

Abstract
Hypertrophy allows the heart to adapt to workload but culminates in later pump failure; how it is achieved remains uncertain. Previously, we showed that hypertrophy is accompanied by activation of cyclin T/Cdk9, which phosphorylates the C-terminal domain of the large subunit of RNA polymerase II, stimulating transcription elongation and pre-mRNA processing; Cdk9 activity was required for hypertrophy in culture, whereas heart-specific activation of Cdk9 by cyclin T1 provoked hypertrophy in mice. Here, we report that αMHC-cyclin T1 mice appear normal at baseline yet suffer fulminant apoptotic cardiomyopathy when challenged by mechanical stress or signaling by the G-protein Gq. At pathophysiological levels, Cdk9 activity suppresses many genes for mitochondrial proteins including master regulators of mitochondrial function (peroxisome proliferator-activated receptor gamma coactivator 1 (PGC-1), nuclear respiratory factor-1). In culture, cyclin T1/Cdk9 suppresses PGC-1, decreases mitochondrial membrane potential, and sensitizes cardiomyocytes to apoptosis, effects rescued by exogenous PGC-1. Cyclin T1/Cdk9 inhibits PGC-1 promoter activity and preinitiation complex assembly. Thus, chronic activation of Cdk9 causes not only cardiomyocyte enlargement but also defective mitochondrial function, via diminished PGC-1 transcription, and a resulting susceptibility to apoptotic cardiomyopathy.
Keywords: cardiac, cyclin-dependent kinase-9, cyclin T, heart failure, mitochondria
Introduction
Hypertrophy (growth by cell enlargement) is the characteristic response to numerous biological signals that impinge on the adult heart including mechanical stress, growth-promoting ligands, and signaling proteins. The hypertrophic phenotype is characterized by increased myocyte size via global increases in RNA and protein content per cell along with, more variably, transcriptional reprogramming that can resemble a ‘fetal' gene program (Olson and Schneider, 2003). Because hypertrophy initially ameliorates wall stress and sustains cardiac output, it has been viewed as salutary adaptation. However, prolonged hypertrophy commonly eventuates in heart failure or sudden death, indicating that this phenotype is not wholly beneficial. If increased mass is construed to predispose the heart to decompensation, blunting hypertrophic growth might prevent or retard heart failure. Alternatively, specific changes of gene expression, such as those impairing myocyte survival, might be the pivotal adverse response, not mass itself.
Besides translation and transcription initiation, cardiac growth is also likely to be governed by transcription elongation (Sano et al, 2002). The hypophosphorylated form of RNA polymerase II (RNAPIIa) is recruited to promoters and starts the elongation phase of transcription only when phosphorylated (IIo). Phosphorylation occurs in the C-terminal domain (CTD), containing 52 copies of a YSPTSPS heptad repeat, and is crucial for productive mRNA synthesis, enabling RNAPII to escape from promoter-proximal pausing and engage factors for pre-mRNA processing (Orphanides and Reinberg, 2002; Pokholok et al, 2002; Shilatifard et al, 2003). In mice, deleting just 13 of the repeats causes growth retardation and embryonic lethality (Litingtung et al, 1999). In mouse myocardium, we showed that RNAPII is phosphorylated as a consequence of diverse hypertrophic stimuli (pressure overload, the small G-protein Gαq, and the calcium-dependent phosphatase calcineurin) and that both principal CTD kinases—cyclin-dependent kinase (Cdk) 7 and Cdk9—become activated in chronic hypertrophy (Sano et al, 2002).
During the transition from transcript initiation to transcript elongation, Cdk7 and Cdk9 are believed to phosphorylate CTD sequentially and, hence, both to mediate mRNA synthesis. Cyclin T/Cdk9 is also known as positive transcription elongation factor-b (P-TEFb); the cyclin H/Cdk7/menage-a-trois (MAT1) complex comprises the kinase subunit of general transcription factor TFIIH and may have a role in mRNA capping. In its capacity as Cdk-activating kinase, cyclin H/Cdk7/MAT1 also phosphorylates Cdks that mediate cell cycling (Rossi et al, 2001). By viral delivery of catalytically inactive kinases to cultured cardiac myocytes, we found that Cdk9 activity is necessary for agonist-induced RNAPII phosphorylation and myocyte growth, with Cdk7 activity dispensable in short-term studies. In all the hypertrophic models, Cdk9 activation involves the dissociation of a noncoding small nuclear RNA, 7SK, which bridges P-TEFb to the protein that in turn inhibits Cdk9 function (Michels et al, 2003; Yik et al, 2003). Activating Cdk9 via knockdown of 7SK RNA or forced expression of cyclin T1 causes myocyte enlargement in culture and transgenic mice, respectively.
These reciprocal loss- and gain-of-function findings implicate Cdk9 as a pivotal regulator of pathophysiological heart growth, yet left unanswered whether increased Cdk9 activity contributes to the transition from hypertrophy to failure (and, if so, how). Here, we report that activation of Cdk9 at the biologically relevant levels found in chronic cardiac disorders causes not only myocyte enlargement but also selective suppression of peroxisome proliferator-activated receptor gamma coactivator 1 (PGC-1), a master regulator of mitochondrial biogenesis and function (Wu et al, 1999; Kelly and Scarpulla, 2004), culminating in mitochondrial defects, enhanced myocyte apoptosis, predisposition to heart failure, and early demise.
Results
RNAPII phosphorylation and Cdk9 activity are increased in human heart failure
First, to test if our prior experimental models could reasonably pertain to human heart disease, we compared ventricular samples from patients with heart failure due to dilated cardiomyopathy (N=8) versus age-matched nondiseased hearts (N=8; Figure 1). Measured by immune complex kinase assays with the recombinant CTD as substrate, Cdk9 activity increased 2.3±0.3-fold in failing samples (P<0.05). All eight diseased samples showed increased kinase activity. As in our mouse studies, Cdk9 activation occurred at unchanged levels of cyclin T1 and Cdk9 protein expression. Cdk7 activity likewise increased significantly (1.6±0.2-fold; P<0.05), associated with higher levels of MAT1, but was elevated in only a minority of samples. Hyperphosphorylation of endogenous human RNAPII was substantiated in failing hearts at the preferred sites within the CTD repeat for both Cdk9 (Ser2) and Cdk7 (Ser5), although Ser2 phosphorylation was seen more consistently.
Figure 1.
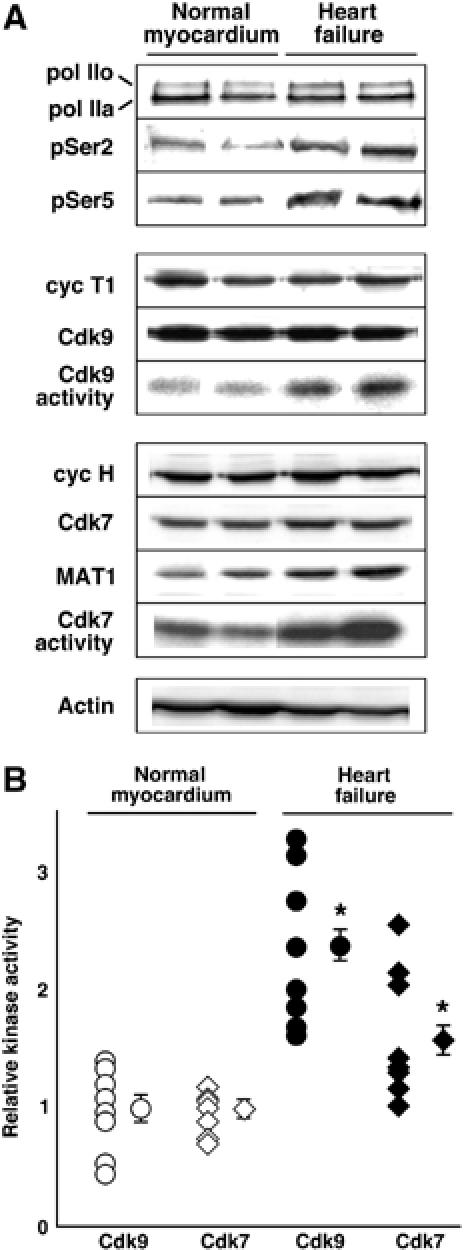
Activation of endogenous human RNAPII CTD kinases (cyclin T/Cdk9 and cyclin H/Cdk7/MAT1) in heart failure. (A) Western blots are shown for RNAPII, the Cdk9 complex, the Cdk7 complex, and total actin as a control; Cdk activities, determined by immune complex kinase assays, are shown as autoradiograms. (B) Individual and group results for Cdk9 and Cdk7 activities. *P<0.05 versus normal myocardium.
Cdk9 activation predisposes hearts to decompensation under stress
Previously, we reported that (1) Cdk9 activity is downregulated during cardiac maturation in part by decreased expression of cyclin T1, (2) cardiomyocyte-restricted expression of cyclin T1 maintains Cdk9 activity at its normal embryonic level, and (3) heart mass and myocyte size increase in proportion to cyclin T1 expression and resulting Cdk9 activity (Sano et al, 2002). As an essential baseline for the potential impact of Cdk9 on cardiac adaptation to stress, a more comprehensive biochemical and functional analysis was undertaken with the αMHC-cyclin T1 mice (Figure 2 and Supplementary Information). Immunoblotting confirmed that CTD phosphorylation was increased by transgenic expression of cyclin T1. Cdk9 levels were also increased, secondarily, by the cyclin T1 transgene. By Doppler echocardiography, cyclin T1 mice had grossly normal left ventricular (LV) systolic function—fractional shortening and peak aortic flow velocity—compared with nontransgenic siblings. Diastolic function at 3 months also was well preserved, unlike αMHC-Gαq mice at that age.
Figure 2.
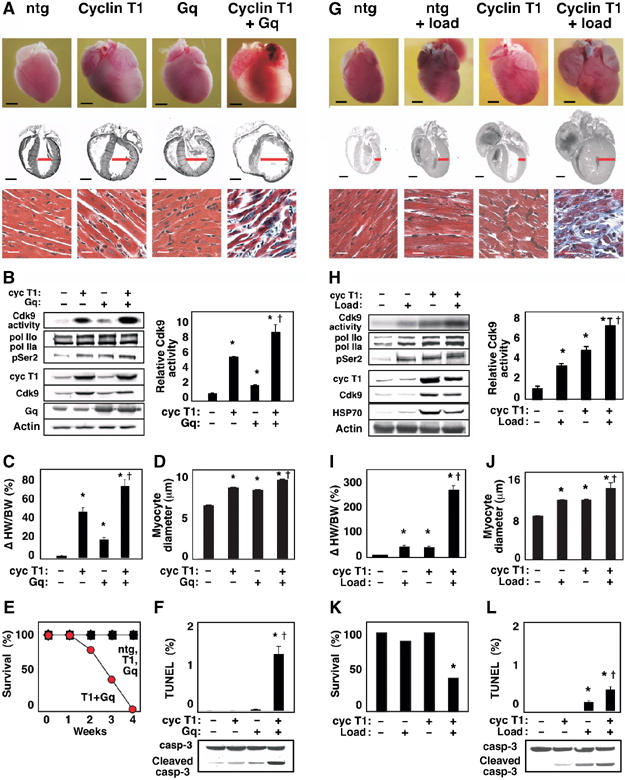
Cdk9 activation by cyclin T1 predisposes to heart failure, in concert with genetic or hemodynamic triggers of hypertrophy. (A–F) Cyclin T1 exacerbates the response to Gq. Mice were heterozygous for the indicated transgenes. Ntg, nontransgenic littermates. (A) Upper and middle rows, dilated cardiomyopathy (age, 2 weeks). Ventricular diameter is denoted in red. Bar, 2 mm. Lower row, Gomori-trichrome stain. Bar, 20 μm. (B) Synergistic activation of Cdk9 (immune complex kinase assay) and Ser2 phosphorylation of endogenous RNAPII (Western blotting). See Figure 1A for details. Right, mean±s.e. (C) Increased heart-weight-to-body-weight ratio. (D) Increased myocyte diameter. (E) Rapid mortality. (F) Increased apoptosis, detected as TUNEL-positive cardiomyocytes and caspase-3 cleavage. (B–D) *P<0.05 versus nontransgenic littermates; †P<0.05 versus cyclin T1 or Gq alone. (G–L) Cyclin T1 exacerbates the response to mechanical stress. (G) Upper and middle rows, ventricular and atrial enlargement in 3-month-old mice after partial transverse aortic occlusion for 21 days. Ventricular wall thickness is denoted in red. Bar, 2 mm. Lower row, Gomori-trichrome stain. Bar, 20 μm. (H) Synergistic activation of Cdk9 (immune complex kinase assay) and Ser2 phosphorylation of RNAPII (Western blot). (I) Increased heart-weight-to-body-weight ratio. (J) Increased myocyte diameter. (K) Increased mortality (21 days after occlusion). (L) Increased apoptosis. TUNEL stain and caspase-3 cleavage, 2 weeks after occlusion. (H–J) *P<0.05 versus nontransgenic control littermates; †P<0.05 versus cyclin T1 or load individually. N=5 for each condition shown (B–D, F, H–J, L).
As a provocative test of whether increased Cdk9 activity is adaptive or adverse, we crossed αMHC-cyclin T1 transgenic mice to the αMHC-Gq line (Figure 2A–F), and subjected the αMHC-cyclin T1 mice to mechanical load (Figure 2G–L). Each of these three states, independently, is a model of compensated concentric hypertrophy, and the mutual exacerbation by Gq plus mechanical stress is well known (Sakata et al, 1998). However, it was conjectural if Cdk9 would interact with either of the established pathways and, if so, with what direction of effect.
Rapid ventricular dilatation, wall thinning, and fibrosis resulted from the cyclin T1 × Gq cross (age, 2 weeks; Figure 2A). Gq itself activates cardiac Cdk9, but activity was further augmented by coinheriting the cyclin T1 transgene (Figure 2B). In double-transgenic mice, the heart-to-body-weight ratios and myocyte cross-sectional diameters increased beyond those provoked by either transgene singly (Figure 2C and D). Invariably, heart failure and death ensued by just 4 weeks of age (Figure 2E). Bigenic mice showed severe myofibril disarray and fibrosis (Figure 2A), plus increased cleavage of caspase-3 and an increased prevalence of TUNEL-positive myocytes, indicating apoptosis: 1.2% versus <0.01% for Gq alone, cyclin T1 alone, or nontransgenic littermates (P<0.001; Figure 2F).
We next tested for a functional interaction of cyclin T1 and mechanical stress. The effect of load and cyclin T1 on heart size was more than additive, with a predominant effect in this context on wall thickening, not dilatation (age, 3 months; duration of load, 21 days; Figure 2G). Both genotypes had received a comparable hemodynamic load, as determined by right-to-left carotid artery flow velocity ratios after constricting the transverse aorta. At 3 weeks after banding, wild-type mice demonstrated a 3.0±0.2-fold increase in Cdk9 activity (P<0.05; Figure 2H) and a 35±6% increase in heart-to-body-weight ratio (P<0.05; Figure 2I). Although baseline Cdk9 activity was already 4.4±0.5-fold higher in cyclin T1 mice than in wild-type littermates (P<0.01), banding elicited a further 1.5±0.2-fold increase (P<0.01; Figure 2H). Like this combined effect on Cdk9, the αMHC-cyclin T1 mice showed even greater increases than nontransgenic littermates in the heart-to-body-weight ratio provoked by load (74.7±0.4%, P<0.001; Figure 2I). Banding increased myocyte diameter from 9.3±0.1 to 12.6±0.1 μm in wild-type mice (P<0.001), and from 12.7±0.1 to 15.1±1.2 μm in cyclin T1 mice (P<0.001; Figure 2J).
Noninvasive echo-Doppler measurements are well suited to perform consecutive longitudinal studies of cardiac performance in mice, allowing each animal to serve as its own control (before and after aortic constriction). Cyclin T1 caused no decrement in baseline systolic function, but potentiated the dysfunction seen after 7 days of load. Peak aortic flow velocity decreased to half the level of nontransgenic controls (before: 109.6±2.2 cm s−1; after: 67.2±3.7 cm s−1; P<0.05). Postoperative lethality was prevalent in αMHC-cyclin T1 mice (62%; N=13) but not the control littermates (11%; N=9; Figure 2K). More-than-additive effects were seen on myocardial fibrosis (Figure 2G). In agreement with the disparate effect of cyclin T1 on load versus Gq macroscopically (increased concentric hypertrophy, not dilatation), cyclin T1 induced a much lower prevalence of apoptosis in banded mice (Figure 2L). Conceivably, load-induced pathways that Gq does not engage, such as gp130 or Akt (Hirota et al, 1999; Howes et al, 2003), might underlie such differences.
Thus, although the baseline phenotype of αMHC-cyclin T1 mice is benign, increased Cdk9 activity predisposes the myocardium to rapid decompensation under stress imposed by the hemodynamic and genetic triggers for hypertrophy we tested.
Cdk9 activation provokes an atypical cardiac gene program
To seek a molecular signature for the susceptibility to heart failure conferred by cyclin T1, we compared microarray expression profiles of ventricular myocardium from αMHC-cyclin T1 mice, αMHC-Gq mice, and nontransgenic mice (not shown). QRT–PCR was then performed to confirm selected findings and survey additional pertinent genes (Figure 3). Since Cdk9 activity promotes transcription elongation, persistent expression of cyclin T1 at the embryonic level might be expected to cause a global increase of cardiac mRNA synthesis. Instead, a more nuanced profile was seen. Even though HSP70 was upregulated more than 10-fold compared with nontransgenic littermates, in accordance with its strict dependence on P-TEFb (Lis et al, 2000; Shim et al, 2002), various subsets of genes were downregulated unexpectedly in αMHC-cyclin T1 hearts. Indeed, despite the fact that αMHC-cyclin T1 caused an increase in cardiac mass even greater than in αMHC-Gq mice, little or no induction occurred for common hypertrophic markers (ANP, BNP, α-skeletal actin, βMHC). Other cardiomyocyte-specific genes were suppressed (αMHC, SERCA2, Ryr2, Cx43). None of several cardiac-specific transcription factors showed any change in expression (Nkx2.5, MEF2c, GATA4, SRF, Tbx5), suggesting that another mechanism must be invoked in modulating the mRNA levels of their downstream targets.
Figure 3.
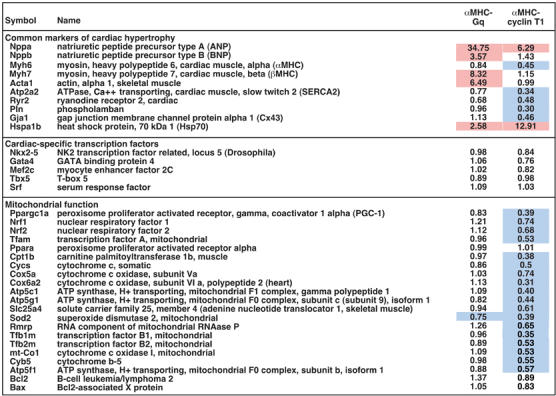
Cyclin T1 preferentially represses genes for mitochondrial function. Ventricular transcript levels were determined by real-time QRT–PCR, and normalized to GAPDH. Results shown are mean changes in gene expression in the hypertrophied hearts of cyclin T1 and Gq transgenic mice, relative to nontransgenic littermates (N=5). Induction (⩾1.5) and repression (⩽0.75) are highlighted in red and blue, respectively.
The largest cluster of genes suppressed by cyclin T1 identified by the microarray survey comprised genes for mitochondrial function: enzymes for respiratory chain complexes, the TCA cycle, and β-oxidation of fatty acids, antioxidant enzymes, mitochondrial creatine kinase, mitochondrial ribosomal proteins, mitochondrial import proteins, mitochondrial RNA processing, and mitochondrial transcription factors (Figure 3 and unpublished results). Impaired mitochondrial function would conceivably explain why cyclin T1 mice decompensate dramatically when challenged with cardiac stress. We speculated that the coordinated suppression of so many genes might result from impaired function or expression of a limiting transcriptional regulator. An especially apt candidate was PGC-1, a master regulator of mitochondrial biogenesis and function (Wu et al, 1999; Kelly and Scarpulla, 2004). PGC-1 induces and coactivates transcription factors NRF-1 and NRF-2, which in turn induce many genes for mitochondrial proteins; furthermore, NRF-1 induces mitochondrial transcription factor A (TFAM), which regulates mitochondrial DNA (mtDNA) replication and transcription. We were unable to detect PGC-1 mRNA in mouse myocardium reliably with microarrays, although the transcript was readily measured using QRT–PCR. By QRT–PCR, cardiac PGC-1 expression was repressed by 60% in αMHC-cyclin T1 mice, compared with nontransgenic littermates (P<0.05; Figure 3). NRF-1, NRF-2, and the NRF-1-dependent gene TFAM were also repressed, by 26% (P<0.05), 32% (P<0.05), and 47% (P<0.05), respectively. Consistent with downregulation of TFAM, transcription of many genes encoded by mtDNA was also suppressed in cyclin T1 transgenic hearts (Cox1, 47%, P<0.05; cytochrome b, 45%, P<0.05; ATP synthase 8, 43%, P<0.05).
Next, we tested if altered gene expression was associated with functional consequences. By transmission electron microscopy, ventricular myocytes in αMHC-cyclin T1 mice had mitochondria containing fewer and less well-organized cristae than did nontransgenic myocytes (Figure 4A). Although citrate synthase (Krebs cycle) was unaffected, many respiratory chain enzymes were significantly decreased in cyclin T1 hearts, such as succinate dehydrogenase (complex II), succinate cytochrome c reductase (complex II+III), NADH dehydrogenase (complex I), NADH cytochrome c reductase (complex I+III), and cytochrome c oxidase (complex IV; Figure 4B). Little or no change occurred in mtDNA copy number (Figure 4C). Although TFAM is essential for both mitochondrial gene transcription and replication, transcription is driven only at high levels of TFAM, with replication active across a wider range (Falkenberg et al, 2002): this dosage effect potentially explains the downregulation of genes for mitochondrial function without a concomitant decrease in mtDNA.
Figure 4.
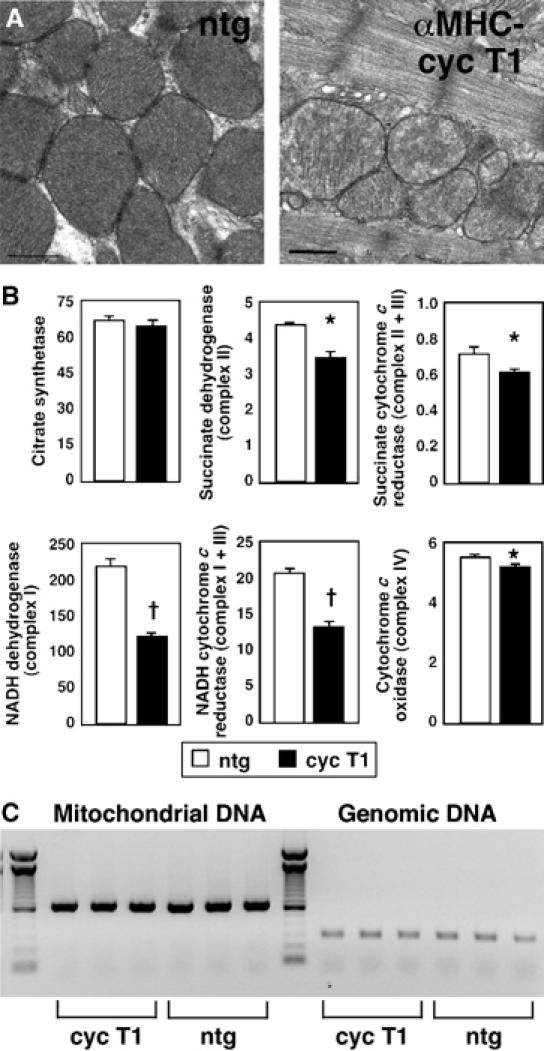
Cyclin T1 disrupts mitochondrial structure and function in mouse myocardium. (A) Abnormal mitochondrial ultrastructure in αMHC-cyclin T1 mice (right), compared with nontransgenic littermate controls (ntg; left). Bar, 0.5 μm. (B) Deficient mitochondrial enzyme activities in αMHC-cyclin T1 mice (filled bars), compared with nontransgenic littermates (open bars; N=8 for both genotypes). (C) MtDNA copy number in ventricular myocardium. DNA from nontransgenic and αMHC-cyclin T1 mice was used to amplify a 648 bp fragment of mtDNA (mt-Cox1) and a 316 bp fragment of genomic DNA (5S ribosomal RNA). Left lane, 100 bp ladder.
Thus, although LV mechanical performance was largely sustained—even at 3 months to 1 year of age—and apoptosis and fibrosis were absent, there exists a latent biological dysfunction in αMHC-cyclin T1 mice. Despite lack of overt abnormalities under basal conditions, the inherent mitochondrial dysfunction might account for the acute decompensation seen when these hearts were subjected to stress.
Loss of PGC-1 mediates the downregulation of genes for mitochondrial proteins by cyclin T/Cdk9
To analyze more directly the effect of Cdk9 on gene expression, we subjected cultured rat cardiomyocytes to adenovirus-mediated gene transfer. Overexpression of cyclin T1 increased Cdk9 activity; increasing Cdk9 protein levels had by itself no effect on kinase activity; expressing Cdk9 and cyclin T1 together synergistically enhanced Cdk9 activity (Figure 5A). Hence, cyclin T1 is limiting, as in mouse myocardium: αMHC-cyclin T1 increased Cdk9 activity, whereas αMHC-Cdk9 did not (Sano et al, 2002).
Figure 5.
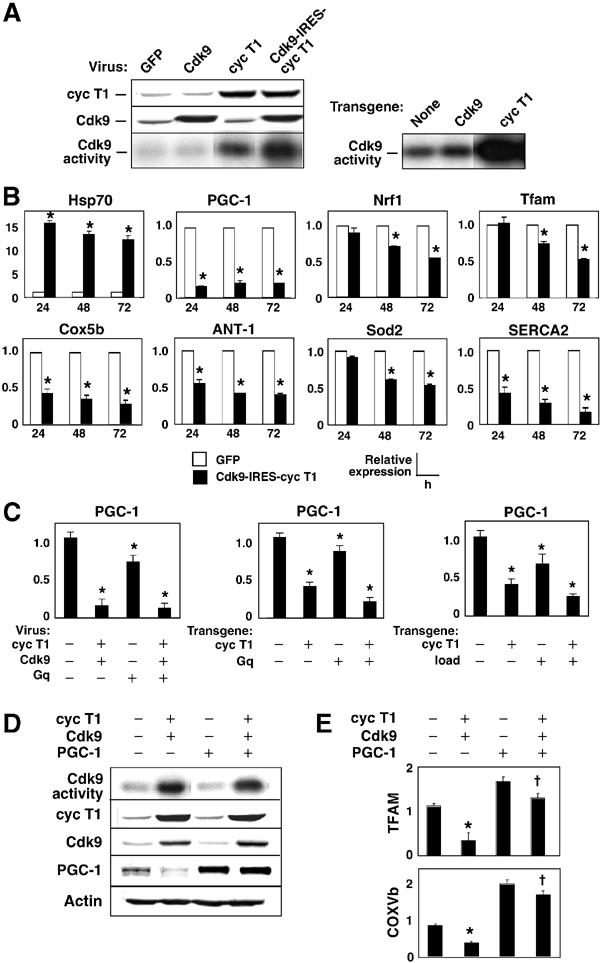
Cyclin T1/Cdk9 suppresses PGC-1, thereby downregulating other genes for mitochondrial function. (A) Left, Western blot and immune complex kinase assays, showing levels of cyclin T1 and Cdk9 48 h after viral gene transfer to cardiac myocytes and their synergistic effect on CTD phosphorylation. Right, Myocardium from wild-type, αMHC-Cdk9 mice, and αMHC-cyclin T1 mice, for comparison. (B) Cyclin T1/Cdk9 suppresses multiple genes for mitochondrial function. RNA from cardiomyocytes 24–72 h after gene transfer was subjected to QRT–PCR analysis. (C) Suppression of PGC-1 mRNA in cultured cardiac myocytes (left) and mouse myocardium (center, right). In vivo, both Gq and mechanical load exacerbate the suppression of PGC-1 by cyclin T1. (D) Cyclin T1/Cdk9 markedly impaired PGC-1 protein expression. Coinfection with PGC-1 virus had no confounding effect on cyclin T1 levels, Cdk9 levels, or Cdk9 kinase activity. (E) Downregulation of genes for mitochondrial function was reversed by supplementing PGC-1. Cells were subjected to virus encoding cyclin T1/Cdk9±virus encoding PGC-1. Gene expression was normalized to GAPDH (Handschin et al, 2003). (B, C (left), E) *P<0.05 versus GFP; †P<0.05 versus cyclin T1/Cdk9. (C (right, middle)) *P<0.05 versus nontransgenic littermates.
To elucidate the relationships among Cdk9, PGC-1, and putative targets of PGC-1 in cardiac myocytes, we analyzed gene expression serially, 24–72 h after gene transfer (Figure 5B). Much as in αMHC-cyclin T1 mice, HSP70 was induced 15-fold by cyclin T1/Cdk9 in culture. PGC-1 mRNA was downregulated by 85% within 24 h, remained suppressed throughout the experiment, and was not suppressed further by coinfection with virus encoding Gq (Figure 5C). Reported targets of PGC-1 (NRF-1, TFAM, Cox5b, cytochrome c, Sod2) decreased more slowly. In mouse myocardium, the cyclin T1 transgene suppressed PGC-1 mRNA expression less completely, and combinations of cyclin T1 plus Gq or after-load suppressed PGC-1 roughly additively, by 75–80% (Figure 5C). Because Gq and load potentiate Cdk9 activity (Figure 2), this may represent a requirement for costimulation or, more simply, a dosage effect.
We speculated that coordinated downregulation of genes for mitochondrial function might result from suppression of PGC-1 by cyclin T1/Cdk9. To test this hypothesis, we first confirmed by immunoblotting that cyclin T1/Cdk9 suppressed PGC-1 protein levels (Figure 5D). Next, we supplemented endogenous PGC-1 by viral gene transfer. Neither cyclin T1/Cdk9 levels nor Cdk9 kinase activity was affected by exogenous PGC-1. Hence, PGC-1 does not act trivially, by interfering with cyclin T1/Cdk9 expression or catalytic activity. As predicted, TFAM and Cox5b were rescued by restoring PGC-1 (Figure 5E). Thus, PGC-1 blocks the suppression of these genes by cyclin T1/Cdk9. In contrast, PGC-1 did not reverse the elevation of HSP70 (not shown).
Cyclin T1/Cdk9 suppresses PGC-1 transcription and preinitiation complex assembly
These expression data—in vivo and in vitro—indicate that cyclin T1/Cdk9 might function, apart from other potential roles, in gene-specific repression. To ascertain how cyclinT1/Cdk9 suppresses PGC-1, we next tested the effect of cyclin T1/Cdk9 on PGC-1 promoter activity, using cotransfection assays in CV-1 cells (Figure 6A and B). We chose to compare constructs driven by the HSP70 promoter (−2509/+231) and PGC-1 promoter (−3541/+93), as these transcripts were induced and repressed, respectively, by cyclin T/Cdk9 in the experiments above. We created 3′ truncations of both, deleted in the promoter-proximal region (HSP70 −2509/+20; PGC-1 −3541/+23), as Cdk9 is believed to be recruited to the promoter-proximal region and to stimulate elongation by phosphorylating RNAPII at this position (Orphanides and Reinberg, 2002; Pokholok et al, 2002; Shilatifard et al, 2003). The default hypothesis for deleting the promoter-proximal region is a defect in the positive response to cyclin T1/Cdk9.
Figure 6.
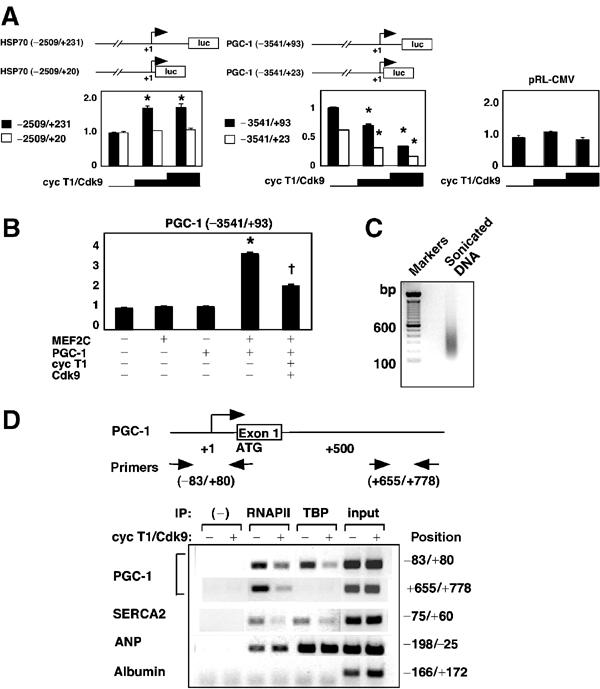
Cyclin T1/Cdk9 selectively suppresses PGC-1 transcription and formation of the PGC-1 preinitiation complex. (A) CV-1 cells were transfected with firefly luciferase reporter genes (0.2 μg) plus pRL-CMV (50 ng), in the presence or absence of cyclin T1 and Cdk9 vectors (0.2 or 0.4 μg). HSP70 and PGC-1 luciferase activities 48 h after transfection, corrected for pRL-CMV, are shown relative to those in the absence of exogenous cyclin T1/Cdk9. *P<0.01 versus baseline luciferase activity; N=6. (B) Cyclin T1/Cdk9 inhibits PGC-1 promoter activity even after forced expression of its activators MEF2C and PGC-1. CV-1 cells were transfected as above, plus MEF2C and PGC-1 vectors where indicated (0.4 μg). *P<0.01 versus baseline luciferase activity; †P<0.01 versus MEF2C- plus PGC-1-induced luciferase activity; N=6. (C, D) Cyclin T1/Cdk9 blocks the recruitment of TBP and RNAPII to the PGC-1 promoter. (C) Ethidium bromide-stained agarose gel of the DNA fragments after sonication. (D) Above, schematic representation of the PGC-1 gene and PCR primers. Below, ChIP assays of promoter occupancy by TBP and RNAPII, measured 24 h after viral delivery of cyclin T1/Cdk9 versus GFP. PCR was performed using chromatin recovered from protein A–agarose in the absence of antibody as the negative control (−) and chromatin from the same lysates prior to protein A–agarose as the ‘input' control.
Cyclin T1/Cdk9 increased activity of the HSP70 promoter 1.6-fold (P<0.01; Figure 6A, left). Despite baseline activity comparable to that of the longer construct, the 3′ truncation had no response to cyclin T1/Cdk9. Thus, the promoter-proximal region +20 to +231 was necessary for upregulation. In contrast, cyclin T1/Cdk9 suppressed the PGC-1 promoter and did not require the promoter-proximal region (Figure 6A, center). Cyclin T1/Cdk9 did not affect the CMV promoter, a constitutive control (Figure 6A, right). The combination of PGC-1 plus the MADS box transcription factor MEF2C increased PGC-1 promoter activity, as reported (Handschin et al, 2003), and cyclin T1/Cdk9 reduced PGC-1 promoter activity even with forced expression of these inducers (Figure 6B). Suppression of the PGC-1 promoter by cyclin T1/Cdk9 was corroborated in cultured cardiomyocytes (not shown).
To investigate how cyclin T1/Cdk9 suppressed the PGC-1 promoter, we next performed chromatin immunoprecipitation (ChIP) assays using cardiomyocytes 24 h after viral delivery of cyclin T1/Cdk9 or GFP alone (Figure 6C and D). After crosslinking, DNA was sonicated to a length of 200–500 bp (Figure 6C). With this method, a gene's transcription is correlated with the presence of RNAPII and TBP on the endogenous gene's promoter. As positive and negative controls, we showed the promoters of genes active in cardiomyocytes to be occupied with TBP and RNAPII under baseline conditions (PGC-1, SERCA2, ANP), whereas the promoter of a representative cardiac-silent gene was unoccupied (albumin). For expressed genes, RNAPII was found in both the promoter and downstream regions, and TBP was found, as predicted, in the promoter only. Cyclin T1/Cdk9 inhibited the recruitment of RNAPII and TBP to the PGC-1 promoter and decreased the level of RNAPII found in the first intron (Figure 6D). Likewise, cyclin T1/Cdk9 suppressed the recruitment of TBP and RNAPII onto the SERCA2 promoter, another gene downregulated by cyclin T1/Cdk9. In contrast, cyclin T1/Cdk9 did not alter occupancy of the ANP promoter by RNAPII or TBP, concordant with the fact that cyclin T1 did not affect ANP levels. Hence, cyclin T1/Cdk9 suppresses PGC-1 by reducing selectively the transcription of the PGC-1 promoter and perturbing selectively the assembly of the PGC-1 preinitiation complex. While unexpected at the inception of our studies, such precision is consistent with recent data for the involvement of cyclin T/Cdk9 with several gene-specific eukaryotic transcription factor complexes (Barboric et al, 2001; Eberhardy and Farnham, 2002). While consistent with a suggested example of Cdk9-dependent repression (de Falco et al, 2000), our results do not exclude inducing a repressor as an intermediate step.
PGC-1 rescues cardiomyocytes from apoptosis induced by Gq plus cyclin T1/Cdk9
As shown in Figure 2, mice with a heart-specific increase in Cdk9 activity develop a lethal apoptotic cardiomyopathy if challenged with mechanical stress or Gq, a mediator of mechanical signal transduction in the heart (Olson and Schneider, 2003). To test this interplay under more acute reductionist conditions, cultured cardiomyocytes were subjected to adenoviral delivery with cyclin T1/Cdk9 in the absence or presence of exogenous Gq. Apoptosis was measured by the hypodiploid (sub-G1) population, caspase-3 activity, and poly(ADP-ribose) poly (PARP) cleavage (Figure 7A). The sub-G1 population was increased in cardiomyocytes expressing cyclin T1/Cdk9 (7.8±0.3%) compared to GFP alone (0.2±0.2%: P<0.05). Likewise, caspase-3 activity was increased (2.2±0.2-fold; P<0.05). Alone, Gq had little effect on apoptosis. However, cotransfecting Gq plus cyclin T1/Cdk9 increased the sub-G1 population to 16.1±0.3%, increased caspase-3 activity 3.0±0.2-fold, and enhanced PARP cleavage, consistent with the synergy in Gq × cyclin T1 bigenic mice. To address the possibility that serum depletion by itself sensitizes cells to the pathways under study, the work was repeated using 5% horse serum, with equivalent results (not shown).
Figure 7.
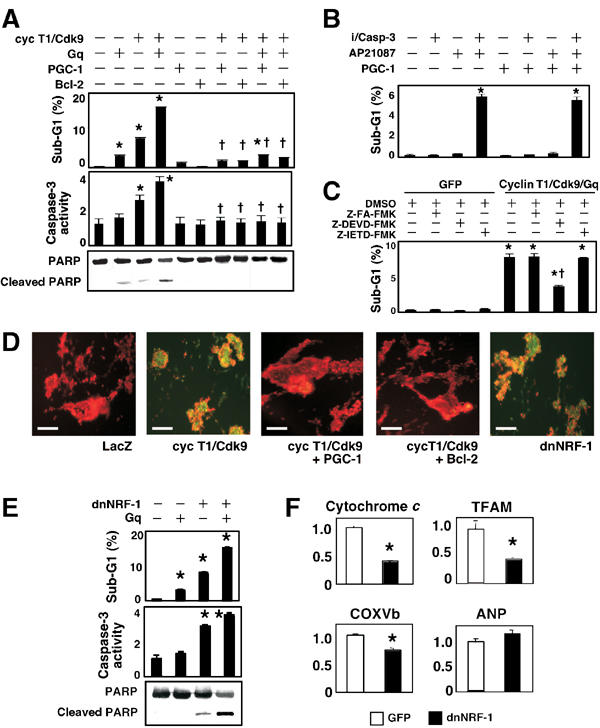
Dysregulation of mitochondrial function by cyclin T1/Cdk9 predisposes cardiomyocytes to apoptosis. After virus delivery, cells were cultured for 48 h and apoptosis was assessed by sub-G1 DNA, caspase-3 activity, PARP cleavage, and ΔΨm. (A) PGC-1 rescues cardiomyocytes from apoptosis caused by cyclin T1/Cdk9±Gq. *P<0.05 versus GFP (−); †P<0.05 versus cyclin T1/Cdk9 plus Gq. (B) PGC-1 fails to block apoptosis induced directly by caspase-3 protease activity. i/Casp-3, dimerization-dependent FKBP12/caspase-3 fusion protein; AP21087, chemical inducer of dimerization. (C) Apoptosis induced by cyclin T1/Cdk9 plus Gq is sensitive to inhibition of caspase-3 (Z-DEVD-FMK) and refractory to inhibition of caspase-8 (Z-IETD-FMK). DMSO, diluent; Z-FA-FMK, negative control. (D–F) Dominant-negative NRF-1 (dnNRF-1) mimics the loss of endogenous PGC-1. (D) Cyclin T1/Cdk9 causes dissipation of ΔΨm, shown as the shift from orange-red to green fluorescence. Note rescue by PGC-1 or Bcl-2, and the equivalent dissipation by dnNRF-1. Bar, 100 μm. (E) Inference with NRF-1 predisposes cardiomyocytes to apoptosis (see panel A for details) and is synergistic with Gq stimulation. (F) Inference with NRF-1 induces downregulation of NRF-1-dependent genes selectively. *P<0.05 versus GFP. †P<0.05 versus cyclin T1/Cdk9 plus Gq.
Based on our expression studies suggesting mitochondrial dysfunction and our ability to rescue representative genes by restoring just PGC-1, we hypothesized that the link from Cdk9 to cardiomyocyte apoptosis might be downregulation of mitochondrial function contingent on partial loss of PGC-1. To test this directly, we supplemented endogenous PGC-1 expression (Figure 7A–C). Apoptosis induced by Gq plus cyclin T1/Cdk9 was completely blocked by adding PGC-1, equal to the effect of Bcl-2, a canonical antiapoptotic gene. We next tested the effect of PGC-1 against caspase-3, using chimeric caspase-3 plus a synthetic dimerizer for its activation (100 nM AP20187) (Shariat et al, 2001). The fusion protein confers dimerizer-dependent caspase-3 activity in cultured cardiomyocytes (not shown) and dimerizer-dependent apoptosis (Figure 7B). As expected from the function of caspase-3 as an executioner caspase—downstream from mitochondria and other death signals—PGC-1 did not impede apoptosis caused directly by caspase-3. Apoptosis provoked by cyclin T1/Cdk9 plus Gq was reduced by a caspase-3 inhibitor, yet not by inhibiting caspase-8, which acts in the ‘extrinsic' death pathway (Figure 7C). Cyclin T1/Cdk9 led to dissipation of the mitochondrial membrane potential (ΔΨm), whereas supplementation of PGC-1 maintained ΔΨm (Figure 7D). Collectively, these findings indicate that cyclin T1/Cdk9 stimulates the ‘intrinsic' mitochondrion-dependent apoptosis pathway, through downregulation of PGC-1.
dnNRF-1 suppresses genes for mitochondrial function and increases cardiomyocytes' susceptibility to apoptosis
If the above interpretation is correct, it follows that one or more mediators of PGC-1 function are essential for normal cardiomyocyte survival. Among the candidates, NRF-1 is especially well posed, and we disrupted NRF-1-dependent transcription in cardiomyocytes by a dominant-negative approach (Figure 7D–F). Since NRF-1 acts as a homodimer, a dominant negative mutant (dnNRF-1) lacking the transactivation domain can suppress endogenous NRF-1 function (Wu et al, 1999; Izumi et al, 2003). As predicted, dnNRF-1 dissipated ΔΨm (Figure 7D) and evoked spontaneous apoptosis, measured by the sub-G1 population (8.0±0.4%; P<0.05), caspase-3 activity (2.9±0.1-fold; P<0.05), and cleavage of PARP (Figure 7E). As found for cyclin T1/Cdk9, dnNRF-1-induced apoptosis was enhanced by costimulation with Gq. dnNRF-1 decreased the expression of NRF-1 target genes (Cox5b, TFAM, cytochrome c; Figure 7F), and did not affect other genes promiscuously, such as ANP. Thus, results using a PGC-1 partner, NRF-1, support the concept that dysregulation of PGC-1 can suffice for the susceptibility to apoptosis conferred by cyclin T1/Cdk9.
Discussion
Cardiac hypertrophy is characterized by the global increase in cell size, protein translation, and mRNA synthesis (Olson and Schneider, 2003). Cyclin T/Cdk9 (P-TEFb) likely plays a role in this myocyte growth response, as a principal mediator of RNAPII CTD phosphorylation—an essential step for productive transcription elongation, whereby RNAPII disengages from promoter-proximal pausing (Komarnitsky et al, 2000; Orphanides and Reinberg, 2002; Pokholok et al, 2002; Zhang et al, 2003). Presently, the most conclusive evidence for an indispensable role of P-TEFb in metazoans comes from RNA interference in Caenorhabditis elegans embryos, indicating that Cdk9 is required in postinitiation pathways for most eukaryotic gene expression (Shim et al, 2002). Loss of the yeast homolog Ctk1 disrupts the recruitment of polyadenylation factors for cotranscriptional 3′-end processing (Ahn et al, 2004). Additional functions might likewise be unmasked by null mutations of the vertebrate kinase. Recent evidence indicates that cyclin T1/Cdk9 can act not merely as a basic transcription elongation factor but also as a regulator with potential selectivity. The best-established example is recruitment of cyclin T1/Cdk9 by the human immunodeficiency virus Tat protein (Price, 2000; Fujinaga et al, 2004); transcriptional regulators that recruit cyclin T1/Cdk9 to specific eukaryotic genes may include Myc (Eberhardy and Farnham, 2002) and the RelA subunit of NF-κB (Barboric et al, 2001).
Data in our study demonstrate the adverse interaction of Cdk9 with known triggers of heart muscle growth, and implicate a PGC-1-dependent mitochondrial program as a likely target of Cdk9. Rather than a global nonspecific activation of genes from enhanced transcription elongation, cyclin T1 provoked heterogeneous responses that were also distinguished readily from the common hypertrophic program of largely ‘fetal' gene induction. αMHC-cyclin T1 hearts did not express the prototypical members of this ensemble, notwithstanding hypertrophic growth. Consistent with its strict dependence on cyclin T/Cdk9 in other settings, HSP70 was upregulated markedly. Conversely, a myriad of genes for mitochondrial function was downregulated by αMHC-cyclin T1 in mouse myocardium and acutely by cyclin T1/Cdk9 in short-term culture.
The mitochondrial content and respiratory capacity of myocardium (and other organs) are regulated by energy demand and modulated by pathophysiological conditions. Because mtDNA encodes only a partial set of the mitochondrially expressed proteins, the function of this organelle requires coordinated expression of nuclear and mitochondrial genomes. Three nuclear transcriptional regulators in particular—NRF-1, NRF-2, and their coactivator PGC-1—are critical to mitochondrial biogenesis and function. In cardiac muscle, transgenic expression of PGC-1 augments mitochondrial biogenesis (Lehman et al, 2000). Conversely, in experimental and clinical heart failure, the expression of genes for mitochondrial oxidative phosphorylation is impaired, downregulation of PGC-1 by load or transgenic expression of a histone deacetylase is accompanied by defects in cardiac expression of mitochondrial proteins (Lehman and Kelly, 2002; Czubryt et al, 2003), and mitochondrial dysfunction causes cardiomyopathy after disrupting genes like Ant1 (Graham et al, 1997), Sod2 (Li et al, 1995), or TFAM (Hansson et al, 2004). Together, these results support the logical inference that impaired mitochondrial function plays a critical role in the pathogenesis of acquired heart failure. Depression of mitochondrial respiratory activity in failing hearts likely results from diminished transcription of specific genes from mitochondrial and nuclear genomes, not simply widespread damage to mtDNA. Although suppression of PGC-1 in heart failure is emerging as a persuasive candidate to explain the dysregulation of downstream nuclear and mitochondria-encoded genes, a unifying molecular basis for the loss of PGC-1 has been elusive.
Our findings link Cdk9 activation—a generalizeable response to hypertrophic triggers—to the deficient expression of genes for mitochondrial function, via impaired PGC-1 transcription: (1) Cdk9 activity is increased in human dilated cardiomyopathy and mouse models of cardiac hypertrophy, states in which mitochondrial function is impaired. (2) Activation of Cdk9 by cyclin T1 in mouse myocardium and cultured cardiomyocytes causes the global suppression of genes for mitochondrial function, concomitant with the loss of PGC-1. (3) Suppression of PGC-1 is an essential target of cyclin T1/Cdk9, shown by the impact of rescuing PGC-1 alone. (4) Cyclin T1/Cdk9 inhibits the PGC-1 promoter, even in the presence of its activators. (5) The repression of PGC-1 by cyclin T1/Cdk9 occurs at or molecularly proximal to the physical recruitment of TBP and RNAPII onto the PGC-1 promoter. Further studies are needed to explicate the manner by which cyclin T/Cdk9 selectively disrupts formation of this preinitiation complex.
Mitochondria play a central role in apoptosis as an essential energy source and a control point in cell death cascades (Danial and Korsmeyer, 2004). Here, we have shown that downregulation of PGC-1 causes susceptibility to apoptosis via the mitochondrial death pathway: (1) Apoptosis induced by cyclin T1/Cdk9 plus Gq is rescued by supplemental PGC-1 or Bcl-2. (2) Cyclin T1/Cdk9 dissipates ΔΨm, which is rescued by PGC-1 or Bcl-2. (3) Interference with the PGC-1 target NRF-1 suffices to dissipate ΔΨm and trigger apoptosis. Although we have not pinpointed the responsible gene(s) downstream of NRF-1 here, several targets of the PGC-1/NRF-1 transcriptional module have proven roles in apoptosis, and more than one may be incriminated subsequently.
In conclusion, Cdk9 may alter gene expression in disease states such as cardiac hypertrophy, where its activity is abnormally increased, affecting a variety of steps in transcriptional control beyond just the global efficiency of transcript elongation. Despite the lack of overt heart failure provoked by cyclin T/Cdk9 under baseline conditions, genetic and physiological provocations both underscore the importance of this kinase in the heart. Impaired transcription of PGC-1 and its targets likely contributes to the transition to heart failure, via mitochondrial dysfunction and, at least in part, the resulting vulnerability to apoptosis.
Materials and methods
Additional methods and details are presented as Supplementary Information.
Human heart samples
Human myocardium was obtained from The Methodist Hospital-DeBakey Heart Center. Tissue procurement was based on patient's informed consent and approved by the institutional review board. Heart failure tissue (idiopathic dilated cardiomyopathy, DCM) was obtained from explanted hearts at the time of therapeutic transplantation. Normal hearts were obtained from unmatched organ donors and victims of motor vehicle accidents.
Western blotting and CTD kinase assays
Sources of antibodies are listed in Supplementary Information. Protein expression was visualized using horseradish peroxidase-conjugated second antibodies and enhanced chemiluminescence (Amersham Biosciences). Cdk9 and Cdk7 activities were assayed using protein A–Sepharose to recover the immune complexes and a recombinant GST-CTD peptide as substrate (Sano et al, 2002).
Mouse models
Cardiac-specific expression of cyclin T1 or Cdk9 was achieved using the mouse αMHC promoter from J Robbins (Sano et al, 2002). αMHC-Gq mice were provided by G Dorn; this is the 25-copy line, with more subtle effects than at higher levels of expression (Adams et al, 1998). Experiments were performed in an isogenic FVB/N background. Pressure-overload hypertrophy was induced by transverse aortic banding (Oh et al, 2003); to ensure the presence of hemodynamic stress, mice were used only if constriction caused a right-to-left carotid flow velocity ratio ⩾4:1.
Recombinant adenoviruses and cell culture
Viruses encoding Cdk9, cyclin T1, and PGC-1 were created using the AdEasy vector system (He et al, 1998; Sano et al, 2002), with and without a 3′ internal ribosome entry site and humanized Renilla GFP. The bicistronic viruses were used, except in fluorescent studies of ΔΨm. Viruses encoding wild-type Gq, Bcl-2, dimerization-dependent caspase-3 (Ad-G/iCasp3), and dnNRF-1 (amino acids 1–342) were kind gifts from J Brown (Adams et al, 2000), L Kirshenbaum (Kirshenbaum and de Moissac, 1997), D Spencer (Shariat et al, 2001), and K Kohno (Izumi et al, 2003). iCasp3 contains two FKBP12 moieties N-terminal to the procaspase domain, harboring the F36V substitution for high-affinity binding to AP20187 (Shariat et al, 2001). Neonatal rat cardiomyocytes were purified and cultured in DMEM/F12 medium (1:1) with 10% horse serum; 24 h after plating, cells were infected at a multiplicity of infection of 20 for 6 h under serum-free conditions, and then cultured in medium without serum. Caspase inhibitors Z-DEVD-FMK (caspase-3), Z-IETD-FMK (caspase-8), and Z-FA-FMK (negative control) were used at 20 μM (BD Biosciences). Typically, viral titer and CMV promoter content were equalized with virus encoding GFP; LacZ was the control for studies of ΔΨm.
Apoptosis
Apoptosis in cultured cardiomyocytes was monitored by hypodiploid DNA, caspase-3 activity, PARP cleavage, and dissipation of ΔΨm, and in myocardium by TUNEL staining and PARP cleavage; see Supplementary Information.
RNA analysis
RNA was isolated using Trizol (Invitrogen) and purified with the RNeasy kit (Qiagen). For expression profiling, samples were labeled with biotinylated nucleotides by reverse transcription, hybridized to Affymetrix MG U74Av2 arrays, and stained with streptavidin–phycoerythrin. Fluorescence intensities were captured with an Affymetrix GeneArray 2500 Scanner, quantified with Affymetrix Microarray Suite 5.0, and analyzed using dChip 1.3 (Harvard University). To confirm and extend the results of microarray hybridization, we subjected the RNA samples to real-time QRT–PCR (ABI Prism 7700, PerkinElmer). TaqMan primers and probes were designed using Primer Express software (version 1.0); see Supplementary Information. For normalization, transcript levels were compared to GAPDH.
Reporter gene assays
CV-1 cells were cultured in DMEM plus 10% FBS and transfected using Effecten (Qiagen). The 5′-flanking sequences of mouse PGC-1 (3.5 kb) and HSP70A1 (2.5 kb) were amplified by high-fidelity PCR from bacterial artificial chromosomes RP23-117M9 and RP23-349B4 respectively (BacPac Resource) and then subcloned into pGL3 (Promega). CMV-driven plasmid expression vectors were kind gifts of the following investigators: Cdk9, A Giordano (de Falco et al, 2000); cyclin T1, R Gaynor (Kwak et al, 1999); PGC-1, D Kelly (Lehman et al, 2000); MEF2C, E Olson (Czubryt et al, 2003). The empty vector pcDNA3 was used to correct DNA content in each experiment. Cells were harvested 48 h after transfection and luciferase activity was assayed using the Dual-Luciferase Reporter Assay System (Promega) and a Monolight 2010 luminometer. Firefly luciferase activity was normalized to that of a cotransfected Renilla luciferase control (pRL-CMV, Promega).
Chromatin immunoprecipitation
ChIP assays were performed using the Chromatin Immunoprecipitation Assay kit (Upstate Biotechnology). Neonatal rat cardiomyocytes were crosslinked 24 h after infection, using 1% formaldehyde (10 min, room temperature). Cells were lysed in 200 μl SDS lysis buffer, incubated for 10 min on ice, and sonicated to obtain DNA fragments averaging ∼200–500 bp in length. Chromatin solutions were precleared using salmon sperm DNA/protein A–agarose and then incubated with the various antibodies. Supernatants without antibody were used to control for total input of chromatin. Immune complexes were eluted, crosslinking was reversed, and DNA was purified using a PCR purification kit (Qiagen). DNA was subjected to PCR with platinum Taq polymerase (Invitrogen).
Statistical analyses
Data, reported as the mean±s.e., were analyzed by ANOVA and Scheffe's test, using a significance level of P<0.05 (StatView, Abacus Concepts).
Supplementary Material
Supplementary Information
Acknowledgments
We thank the investigators cited for kind gifts of reagents or mice: W Boerwinkle, L Shirley, D Nishimura, M Ramirez, Q Xiang, I Callahan, T Pham, A Evans, and J Pocius for technical assistance; H Taegmeyer, T Wada, H Tanaka, J Wong, and N Shimizu for discussions; and the staff of Baylor's Microarray and Integrated Microscopy Core Facilities. This work was supported by NIH grants and the MD Anderson Foundation Professorship (MDS).
References
- Adams JW, Pagel AL, Means CK, Oksenberg D, Armstrong RC, Brown JH (2000) Cardiomyocyte apoptosis induced by Galphaq signaling is mediated by permeability transition pore formation and activation of the mitochondrial death pathway. Circ Res 87: 1180–1187 [DOI] [PubMed] [Google Scholar]
- Adams JW, Sakata Y, Davis MG, Sah VP, Wang YB, Liggett SB, Chien KR, Brown JH, Dorn GW (1998) Enhanced G alpha q signaling: a common pathway mediates cardiac hypertrophy and apoptotic heart failure. Proc Natl Acad Sci USA 95: 10140–10145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn SH, Kim M, Buratowski S (2004) Phosphorylation of serine 2 within the RNA polymerase II C-terminal domain couples transcription and 3′ end processing. Mol Cell 13: 67–76 [DOI] [PubMed] [Google Scholar]
- Barboric M, Nissen RM, Kanazawa S, Jabrane-Ferrat N, Peterlin BM (2001) NF-kappaB binds P-TEFb to stimulate transcriptional elongation by RNA polymerase II. Mol Cell 8: 327–337 [DOI] [PubMed] [Google Scholar]
- Czubryt MP, McAnally J, Fishman GI, Olson EN (2003) Regulation of peroxisome proliferator-activated receptor gamma coactivator 1 alpha (PGC-1 alpha) and mitochondrial function by MEF2 and HDAC5. Proc Natl Acad Sci USA 100: 1711–1716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danial NN, Korsmeyer SJ (2004) Cell death: critical control points. Cell 116: 205–219 [DOI] [PubMed] [Google Scholar]
- de Falco G, Bagella L, Claudio PP, De Luca A, Fu Y, Calabretta B, Sala A, Giordano A (2000) Physical interaction between CDK9 and B-Myb results in suppression of B-Myb gene autoregulation. Oncogene 19: 373–379 [DOI] [PubMed] [Google Scholar]
- Eberhardy SR, Farnham PJ (2002) Myc recruits P-TEFb to mediate the final step in the transcriptional activation of the cad promoter. J Biol Chem 277: 40156–40162 [DOI] [PubMed] [Google Scholar]
- Falkenberg M, Gaspari M, Rantanen A, Trifunovic A, Larsson NG, Gustafsson CM (2002) Mitochondrial transcription factors B1 and B2 activate transcription of human mtDNA. Nat Genet 31: 289–294 [DOI] [PubMed] [Google Scholar]
- Fujinaga K, Irwin D, Huang Y, Taube R, Kurosu T, Peterlin BM (2004) Dynamics of human immunodeficiency virus transcription: P-TEFb phosphorylates RD and dissociates negative effectors from the transactivation response element. Mol Cell Biol 24: 787–795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham BH, Waymire KG, Cottrell B, Trounce IA, MacGregor GR, Wallace DC (1997) A mouse model for mitochondrial myopathy and cardiomyopathy resulting from a deficiency in the heart/muscle isoform of the adenine nucleotide translocator. Nat Genet 16: 226–234 [DOI] [PubMed] [Google Scholar]
- Handschin C, Rhee J, Lin J, Tarr PT, Spiegelman BM (2003) An autoregulatory loop controls peroxisome proliferator-activated receptor gamma coactivator 1alpha expression in muscle. Proc Natl Acad Sci USA 100: 7111–7116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansson A, Hance N, Dufour E, Rantanen A, Hultenby K, Clayton DA, Wibom R, Larsson NG (2004) A switch in metabolism precedes increased mitochondrial biogenesis in respiratory chain-deficient mouse hearts. Proc Natl Acad Sci USA 101: 3136–3141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He TC, Zhou SB, daCosta LT, Yu J, Kinzler KW, Vogelstein B (1998) A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci USA 95: 2509–2514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota H, Chen J, Betz UA, Rajewsky K, Gu Y, Ross J Jr, Muller W, Chien KR (1999) Loss of a gp130 cardiac muscle cell survival pathway is a critical event in the onset of heart failure during biomechanical stress. Cell 97: 189–198 [DOI] [PubMed] [Google Scholar]
- Howes AL, Arthur JF, Zhang T, Miyamoto S, Adams JW, Dorn IG, Woodcock EA, Brown JH (2003) Akt-mediated cardiomyocyte survival pathways are compromised by G alpha q-induced phosphoinositide 4,5-bisphosphate depletion. J Biol Chem 278: 40343–40351 [DOI] [PubMed] [Google Scholar]
- Izumi H, Ohta R, Nagatani G, Ise T, Nakayama Y, Nomoto M, Kohno K (2003) p300/CBP-associated factor (P/CAF) interacts with nuclear respiratory factor-1 to regulate the UDP-N-acetyl-alpha-D-galactosamine: polypeptide N-acetylgalactosaminyltransferase-3 gene. Biochem J 373: 713–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly DP, Scarpulla RC (2004) Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes Dev 18: 357–368 [DOI] [PubMed] [Google Scholar]
- Kirshenbaum LA, de Moissac D (1997) The bcl-2 gene product prevents programmed cell death of ventricular myocytes. Circulation 96: 1580–1585 [DOI] [PubMed] [Google Scholar]
- Komarnitsky P, Cho EJ, Buratowski S (2000) Different phosphorylated forms of RNA polymerase II and associated mRNA processing factors during transcription. Genes Dev 14: 2452–2460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak YT, Ivanov D, Guo J, Nee E, Gaynor RB (1999) Role of the human and murine cyclin T proteins in regulating HIV-1 tat-activation. J Mol Biol 288: 57–69 [DOI] [PubMed] [Google Scholar]
- Lehman JJ, Barger PM, Kovacs A, Saffitz JE, Medeiros DM, Kelly DP (2000) Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. J Clin Invest 106: 847–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehman JJ, Kelly DP (2002) Transcriptional activation of energy metabolic switches in the developing and hypertrophied heart. Clin Exp Pharmacol Physiol 29: 339–345 [DOI] [PubMed] [Google Scholar]
- Li Y, Huang TT, Carlson EJ, Melov S, Ursell PC, Olson JL, Noble LJ, Yoshimura MP, Berger C, Chan PH, Wallace DC, Epstein CJ (1995) Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat Genet 11: 376–381 [DOI] [PubMed] [Google Scholar]
- Lis JT, Mason P, Peng J, Price DH, Werner J (2000) P-TEFb kinase recruitment and function at heat shock loci. Genes Dev 14: 792–803 [PMC free article] [PubMed] [Google Scholar]
- Litingtung Y, Lawler AM, Sebald SM, Lee E, Gearhart JD, Westphal H, Corden JL (1999) Growth retardation and neonatal lethality in mice with a homozygous deletion in the C-terminal domain of RNA polymerase II. Mol Gen Genet 261: 100–105 [DOI] [PubMed] [Google Scholar]
- Michels AA, Nguyen VT, Fraldi A, Labas V, Edwards M, Bonnet F, Lania L, Bensaude O (2003) MAQ1 and 7SK RNA interact with CDK9/cyclin T complexes in a transcription-dependent manner. Mol Cell Biol 23: 4859–4869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh H, Wang SC, Prahash A, Sano M, Moravec CS, Taffet GE, Michael LH, Youker KA, Entman ML, Schneider MD (2003) Telomere attrition and Chk2 activation in human heart failure. Proc Natl Acad Sci USA 100: 5378–5383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson EN, Schneider MD (2003) Sizing up the heart: development redux in disease. Genes Dev 17: 1937–1956 [DOI] [PubMed] [Google Scholar]
- Orphanides G, Reinberg D (2002) A unified theory of gene expression. Cell 108: 439–451 [DOI] [PubMed] [Google Scholar]
- Pokholok DK, Hannett NM, Young RA (2002) Exchange of RNA polymerase II initiation and elongation factors during gene expression in vivo. Mol Cell 9: 799–809 [DOI] [PubMed] [Google Scholar]
- Price DH (2000) P-TEFb, a cyclin-dependent kinase controlling elongation by RNA polymerase II. Mol Cell Biol 20: 2629–2634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi DJ, Londesborough A, Korsisaari N, Pihlak A, Lehtonen E, Henkemeyer M, Makela TP (2001) Inability to enter S phase and defective RNA polymerase II CTD phosphorylation in mice lacking Mat1. EMBO J 20: 2844–2856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakata Y, Hoit BD, Liggett SB, Walsh RA, Dorn G (1998) Decompensation of pressure-overload hypertrophy in G alpha q-overexpressing mice. Circulation 97: 1488–1495 [DOI] [PubMed] [Google Scholar]
- Sano M, Abdellatif M, Oh H, Xie M, Bagella L, Giordano A, Michael LH, DeMayo FJ, Schneider MD (2002) Activation and function of cyclin T-Cdk9 (positive transcription elongation factor-b) in cardiac muscle-cell hypertrophy. Nat Med 8: 1310–1317 [DOI] [PubMed] [Google Scholar]
- Shariat SF, Desai S, Song W, Khan T, Zhao J, Nguyen C, Foster BA, Greenberg N, Spencer DM, Slawin KM (2001) Adenovirus-mediated transfer of inducible caspases: a novel ‘death switch' gene therapeutic approach to prostate cancer. Cancer Res 61: 2562–2571 [PubMed] [Google Scholar]
- Shilatifard A, Conaway RC, Conaway JW (2003) The RNA polymerase II elongation complex. Annu Rev Biochem 72: 693–715 [DOI] [PubMed] [Google Scholar]
- Shim EY, Walker AK, Shi Y, Blackwell TK (2002) CDK-9/cyclin T (P-TEFb) is required in two postinitiation pathways for transcription in the C. elegans embryo. Genes Dev 16: 2135–2146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC, Spiegelman BM (1999) Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 98: 115–124 [DOI] [PubMed] [Google Scholar]
- Yik JH, Chen R, Nishimura R, Jennings JL, Link AJ, Zhou Q (2003) Inhibition of P-TEFb (CDK9/Cyclin T) kinase and RNA polymerase II transcription by the coordinated actions of HEXIM1 and 7SK snRNA. Mol Cell 12: 971–982 [DOI] [PubMed] [Google Scholar]
- Zhang F, Barboric M, Blackwell TK, Peterlin BM (2003) A model of repression: CTD analogs and PIE-1 inhibit transcriptional elongation by P-TEFb. Genes Dev 17: 748–758 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information