteria |
| COG2824 | Uncharacterized Zn ribbon-containing protein involved in phosphonate metabolism (phnA) | (V. cholerae), (γ-Proteobacteria) | 86.0 | 54.0 | 3.74 | 1.416 | 0.6302 | 72 | Uncertain; a poor-quality tree with LBA, although multip le HGT cannot be ruled out |
| COG0720 | 6-Pyruvoyl-tetrahydropterin synthase (BS_ykvK) | (H. influenzae, P. multocida), (γ-Proteobacteria) | 72.4 | 17.5 | 3.71 | 1.035 | 1.0000 | 93 | XGD in Haemophilus and Pasteurella; Haemophilus and Pasteure lla belong to a heterogeneous bacterial cluster, suggesting multiple HGT |
| COG0048 | Ribosomal protein S12 (rpsL) | (R. prowazekii, R. conorii), (α-Proteobacteria) | 142.5 | 10.4 | 3.02 | 0.212 | 0.8969 | 87 | Uncertain; very long branches of Rickettsia and eukaryotes; possible HGT from eukaryotes to Rickettsia, but LBA also possible. |
| COG0780 | Enzyme related to GTP, cyclohydrolase 1 (BS_ykvM) | (R. prowazekii, R. conorii), (α-Proteobacteria) | 90.4 | 7.0 | 2.98 | 0.648 | 1.0000 | 100 | XGD either in Rickettsia or in the rest of the α-Proteobacteria; HGT from γ-Proteobacteria to Rickettsia or from an uncertain bacterial source to other α-Proteobacteria |
| COG | Function (gene) | Proposed split | FCa | FTb | DT | v | Tree-Puzzle ELWc | Boot-strap support (%)d | Conclusions |
| COG0100 | Ribosomal protein S11 (rpsK) | (R. prowazekii, R. conorii), (α-Proteobacteria) | 122.9 | 13.7 | 2.69 | 0.234 | 0.9015 | 88 | XGD(?) from γ-Proteobacteria to Rickettsia; long branch of Rickettsia-LBAf also possible |
| COG0060 | Isoleucyl-tRNA synthetase (ileS) | (R. prowazekii, R. conorii), (α-Proteobacteria) | 629.3 | 90.7 | 2.67 | 0.740 | 1.0000 | 100 | XGD from eukaryotes to Rickettsia |
| COG0525 | Valyl-tRNA synthetase (valS) | (R. prowazekii, R. conorii), (α-Proteobacteria) | 86.8 | 16.0 | 2.57 | 0.830 | 1.0000 | 100 | XGD; HGT from archaea to Rickettsia and certain Actinobacteria |
| COG0018 | Arginyl-tRNA synthetase (argS) | (C. crescentus), (α-Proteobacteria) | 267.8 | 144.4 | 2.44 | 1.020 | 1.0000 | 100 | XGD from eukaryotes to bacterium, with subsequent dissemination among diverse bacteria via HGT |
| COG0124 | Histidyl-tRNA synthetase (hisS) | (R. prowazekii, R. conorii), (α-Proteobacteria) | 70.9 | 14.1 | 2.14 | 1.037 | 1.0000 | 100 | XGD from eukaryotes to bacteria, probable horizontal dissemination among diverse bacteria, inclu ding nonrickettsial α-Proteobacteria |
| COG0276 | Protoheme ferrolyase (ferrochelatase) (hemH) | (R. prowazekii, R. conorii), (α-Proteobacteria) | 64.1 | 15.1 | 2.03 | 1.082 | 1.0000 | 100 | XGD from eukaryotes with subsequent dissemination among diverse bacteria via HGT; possible additional XGD between γ- and α-Proteobacteria |
| COG0099 | Ribosomal protein S13 (rpsM) | (R. prowazekii, R. conorii), (α-Proteobacteria) | 42.4 | 4.2 | 1.99 | 0.277 | NAe | 52 | Acceleration of evolution; long branch of Rickettsia; no suspicion of HGT |
| COG0527 | Aspartokinase (lysC) | (R. prowazekii, R. conorii), (α-Proteobacteria) | 157.9 | 8.8 | 1.91 | 0.655 | 0.2774 | 0 | Uncertain; very long branch of Rickettsia |
| COG0015 | Adenylosuccinate lyase (purB) | (C. acetobutylicum), (Bacillus-Clostridium group) | 188.8 | 76.5 | 5.11 | 0.454 | 1.0000 | 100 | XGD from a eukaryote to clostridia and Fusobacterium |
| COG0461 | Orotate phosphoribosyl-transferase (pyrE) | (C. acetobutylicum), Bacillus-Clostridium group) | 49.8 | 19.1 | 3.02 | 0.866 | 1.0000 | 100 | XGD; γ-Proteobacteria to C. acetobutylicum (other clostridia partition into the gram-positive cluster) |
| COG0482 | tRNA (5-methylamino-methyl-2-thiouridylate) methyltransferase (trmU) | (C. acetobutylicum), (Bacillus-Clostridium group) | 60.7 | 12.7 | 2.39 | 0.492 | 1.0000 | 100 | XGD in Clostridium; Clostridium belongs to a diverse c luster of bacteria, suggesting multiple HGT |
| COG0060 | Isoleucyl-tRNA synthetase (ileS) | (C. acetobutylicum), (Bacillus-Clostridium group) | 65.8 | 21.1 | 2.32 | 0.818 | 1.0000 | 100 | XGD via ancient HGT from eukaryotes to several diverse bacterial clades including Clostridium; probable multiple HGT among these bacteria |
| COG0082 | Chorismate synthase (aroC) | (C. acetobutylicum), (Bacillus-Clostridium group) | 64.8 | 17.2 | 2.25 | 0.778 | 1.0000 | 100 | XGD via HGT from archaea to clostridia and Fusobacterium |
| COG0124 | Histidyl-tRNA synthetase (hisS) | (C. acetobutylicum), (Bacillus-Clostridium group) | 103.2 | 39.3 | 2.03 | 1.195 | 1.0000 | 100 | XGD via ancient HGT from archaea or eukaryotes to several diverse bacterial clades, including some Clostridium species; probable multiple HGT among these bacteria |
| COG0396 | ABC-type transport system involved in Fe-S cluster assembly ATPase component (sufC) | (C. acetobutylicum), (Bacillus-Clostridium group) | 46.6 | 12.4 | 1.93 | 0.422 | 0.8552 | 99 | XGD in Clostridium; Clostridium belongs t o a diverse cluster of bacteria and archaea, suggesting multiple HGT |
| COG2739 | Uncharacterized protein (BS_ylxM) | (C. acetobutylicum), (Bacillus-Clostridium group) | 30.3 | 8.6 | 1.82 | 0.813 | 0.4673 | 63 | Possible XGD in Clostridium and/or Ureaplasma; however, LBA cannot be ruled out |
| COG0343 | Queuine/archaeosine tRNA ribosyltransferase (tgt) | (C. acetobutylicum), (Bacillus-Clostridium group) | 59.2 | 14.4 | 1.81 | 0.423 | 0.0333 | 65 | XGD from the Aquifex lineage to Clostridium; however, given the long Clostrid ium branch, acceleration of evolution without HGT cannot be ruled out |
| COG0495 | Leucyl-tRNA synthetase (leuS) | (C. acetobutylicum), (Bacillus-Clostridium group) | 66.3 | 14.6 | 1.78 | 0.461 | 1.0000 | 100 | XGD in clostridia; probable HGT from the Deinococcus lineage |
Fc, F-statistics for the data fit to one of the evolutionary models.
FT, F-statistics for the data fit to the model, involving split of the group.
ELW, expected likelihood weight for the tree topology involving split versus topologies in which the corresponding lineage remains monophyletic as reported by Tree-Puzzle (42).
Bootstrap support for the branch separating two groups in the limited tree analysis.
NA, not applicable.
LBA, long branch attraction.
Table 4 lists the top 10 COGs with the most pronounced deviation from the clock-like model (i.e., the greatest DT value) for each of the bacterial lineages analyzed. These COGS were further subjected to complete phylogenetic analysis, including statistical tests (see Materials and Methods), in order to determine the nature of the anomalies detected. Upon detailed inspection of the trees produced with the three methods, the cases were classified into those that most likely reflected HGT resulting in XGD, those that were best explained by accelerated evolution in a particular lineage, and those for which the evolutionary scenario remained uncertain. Of the 30 cases analyzed, only 5 did not appear to be resolved well enough to reach conclusions on the evolutionary mechanism; one case involved acceleration of evolution, and the remaining cases seemed to be best explained by invoking HGT, mostly resulting in XGD. The statistical tests comparing the likelihoods of tree topologies with different positions of the corresponding branches validated the anomalous clustering and thus supported the case for HGT (Table 4). In functional terms, the majority of the inferred cases of HGT involved metabolic enzymes, in agreement with the complexity hypothesis, which postulated that genes coding for proteins that are not involved in tight interactions with other proteins as subunits of macromolecular complexes are more readily subject to HGT (25). It is also noteworthy that 5 of the top 30 cases included amianoacyl-tRNA synthetases, a category of enzymes which appears to be particularly prone to HGT (49, 50).
In COG0060 (isoleucyl-tRNA synthetase, IleS), both Rickettsia species and C. acetobutylicum are separated from their sister groups (α-Proteobacteria and gram-positive bacteria, respectively) and partition into the archaeon-eukaryote part of the tree (Fig. 5A). No alternative topology placing Rickettsia with other α-Proteobacteria and/or Clostridium with bacilli showed a detectable degree of support in statistical tests. The apparent origin of many bacterial IleRS proteins via HGT from eukaryotes is well documented (10, 49, 50); the resulting inconsistency of gene-to-gene distances is readily captured by the analysis presented here.
FIG. 5.

Maximum-likelihood trees for detected cases of probable XGD. Colors indicate species belonging to the bacterial lineage(s) for which a significant deviation from the clock-like model was detected, namely, α-Proteobacteria and gram-positive bacteria (A), gram-positive bacteria (B), α-Proteobacteria (C), and γ-Proteobacteria (D). The triangles represent collapsed clades (the numbers of proteins are indicated in brackets). (A) COG0060 (isoleucyl-tRNA synthetase, IleS). The following proteins and species are included: RP617 of R. prowazekii, RC0953 of R. conorii, CC0701of C. crescentus, BMEII1043 of B. melitensis, AGc1230 of A. tumefaciens, SMc00908 of S. meliloti, mlr8250 of M. loti, CAC3038 of C. acetobutylicum, SA1036 of S. aureus, BS_ileS of B. subtilis, BH2545 of B. halodurans, lin2127 of L. innocua, L0350 of L. lactis, SPy1513 of S. pyogenes, SP1659 of S. pneumoniae, FN0067 of F. nucleatum, TM1361 of Thermotoga maritima, aq_305 of Aquifex aeolicus, DR1335 of Deinococcus radiodurans, BB0833 of Borrelia burgdorferi, and TP0452 of Treponema pallidum. (B) COG1217 (predicted membrane GTPase involved in stress response, TypA). The following proteins and species are included: CAC1684 of C. acetobutylicum, SA0959 of S. aureus, BS_ylaG of B. subtilis, BH2632 of B. halodurans, lin1055 of L. innocua, L0370 of L. lactis, SPy1527 of S. pyogenes, SP0681 of S. pneumoniae, FN0634 of F. nucleatum, and DR1198 of D. radiodurans. (C) COG1282 (NAD/NADP transhydrogenase beta subunit, PntB). The following proteins and species are included: RP074 of R. prowazekii, RC0104 of R. conorii, CC3303 of C. crescentus, BMEII0325 of B. melitensis, AGc4531 of A. tumefaciens, SMc03938 of S. meliloti, mlr5184 of M. loti, slr1434 of Synechocystis sp., and all3408 of Nostoc sp. (D) COG1526 (uncharacterized protein required for formate dehydrogenase activity, FdhD). The following proteins and species are included: FdhD of E. coli K-12, ZfdhD of E. coli O157:H7, ECs4821 of E. coli O157:H7 EDL933, STM4038 of S. enterica serovar Typhimurium, YPO4060 of Y. pestis, HI0005 of H. influenzae, PM0410 of P. multocida, VC1519 of V. cholerae, PA5180 of Pseudomonas aeruginosa, RSp1050 and RSc2367 of Ralstonia solanacearum, CC1242 of C. crescentus, and Cj1508c of Campylobacter jejuni.
COG1217 (predicted membrane GTPase involved in stress response, TypA) is another case of the C. acetobutylicum sequence being separated from the sequences of the rest of the Bacillus-Clostridium group. The C. acetobutylicum protein clusters with the protein from Fusobacterium nucleatum (an arrangement which is observed in phylogenetic trees of many genes [Wolf, unpublished data]) and is separated from its cognate group by the cyanobacterial, actinobacterial, and Deinococcus branches (Fig. 5B). Although the support for this topology was not overwhelming (expected likelihood weight, 0.67 to 0.89 for the topology shown in Fig. 5B versus 0.10 to 0.29 for the alternative topology placing both Clostridium and Fusobacterium at the base of the gram-positive bacteria), HGT from the Fusobacterium lineage to Clostridium resulting in XGD seems to be the best interpretation of this tree.
In COG1282 (NAD/NADP transhydrogenase beta subunit, PntB) the Agrobacterium sequence clusters with γ-Proteobacteria and Neisseria, whereas the rest of the α-Proteobacteria cluster with Ralstonia (Fig. 5C). No alternative topology is viable according to the tests performed. Thus, in Agrobacterium, the original α-proteobacterial gene apparently has been displaced with the γ-proteobacterial ortholog.
COG1526 (uncharacterized protein required for formate dehydrogenase activity, FdhD) exemplifies the frequently observed separation of Vibrio from the rest of the γ-Proteobacteria (Fig. 5D). In this case, the Vibrio protein is closely related to the Ralstonia ortholog, and the Vibrio-Ralstonia clade joins the gram-positive branch instead of the proteobacterial branch. No topology joining Vibrio with other γ-Proteobacteria is supported. In this case, at least two HGT events have to be inferred: displacement of the original proteobacterial gene in either the Vibrio or the Ralstonia lineage by the ortholog from gram-positive bacteria and a secondary HGT between Vibrio and Ralstonia.
We also examined whether the presence a molecular clock violation in a COG is correlated for the three bacterial lineages studied. In the set of 108 conserved COGs, the Pearson correlation coefficients were −0.02 between γ- and α-Proteobacteria, −0.11 between γ-Proteobacteria and gram-positive bacteria, and 0.17 between α-Proteobacteria and gram-positive bacteria. The distribution of the number of anomalies detected in each COG (range, 0 to 3) nearly perfectly agreed with the expectation under the independence hypothesis [P(χ2) > 0.5]. Thus, somewhat counterintuitively, HGT appeared to occur independently in different lineages, and there was no obvious HGT propensity that could be considered a characteristic of evolution of an entire COG. Nor did we detect any significant connection between the violations of the molecular clock detected and the relative evolution rates. The distributions of v values were very similar for split and nonsplit sets, and the difference between the means was insignificant for all three groups according to the t test (data not shown).
General discussion and conclusions.
Comparative genomics allows researchers to restate old questions of evolutionary biology on a grander scale and, perhaps, in a more biologically meaningful way. Thus, rather than analyzing the molecular clock for one or a few selected protein families, it is possible to address the issue at the level of complete genomes and to ask what fraction of the genes follow the clock-like model of evolution and which genes demonstrably deviate from it. Here we describe a simple theoretical framework that allowed us to classify orthologous sets of bacterial genes (COGs) into these two categories. A similar relative rate test was described by Syvanen in his analysis of the acceleration of evolution of rRNA in eukaryotes (45).
We found that for several hundred COGs analyzed representing three well-defined bacterial lineages, the α-Proteobacteria, the γ-Proteobacteria, and the Bacillus-Clostridium group, the clock-like null hypothesis could not be rejected for ∼70% of the COGs, whereas the rest showed substantial anomalies. It should be noted that the null hypothesis employed here is, in fact, a soft clock, which, strictly speaking, does not require constancy of evolutionary rates for the genes analyzed. All that is required is that the rate distribution remains constant (i.e., all genes are allowed to accelerate or decelerate synchronously) (21). Notably, we also found that, within the set of 108 COGs that were represented by a single ortholog in all species analyzed from the three lineages, the relative evolutionary rates were strongly correlated among the lineages. This observation emphasizes the general validity of the soft genomic clock.
We also analyzed the nature of the observed anomalies and found that, for the most conspicuous anomalies, the majority were most readily explained by HGT from phylogenetically distant lineages. Importantly, this is a conservative estimate because we analyzed only a special set of well-behaved COGs, which contained exactly one ortholog from each of the species included. HGT events have been classified into three broad categories: (i) acquisition of genes new to the recipient lineage, (ii) acquisition of paralogs of resident genes, and (iii) XGD, in which a resident gene is displaced by an ortholog from a different lineage (30). The present analysis was designed to identify only cases of XGD (although in some exceptional situations we obtained indications of paralog acquisition where the COG analyzed seemed to contain hidden paralogs). The results suggest that XGD occurs during evolution of ∼10 to 15% of the bacterial genes. This relatively low fraction of HGT among single-ortholog bacterial genes is compatible with the notion that, at least within well-defined clades, such as the γ-Proteobacteria, the α-Proteobacteria, and the Bacillus-Clostridium group, these genes may be combined to produce organismal phylogenies, preferably after exclusion of the genes with detected HGT (9, 33, 51). However, the fraction of likely HGT detected here is considerably greater than that recently reported for single-ortholog gene sets from γ-Proteobacteria (33). It seems likely that the difference is a consequence of the criterion used for selection of orthologous gene sets in the latter study, in which only genes that are highly conserved within γ-Proteobacteria were examined. This criterion probably resulted in exclusion of orthologous sets deviating from the clock-like behavior.
An unexpected finding of this study is the lack of significant correlation among the three bacterial lineages analyzed with respect to the deviations from the clock model and the probable occurrence of XGD. An observation of such a deviation in any one of the lineages was a poor predictor of deviations in other lineages. This result seems to be poorly compatible with the complexity hypothesis (25) and similar notions concerning the dependence of HGT on biological function and is more in line with the ideas on random scatter and lineage-specific trends of HGT events (55). It should be noted that we analyzed a limited gene set and only one form of HGT (XGD). Functional correlates are likely to emerge in larger-scale studies, but our present results indicate that these connections are far from being absolute.
The approach described here allows rapid identification of orthologous gene sets whose evolution significantly deviates from the soft clock model. Interpretation of these deviations as lineage-specific acceleration of evolution, XGD, or a combination of the two requires detailed phylogenetic analysis. Nevertheless, we believe that this methodology has its own advantages and could be useful in the study of genome-wide evolutionary trends. In particular, this approach allows workers to detect significant deviations from the clock-like model of evolution in a particular lineage without using any information on species outside that lineage, such as the (often unknown) source of the potential HGT. More practically, the procedure described here could be suitable for removing anomalous COGs from multigene sets employed for construction of organismal phylogenies.
Acknowledgments
We thank Eva Czabarka and Georgy Karev (National Center for Biotechnology Information) for helpful discussions on the statistical analysis of the data.
P.S.N., M.S.G., and A.A.M. were partially supported by grants from the Howard Hughes Medical Institute (grant 55000309), the programs “Molecular and Cellular Biology” and “Origin and Evolution of the Biosphere” of the Russian Academy of Sciences, and the Fund for Support of Russian Science (MSG).
REFERENCES
- 1.Adachi, J., and M. Hasegawa. 1992. MOLPHY: programs for molecular phylogenetics. Institute of Statistical Mathematics, Tokyo, Japan.
- 2.Aravind, L., R. L. Tatusov, Y. I. Wolf, D. R. Walker, and E. V. Koonin. 1998. Evidence for massive gene exchange between archaeal and bacterial hyperthermophiles. Trends Genet. 14:442-444. [DOI] [PubMed] [Google Scholar]
- 3.Brochier, C., E. Bapteste, D. Moreira, and H. Philippe. 2002. Eubacterial phylogeny based on translational apparatus proteins. Trends Genet. 18:1-5. [DOI] [PubMed] [Google Scholar]
- 4.Brochier, C., H. Philippe, and D. Moreira. 2000. The evolutionary history of ribosomal protein RpS14: horizontal gene transfer at the heart of the ribosome. Trends Genet. 16:529-533. [DOI] [PubMed] [Google Scholar]
- 5.Bromham, L., and D. Penny. 2003. The modern molecular clock. Nat. Rev. Genet. 4:216-224. [DOI] [PubMed] [Google Scholar]
- 6.Brown, J. R. 2003. Ancient horizontal gene transfer. Nat. Rev. Genet. 4:121-132. [DOI] [PubMed] [Google Scholar]
- 7.Clarke, G. D., R. G. Beiko, M. A. Ragan, and R. L. Charlebois. 2002. Inferring genome trees by using a filter to eliminate phylogenetically discordant sequences and a distance matrix based on mean normalized BLASTP scores. J. Bacteriol. 184:2072-2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cutler, D. J. 2000. Understanding the overdispersed molecular clock. Genetics 154:1403-1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Daubin, V., N. A. Moran, and H. Ochman. 2003. Phylogenetics and the cohesion of bacterial genomes. Science 301:829-832. [DOI] [PubMed] [Google Scholar]
- 10.Doolittle, R. F., and J. Handy. 1998. Evolutionary anomalies among the aminoacyl-tRNA synthetases. Curr. Opin. Genet. Dev. 8:630-636. [DOI] [PubMed] [Google Scholar]
- 11.Doolittle, W. F. 1999. Lateral genomics. Trends Cell Biol. 9:M5-M8. [PubMed] [Google Scholar]
- 12.Doolittle, W. F. 1999. Phylogenetic classification and the universal tree. Science 284:2124-2129. [DOI] [PubMed] [Google Scholar]
- 13.Fay, J. C., and C. I. Wu. 2001. The neutral theory in the genomic era. Curr. Opin. Genet. Dev. 11:642-646. [DOI] [PubMed] [Google Scholar]
- 14.Felsenstein, J. 2004. Inferring phylogenies. Sinauer Associates, Sunderland, Mass.
- 15.Felsenstein, J. 1996. Inferring phylogenies from protein sequences by parsimony, distance, and likelihood methods. Methods Enzymol. 266:418-427. [DOI] [PubMed] [Google Scholar]
- 16.Fitch, W. M. 1970. Distinguishing homologous from analogous proteins. Syst. Zool. 19:99-106. [PubMed] [Google Scholar]
- 17.Gamieldien, J., A. Ptitsyn, and W. Hide. 2002. Eukaryotic genes in Mycobacterium tuberculosis could have a role in pathogenesis and immunomodulation. Trends Genet. 18:5-8. [DOI] [PubMed] [Google Scholar]
- 18.Gillespie, J. H. 1991. The causes of molecular evolution. Oxford University Press, Oxford, United Kingdom.
- 19.Gillespie, J. H. 1984. The molecular clock may be an episodic clock. Proc. Natl. Acad. Sci. USA 81:8009-8013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gogarten, J. P., W. F. Doolittle, and J. G. Lawrence. 2002. Prokaryotic evolution in light of gene transfer. Mol. Biol. Evol. 19:2226-2238. [DOI] [PubMed] [Google Scholar]
- 21.Grishin, N. V., Y. I. Wolf, and E. V. Koonin. 2000. From complete genomes to measures of substitution rate variability within and between proteins. Genome Res. 10:991-1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hansmann, S., and W. Martin. 2000. Phylogeny of 33 ribosomal and six other proteins encoded in an ancient gene cluster that is conserved across prokaryotic genomes: influence of excluding poorly alignable sites from analysis. Int. J. Syst. Evol. Microbiol. 50:1655-1663. [DOI] [PubMed] [Google Scholar]
- 23.Huang, X. 1994. On global sequence alignment. Comput. Appl. Biosci. 10:227-235. [DOI] [PubMed] [Google Scholar]
- 24.Iyer, L. M., E. V. Koonin, and L. Aravind. 2004. Evolution of bacterial RNA polymerase: implications for large-scale bacterial phylogeny, domain accretion, and horizontal gene transfer. Gene 335:73-88. [DOI] [PubMed] [Google Scholar]
- 25.Jain, R., M. C. Rivera, and J. A. Lake. 1999. Horizontal gene transfer among genomes: the complexity hypothesis. Proc. Natl. Acad. Sci. USA 96:3801-3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kimura, M. 1983. The neutral theory of molecular evolution. Cambridge University Press, Cambridge, United Kingdom.
- 27.Koonin, E. V. 2003. Horizontal gene transfer: the path to maturity. Mol. Microbiol. 50:725-727. [DOI] [PubMed] [Google Scholar]
- 28.Koonin, E. V., N. D. Fedorova, J. D. Jackson, A. R. Jacobs, D. M. Krylov, K. S. Makarova, R. Mazumder, S. L. Mekhedov, A. N. Nikolskaya, B. S. Rao, I. B. Rogozin, S. Smirnov, A. V. Sorokin, A. V. Sverdlov, S. Vasudevan, Y. I. Wolf, J. J. Yin, and D. A. Natale. 2004. A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. Genome Biol. 5:R7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koonin, E. V., and M. Y. Galperin. 2002. Sequence-evolution-function. Computational approaches in comparative genomics. Kluwer Academic Publishers, New York, N.Y. [PubMed]
- 30.Koonin, E. V., K. S. Makarova, and L. Aravind. 2001. Horizontal gene transfer in prokaryotes: quantification and classification. Annu. Rev. Microbiol. 55:709-742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koski, L. B., and G. B. Golding. 2001. The closest BLAST hit is often not the nearest neighbor. J. Mol. Evol. 52:540-542. [DOI] [PubMed] [Google Scholar]
- 32.Lawrence, J. G., and H. Hendrickson. 2003. Lateral gene transfer: when will adolescence end? Mol. Microbiol. 50:739-749. [DOI] [PubMed] [Google Scholar]
- 33.Lerat, E., V. Daubin, and N. A. Moran. 2003. From gene trees to organismal phylogeny in prokaryotes: the case of the gamma-proteobacteria. PLoS Biol. 1:E19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Makarova, K. S., V. A. Ponomarev, and E. V. Koonin. 2001. Two C or not two C: recurrent disruption of Zn-ribbons, gene duplication, lineage-specific gene loss, and horizontal gene transfer in evolution of bacterial ribosomal proteins. Genome Biol. 2:RESEARCH 0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Myers, E. W., and W. Miller. 1988. Optimal alignments in linear space. Comput. Appl. Biosci. 4:11-17. [DOI] [PubMed] [Google Scholar]
- 36.Nelson, K. E., R. A. Clayton, S. R. Gill, M. L. Gwinn, R. J. Dodson, D. H. Haft, E. K. Hickey, J. D. Peterson, W. C. Nelson, K. A. Ketchum, L. McDonald, T. R. Utterback, J. A. Malek, K. D. Linher, M. M. Garrett, A. M. Stewart, M. D. Cotton, M. S. Pratt, C. A. Phillips, D. Richardson, J. Heidelberg, G. G. Sutton, R. D. Fleischmann, J. A. Eisen, C. M. Fraser, et al. 1999. Evidence for lateral gene transfer between Archaea and bacteria from genome sequence of Thermotoga maritima. Nature 399:323-329. [DOI] [PubMed] [Google Scholar]
- 37.Notredame, C., D. G. Higgins, and J. Heringa. 2000. T-Coffee: a novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 302:205-217. [DOI] [PubMed] [Google Scholar]
- 38.Ochman, H., J. G. Lawrence, and E. A. Groisman. 2000. Lateral gene transfer and the nature of bacterial innovation. Nature 405:299-304. [DOI] [PubMed] [Google Scholar]
- 39.Ohta, T., and J. H. Gillespie. 1996. Development of neutral and nearly neutral theories. Theor. Popul. Biol. 49:128-142. [DOI] [PubMed] [Google Scholar]
- 40.Ponting, C. P., L. Aravind, J. Schultz, P. Bork, and E. V. Koonin. 1999. Eukaryotic signalling domain homologues in archaea and bacteria. Ancient ancestry and horizontal gene transfer. J. Mol. Biol. 289:729-745. [DOI] [PubMed] [Google Scholar]
- 41.Ragan, M. A. 2001. On surrogate methods for detecting lateral gene transfer. FEMS Microbiol. Lett. 201:187-191. [DOI] [PubMed] [Google Scholar]
- 42.Schmidt, H. A., K. Strimmer, M. Vingron, and A. von Haeseler. 2002. TREE-PUZZLE: maximum likelihood phylogenetic analysis using quartets and parallel computing. Bioinformatics 18:502-504. [DOI] [PubMed] [Google Scholar]
- 43.Sicheritz-Ponten, T., and S. G. Andersson. 2001. A phylogenomic approach to microbial evolution. Nucleic Acids Res. 29:545-552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sonnhammer, E. L., and E. V. Koonin. 2002. Orthology, paralogy and proposed classification for paralog subtypes. Trends Genet. 18:619-620. [DOI] [PubMed] [Google Scholar]
- 45.Syvanen, M. 2002. Rates of ribosomal RNA evolution are uniquely accelerated in eukaryotes. J. Mol. Evol. 55:85-91. [DOI] [PubMed] [Google Scholar]
- 46.Tatusov, R. L., N. D. Fedorova, J. D. Jackson, A. R. Jacobs, B. Kiryutin, E. V. Koonin, D. M. Krylov, R. Mazumder, S. L. Mekhedov, A. N. Nikolskaya, B. S. Rao, S. Smirnov, A. V. Sverdlov, S. Vasudevan, Y. I. Wolf, J. J. Yin, and D. A. Natale. 2003. The COG database: an updated version includes eukaryotes. BMC Bioinformatics 4:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tatusov, R. L., E. V. Koonin, and D. J. Lipman. 1997. A genomic perspective on protein families. Science 278:631-637. [DOI] [PubMed] [Google Scholar]
- 48.Tatusov, R. L., D. A. Natale, I. V. Garkavtsev, T. A. Tatusova, U. T. Shankavaram, B. S. Rao, B. Kiryutin, M. Y. Galperin, N. D. Fedorova, and E. V. Koonin. 2001. The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res. 29:22-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Woese, C. R., G. J. Olsen, M. Ibba, and D. Soll. 2000. Aminoacyl-tRNA synthetases, the genetic code, and the evolutionary process. Microbiol. Mol. Biol. Rev. 64:202-236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wolf, Y. I., L. Aravind, N. V. Grishin, and E. V. Koonin. 1999. Evolution of aminoacyl-tRNA synthetases—analysis of unique domain architectures and phylogenetic trees reveals a complex history of horizontal gene transfer events. Genome Res. 9:689-710. [PubMed] [Google Scholar]
- 51.Wolf, Y. I., I. B. Rogozin, N. V. Grishin, and E. V. Koonin. 2002. Genome trees and the tree of life. Trends Genet. 18:472-479. [DOI] [PubMed] [Google Scholar]
- 52.Wolf, Y. I., I. B. Rogozin, N. V. Grishin, R. L. Tatusov, and E. V. Koonin. 2001. Genome trees constructed using five different approaches suggest new major bacterial clades. BMC Evol. Biol. 1:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wolf, Y. I., I. B. Rogozin, and E. V. Koonin. 2004. Coelomata and not ecdysozoa: evidence from genome-wide phylogenetic analysis. Genome Res. 14:29-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yang, Z. 1997. PAML: a program package for phylogenetic analysis by maximum likelihood. Comput Appl Biosci. 13:555-556. [DOI] [PubMed] [Google Scholar]
- 55.Zhaxybayeva, O., P. Lapierre, and J. P. Gogarten. 2004. Genome mosaicism and organismal lineages. Trends Genet. 20:254-260. [DOI] [PubMed] [Google Scholar]
- 56.Zuckerkandl, E., and L. Pauling. 1962. Molecular evolution, p. 189-225. In M. Kasha and B. Pullman (ed.), Horizons in biochemistry. Academic Press, New York, N.Y.
- 57.Zuckerkandl, E., and L. Pauling. 1965. Molecules as documents of evolutionary history. J. Theor. Biol. 8:357-366. [DOI] [PubMed] [Google Scholar]