

Abstract
Basic leucine zipper proteins Jun and Fos form the dimeric transcription factor AP-1, essential for cell differentiation and immune and antioxidant defenses. AP-1 activity is controlled, in part, by the redox state of critical cysteine residues within the basic regions of Jun and Fos. Mutation of these cysteines contributes to oncogenic potential of Jun and Fos. How cells maintain the redox-dependent AP-1 activity at favorable levels is not known. We show that the conserved coactivator MBF1 is a positive modulator of AP-1. Via a direct interaction with the basic region of Drosophila Jun (D-Jun), MBF1 prevents an oxidative modification (S-cystenyl cystenylation) of the critical cysteine and stimulates AP-1 binding to DNA. Cytoplasmic MBF1 translocates to the nucleus together with a transfected D-Jun protein, suggesting that MBF1 protects nascent D-Jun also in Drosophila cells. mbf1-null mutants live shorter than mbf1+ controls in the presence of hydrogen peroxide (H2O2). An AP-1-dependent epithelial closure becomes sensitive to H2O2 in flies lacking MBF1. We conclude that by preserving the redox-sensitive AP-1 activity, MBF1 provides an advantage during oxidative stress.
Keywords: AP-1, coactivator, Drosophila, MBF1, oxidative stress
Introduction
Organisms from the most primitive prokaryotes to mammals have evolved a number of mechanisms to maintain cellular redox balance and thus evade oxidative stress caused by naturally arising reactive oxygen species (ROS). These mechanisms include low-molecular radical scavengers and antioxidant enzymes. Studies in yeast and mammalian cell lines have identified regulatory pathways of antioxidant defense. These involve protein kinases, such as JNK and ERK, that activate transcription factors, which turn on stress response genes (Davis, 2000). JNK signaling is required for oxidative stress defense also in the fruit fly Drosophila melanogaster, which serves as a model to characterize it genetically (Stronach and Perrimon, 1999; Wang et al, 2003).
Among the key transcription factors activated by the JNK and ERK kinases are the basic region leucine zipper (bZIP) proteins of the Jun and Fos families (Karin, 1995). Related bZIP proteins combine through their leucine zippers to yield an array of DNA-binding dimers, known as AP-1 (activator protein-1). AP-1 activity can be induced by signals as diverse as growth factors, peptidic hormones and neurotransmitters, microbial infections and physical and chemical stresses. In response, AP-1 triggers a spectrum of cellular processes such as apoptosis or proliferation, differentiation and mobilization of defense against stress (Shaulian and Karin, 2002).
Jun and Fos not only activate the cellular defense against oxidative challenge, but also sense redox imbalances: their activity depends upon their redox state. This appears to be an important and universal principle, since also other transcription regulators, such as NF-κB or p53, are sensitive to oxidation (Marshall et al, 2000). The sensitivity is conferred by reactive sulfhydryl groups of cysteine residues. Abate et al (1990) have shown that specific cysteines within the basic regions of c-Jun and c-Fos are responsible for oxidative inhibition of AP-1 DNA-binding capacity in vitro, which can be restored with reducing agents. In contrast, Jun and Fos mutants with serine replacing the critical cysteine residues bind DNA regardless of redox conditions. Thus the hypersensitive cysteine introduces a new element of regulation into AP-1. That such regulation is indeed necessary for normal cell functioning is obvious from consequences of the critical cysteine mutation into serine. Exactly such substitution is found in the viral transforming protein v-Jun (Bohmann et al, 1987; Maki et al, 1987), where it contributes to the oncogenic activity synergistically with other mutations (Morgan et al, 1994). Similarly, the corresponding cysteine-to-serine substitution leads to a transforming activity of Fos (Okuno et al, 1993). In both cases, the mutant AP-1 forms apparently escape the redox regulation.
Paradoxically, while AP-1 mobilizes antioxidant defense, it is at the same time sensitive to oxidation. This suggests that during redox imbalances in vivo, something must protect Jun and Fos from oxidative damage. A nuclear protein Ref-1 (redox factor 1), also implicated in DNA repair, has been shown to reactivate AP-1 by a thioredoxin-dependent reduction of the critical cysteine residues (Xanthoudakis and Curran, 1992; Xanthoudakis et al, 1992; Hirota et al, 1997; Ordway et al, 2003). While Ref-1 mediates a reparative reduction of once inactivated nuclear AP-1, it seems logical that some factor should also prevent oxidation of newly synthesized Jun and Fos, particularly during oxidative stress.
A good candidate to perform this protective role is the multiprotein bridging factor 1 (MBF1). MBF1, also known as endothelial differentiation-related factor 1 (EDF1), primarily resides in the cytoplasm and can relocate to the nucleus upon external stimuli (Mariotti et al, 2000). MBF1 acts as a coactivator of the bZIP protein GCN4, the closest relative of Jun in the budding yeast. MBF1 interacts directly with both GCN4 and the TATA-binding protein (TBP), implying that it interconnects the bZIP factor with the basal transcription machinery (Takemaru et al, 1998). MBF1 has also been shown to bind human c-Jun (Kabe et al, 1999) and stimulate c-Jun-dependent transcription (Busk et al, 2003). Unlike other AP-1 interacting proteins, MBF1 is unique in that it binds to the basic region where the redox-sensitive cysteines are located.
We have chosen the fruit fly Drosophila to examine the relationships between AP-1 and MBF1. Compared to four Fos and three Jun paralogs in mammals, Drosophila is a simple model with only one D-Fos and one D-Jun protein (Kockel et al, 2001). In this study, we show that through a direct interaction with D-Jun, MBF1 protects the critical cysteine residue from oxidation and stimulates AP-1 binding to DNA. A mutation removing mbf1 causes sensitivity to oxidative stress in vivo and compromises an AP-1-dependent process of epithelial tissue closure. Studies of MBF1 therefore open an avenue to learn more about AP-1 regulation and function.
Results
Critical cysteine residues confer D-Jun and D-Fos sensitivity to oxidation
Drosophila D-Jun and D-Fos proteins have cysteine residues in the same positions as the subunits of human AP-1, which rapidly loses DNA-binding activity upon oxidation (Figure 1A). To see whether Drosophila AP-1 also undergoes oxidative inactivation, we tested the binding of bacterially expressed D-Jun and D-Fos bZIP domains to an AP-1 site using electrophoresis mobility shift assays under disparate redox conditions. Both D-Jun and D-Fos were truncated such that each protein harbored a single cysteine within the basic region, and were designated J and F, respectively (Figure 1B). Their DNA-binding properties were compared with those of point mutants, JS and FS, in which the critical cysteine residues had been replaced with serine (Figure 1A). Like their human orthologs, J and F proteins were unable to bind DNA in the absence of dithiothreitol (DTT; Figure 2A, lane 1). Because neither protein bound to DNA alone in our conditions, no binding occurred also when the cysteine was mutated in either D-Fos (Figure 2A, lane 4) or D-Jun (not shown). Only when both proteins were mutated, their complex with the AP-1 site could be detected in the absence of DTT (lane 7). Addition of 1 mM DTT allowed weak binding of the J/F dimer (lane 2); the binding of the mutant proteins under the same conditions was stronger (lanes 5 and 8). Increasing DTT concentration to 10 mM enhanced the binding of dimers in which at least one protein contained cysteine (lanes 3 and 6). These results show that Drosophila Jun and Fos are readily inactivated by oxidation of the critical cysteine residues, and suggest existence of factor(s) that maintain the activity of these proteins under oxidative conditions.
Figure 1.
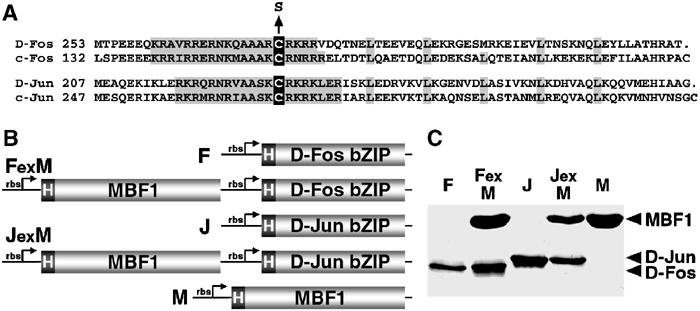
Recombinant D-Fos, D-Jun and MBF1 proteins used in this study. (A) Drosophila Fos and Jun truncated proteins, aligned with human c-Fos and c-Jun. Conserved basic regions and leucine residues of the bZIP domains are shaded; the critical cysteines are in black boxes. These cysteines were mutated to serine to produce redox-insensitive D-Fos and D-Jun forms (FS and JS). The cysteines C-terminal to the leucine zipper were replaced with stop codons in all D-Fos and D-Jun constructs. (B) His-tagged D-Fos (F), D-Jun (J) and MBF1 (M) were expressed in E. coli either separately or coexpressed from bicistronic plasmid constructs (FexM and JexM). rbs, ribosome-binding site. (C) AP-1 and MBF1 proteins were expressed from constructs shown in (B), affinity purified using the His tag, separated on a reducing SDS–polyacrylamide gel and stained with Coomassie blue.
Figure 2.
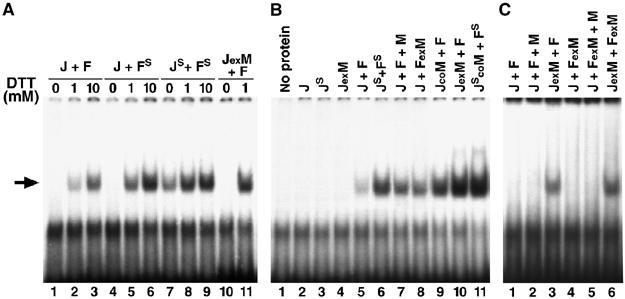
Effects of oxidation and MBF1 on the binding of D-Fos and D-Jun to an AP-1 site. Gel retardation assays were performed with Jun and Fos, each containing the single critical cysteine (J, F), and with the serine mutants (JS, FS). MBF1 was added separately (J+F+M), coexpressed with Jun (JexM) or Fos (FexM) from a bicistronic plasmid, or copurified from mixed E. coli cultures each expressing one protein (JcoM). The arrow shows the AP-1/DNA complex. (A) Binding of freshly purified proteins under oxidative (no DTT) or reducing conditions revealed that Drosophila AP-1 activity depends on the redox state of the critical cysteine residues in its DNA-binding domain. (B) Binding of freshly purified proteins in the presence of 1 mM DTT showed that weak AP-1 activity (lane 5) was greatly enhanced by coexpression of Jun with MBF1 (lane 10). Both Jun and Fos were required for the binding. (C) The assay conditions were as in (B) except that the proteins were aged for 5 days in solution at 4°C. No binding was observed unless MBF1 had been coexpressed with Jun.
MBF1 ensures AP-1 binding to DNA through interaction with D-Jun
To study whether Drosophila MBF1 supports AP-1 activity, we first examined the effect of MBF1 on the DNA-binding activity of AP-1 (Figure 2). When MBF1 was added to the electrophoresis mobility shift assay with bacterially produced J and F, it only mildly stimulated DNA binding (Figure 2B, compare lanes 5 and 7). MBF1 exerted a stronger effect when copurified with J from pooled bacterial cultures, each expressing only one protein (lane 9), and the strongest effect was observed when MBF1 was coexpressed with J in Escherichia coli using a bicistronic construct (lane 10). The increased DNA-binding activity was not due to a higher yield of the coexpressed proteins (Figure 1C). Thus, MBF1 had to be in contact with D-Jun already within the E. coli cells or at least during the purification steps in order to ensure robust AP-1 activity; later addition of MBF1 was not sufficient. MBF1 could also stimulate the DNA-binding activity of the mutant JS/FS complex (Figure 2B, lanes 6 and 11), which is only partially sensitive to oxidative condition (Figure 2A, lanes 7 and 8). This result suggests that MBF1 has a more general positive effect on D-Jun that is not entirely mediated by keeping the cysteines reduced.
To further test the function of MBF1 in preserving AP-1 activity, we artificially ‘aged' purified proteins for several days at 4°C. Upon such treatment, AP-1 completely lost its DNA-binding activity, suggesting inactivation due to oxidation and/or denaturation (compare Figure 2B, lane 5, with Figure 2C, lane 1). However, when D-Jun was coexpressed with MBF1, it was able to form active AP-1 even after the aging treatment (Figure 2C, lane 3). Once lost, the AP-1 activity could not be restored by subsequent addition of MBF1 (Figure 2C, lane 2). MBF1 showed its protective effect only on D-Jun; aged AP-1 proteins failed to bind DNA when MBF1 was either added or coexpressed with D-Fos (Figure 2C, lanes 2, 4 and 5). However, D-Fos was still functional, because no DNA binding occurred without it in our assays (Figure 2B, lanes 2–4). These results indicate that MBF1 prevents the deterioration of AP-1 activity specifically through acting on D-Jun.
MBF1 binds the basic region of D-Jun and protects the critical cysteine from oxidation
The enhanced DNA-binding activity of AP-1 in the presence of MBF1 suggests that MBF1 may protect AP-1 from oxidation through a direct contact. To test for interaction between MBF1 and AP-1 proteins, we performed GST pull-down assays with the hexahistidine-tagged D-Jun and D-Fos bZIP domains (Figure 1) and a GST-MBF1 fusion. Figure 3A shows that MBF1 specifically bound the D-Jun but not the D-Fos bZIP region. The failure to bind D-Fos likely was not due to D-Fos deterioration, since this protein was active in our electrophoresis mobility shift assays. Although we have not mapped the exact amino acids required for MBF1 binding in the D-Jun bZIP peptide, we surmise that they include the basic residues near the critical cysteine (Figure 3B), because these basic residues in GCN4 are required for yeast MBF1 binding (Takemaru et al, 1998). A very similar basic motif in the nuclear receptor Ftz-F1 (Figure 3B) is necessary for the binding of insect MBF1 (Takemaru et al, 1997).
Figure 3.
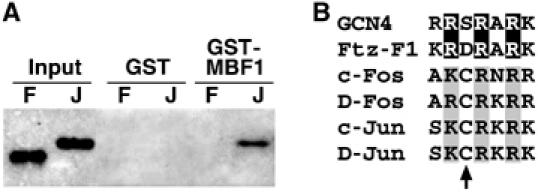
MBF1 binds a conserved basic motif. (A) GST pull-down assay showed a direct interaction of a GST-MBF1 fusion protein with the His-tagged D-Jun bZIP domain (J) but not with the same region of D-Fos (F). (B) Alignment of basic regions in the yeast bZIP factor GCN4, a nuclear receptor Ftz-F1, and the human and Drosophila AP-1 family members. All proteins except D-Fos have been shown to bind MBF1. The black boxes indicate arginine residues in GCN4 and Ftz-F1, known to be required for MBF1 binding; the corresponding basic residues in Fos and Jun are shaded. The arrow points to the critical redox-sensitive cysteine in AP-1 proteins.
To test whether MBF1 prevents oxidation of the redox-sensitive cysteine of D-Jun, the D-Jun bZIP domain expressed in E. coli either alone or with MBF1 (Figure 1C) was subjected to MALDI-TOF mass analysis. Figure 4 shows that D-Jun coexpressed with MBF1 remained in the reduced state. In contrast, when expressed alone, a majority of D-Jun increased its mass by 222.6 Da, an increment corresponding to S-cystenyl cystenylation (i.e. cystenyl cyteine attached to D-Jun via a mixed disulfide bond). Consistently, the modified form retained a single reactive SH group per molecule as revealed by monoalkylation with iodoacetamide (data not shown). To confirm that S-cystenyl cystenylation indeed concerned the critical cysteine residue, we show that no such modification occurred in the D-Jun bZIP domain harboring the cysteine-to-serine substitution (JS), expressed in the absence of MBF1 (Figure 4). MBF1 therefore functions to protect the critical cysteine from oxidative modification.
Figure 4.
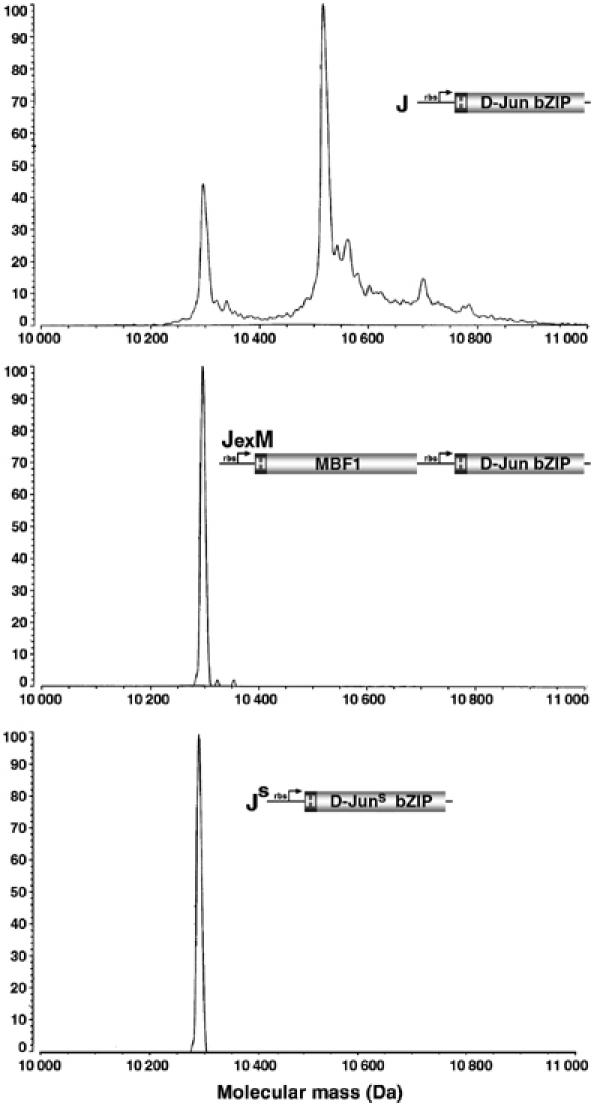
MBF1 prevents an oxidative modification of D-Jun. The His-tagged bZIP domain of D-Jun (J) was expressed in E. coli either alone (top) or coexpressed with MBF1 (JexM); its cysteine-to-serine mutant (JS) was expressed alone (bottom). The purified proteins (see Figure 1C) were subjected to MALDI-TOF mass analysis. D-Jun expressed alone shows a mass increment of 222.6 Da, corresponding to S-cystenyl cystenylation. The mass of His-tagged MBF1 is around 18 kDa.
MBF1 expression and nuclear translocation with D-Jun
Since oxidative stress can occur at any time, MBF1 should be expressed constantly in order to prevent AP-1 oxidation. We determined the developmental profile of MBF1 expression using a specific polyclonal antibody that detected a single band of the expected size (16 kDa) on Western blots. The presence of the MBF1 protein started in the embryo with a strong maternal contribution and was maintained throughout embryogenesis, with zygotic translation ensuing 5–7 h after egg laying (Figure 5A). Expression continued for the entire postembryonic life without temporal fluctuations (Figure 5B). Among tissues exhibiting high MBF1 levels were the central nervous system, imaginal discs and gonads, but not the fat body (Figure 5C–E).
Figure 5.
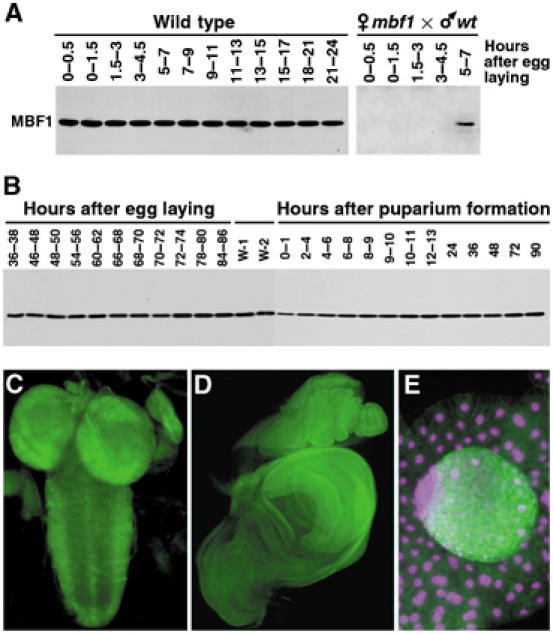
Expression pattern of MBF1 in Drosophila. (A) Western blot showing constitutive MBF1 expression during embryogenesis. Embryos from mbf1 mutant mothers were devoid of all maternal MBF1 protein. Zygotic expression from the paternal wild-type chromosome began between 5 and 7 h after egg laying (right panel). (B) Western blot showing MBF1 expression throughout the postembryonic life. W-1 and W-2, wandering stages of larvae. (C–E) Sites of high MBF1 expression during larval life were the central nervous system (C, second instar), imaginal discs (D, late third instar) and the testis (E, center), but not the fat body (E, surrounding tissue). MBF1 was detected with a specific polyclonal antibody, and DAPI was used for DNA staining in (E). Magenta is used as colorblind friendly (http://jfly.iam.u-tokyo.ac.jp/color/text.html).
Although MBF1 interacts with nuclear partners, previous data (Kabe et al, 1999; Mariotti et al, 2000; Liu et al, 2003) have shown that MBF1 is primarily a cytoplasmic protein, suggesting that MBF1 may cotransport with interacting transcription factors to the nucleus. We have tested whether MBF1 translocates to the nucleus with D-Jun in Drosophila cells. As shown in Figure 6, the MBF1 protein resides predominantly in the cytoplasm of both embryonic (S2) and imaginal disc (Cl.8+) cells, cultured under control conditions. In contrast, MBF1 moved to the nucleus in S2 cells that had been transfected with a plasmid expressing D-Jun-His, the entire D-Jun protein with a C-terminal hexahistidine tag (Figure 6A). As revealed with an antibody against His tag, the D-Jun-His protein also accumulated in the nucleus. Co-immunoprecipitation of MBF1 and D-Jun-His from these transfected cells with the His-tag antibody (Figure 6B) suggested that MBF1 translocated to the nucleus upon interaction with D-Jun. Such an interaction likely occurs through MBF1 binding to the bZIP domain of D-Jun, demonstrated in the GST pull-down assay (Figure 3). Transfection of Cl.8+ (Figure 6C) or S2 cells (not shown) with only the His-tagged bZIP domain of D-Jun confirmed that this domain alone was sufficient for the nuclear translocation of MBF1. These data suggest that MBF1 migrates to the nucleus in complex with the newly synthesized D-Jun protein.
Figure 6.
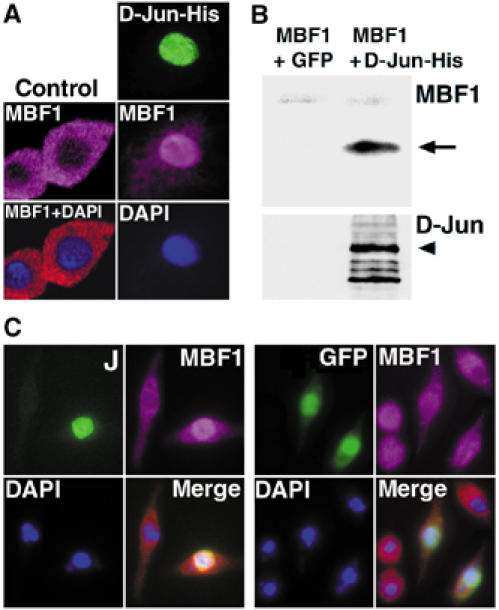
MBF1 and D-Jun form a complex and cotranslocate to the nucleus in Drosophila cells. (A) Drosophila S2 cells showed translocation of the endogenous cytoplasmic MBF1 protein (control) to the nucleus upon misexpression of the whole His-tagged D-Jun protein (right column). (B) Upon cotransfection of S2 cells with MBF1 and D-Jun-His (but not GFP), MBF1 was recovered from the cell lysate together with the D-Jun-His protein by using an anti-His-tag monoclonal antibody. The Western blot was first probed with anti-MBF1, then stripped and re-probed with a D-Jun antiserum (bottom). (C) Expression of a His-tagged bZIP domain of D-Jun in Cl.8+ cells was sufficient for nuclear translocation of the MBF1 protein (left panel); expression of nuclear GFP had no effect on MBF1 localization. Cells were fixed and stained 36 h after transfection. Staining with anti-His tag was visualized with DTAF (FITC)-conjugated secondary antibody. Anti-MBF1 was detected with a Cy3-conjugated secondary antibody, shown as magenta that is friendly to colorblind people (http://jfly.iam.u-tokyo.ac.jp/color/). GFP was visualized with direct fluorescence.
Null mbf1 mutants are sensitive to oxidative stress
To study the role of MBF1 in vivo, we generated deletions in the Drosophila mbf1 gene through P-element transposon insertion and its subsequent imprecise excision (Figure 7A). The molecular lesions of four deletion alleles (mbf11 through mbf14) spanned from the original P-element insertion site toward the coding region, affecting mbf1 but no other gene (Figure 7A and B). Except for the mbf11 allele that had a shortened mRNA, all alleles failed to produce the mbf1 transcript (Figure 7C). All four alleles, either homozygous or hemizygous over a Df(3L)st7P deficiency that includes mbf1, were totally devoid of the MBF1 protein (e.g. mbf12; Figure 7D). Despite the complete absence of the MBF1 protein, all four alleles were viable and fertile under standard laboratory conditions. We used mbf12, which had the longest deletion of 2082 bp, in all experiments described hereafter. A rescue construct that includes a 4.6 kb genomic mbf1 fragment in a P-element vector restored production of the MBF1 protein at all stages examined (Figure 7D and data not shown).
Figure 7.
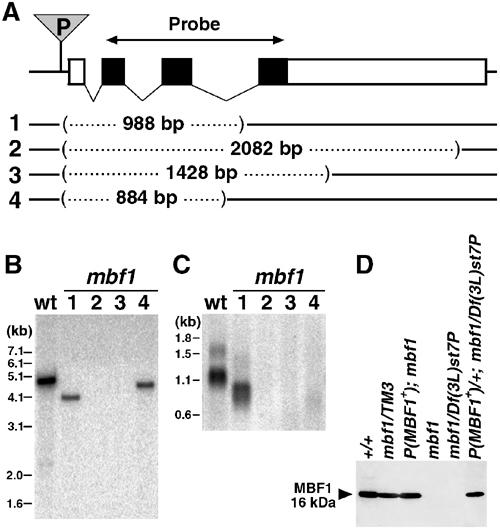
mbf1 mutant flies are molecularly null. (A) Map of the mbf1 gene located on chromosome 3L with a P-element insertion (P) 21 bp upstream of the first exon and four imprecise P excisions (1–4). Black boxes denote coding exons of mbf1, used as a hybridization probe in (B, C); untranslated regions are open. The dotted lines represent DNA deleted in mbf1 alleles mbf11 to mbf14 (1–4). Centromere is to the right. (B) Southern blot of genomic DNA from wild-type (wt) and homozygous mbf1 mutant flies shows that mbf12and mbf13 lack the coding region of mbf1. (C) Northern hybridization of mRNA shows the complete loss of both transcripts (1.6 and 1.1 kb) in mbf12 to mbf14 homozygotes. (D) Western blot analysis of the MBF1 protein from adult flies confirms that mbf12 is a null allele. A transgenic construct P(mbf1+) restores the production of the MBF1 protein. TM3 is a third chromosome balancer; Df(3L)st7P is a deficiency including mbf1.
The ability of MBF1 to prevent oxidation of D-Jun suggests that the mbf1 gene might have an important function during stress, when AP-1 triggers various stress responses. To test the possibility that the loss of mbf1 affects oxidant tolerance in Drosophila, we compared the survival of the mbf12 mutants with mbf1+ animals (P(w+mbf1+)/P(w+,mbf1+); mbf12) in the presence of hydrogen peroxide (H2O2) as a source of oxidative stress. When placed on diet containing 0.1 or 0.3% H2O2 as first instar larvae, mbf12 animals reached adulthood about 3.5 times less frequently than the mbf1+ strain (Figure 8A). To test oxidative stress tolerance in adults, we exposed males of equal size and age to 0.5% H2O2 and percent surviving was scored at regular intervals (Figure 8B). The median survival time of the mbf12 homozygotes was 67 h, compared to 93 h for the mbf1+ strain. mbf1− hemizygotes obtained from Df(3L)st7P/+ mothers, crossed with mbf12 males, lived on average 64 h on 0.5% H2O2. The sensitivity was therefore not caused by another mutation on the mbf1− chromosome or by a maternal effect. Flies possessing four doses of mbf1+ (two endogenous and two transgenic) were more resistant to H2O2 than animals with two copies (Figure 8B). When catalase activity was inhibited prior to H2O2 treatment by feeding flies with 5 mM aminotriazole, the effect of H2O2 was greatly enhanced; the lifespan of mbf1 mutants was less than 60% that of the rescued flies (Figure 8B). A similar enhancement was observed when flies were pretreated with 5 mM buthionine-sulfoximine, a drug that reduces the free radical scavenging capacity by depleting the endogenous pool of glutathione (data not shown). Together, these results show that the loss of MBF1 renders animals sensitive to ROS.
Figure 8.
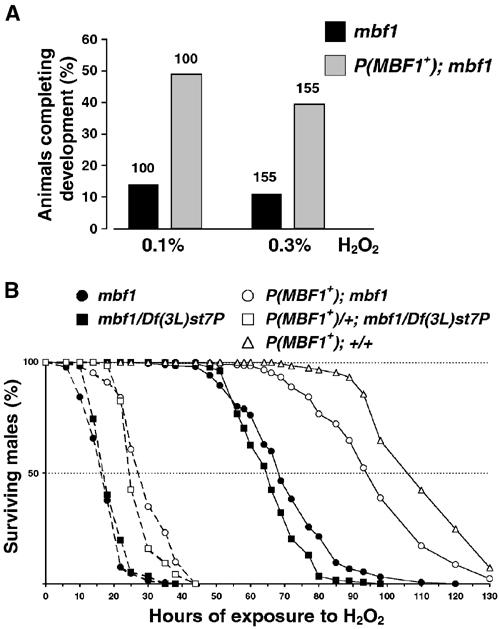
MBF1-deficient mutants are sensitive to oxidative stress. (A) Equal numbers of first instar larvae of the mbf1 mutant and rescued lines were placed on diet containing indicated concentrations of H2O2 and numbers of emerging adults were counted. Values above bars indicate the numbers of larvae. (B) Adult males (30 per vial) were exposed to H2O2 either directly (solid lines) or after reducing their catalase activity by feeding 5 mM aminotriazole for 60 h (broken lines), and their lifetime was recorded. The open symbols denote flies possessing at least one copy of mbf1+. Line P(mbf1+)/P(mbf1+); +/+ contains four copies of mbf1+. The numbers of flies tested per genotype ranged between 120 and 540, and the resistance of each genotype was tested at least three times.
An AP-1-dependent developmental process becomes sensitive to oxidative stress in mbf1 mutant background
In addition to triggering antioxidant responses, AP-1 has various developmental functions including formation of the adult thorax (reviewed by Kockel et al, 2001). D-Fos is required for fusion of the wing imaginal discs at the dorsal midline (Riesgo-Escovar and Hafen, 1997a; Zeitlinger and Bohmann, 1999). To see whether also D-Jun is required for thorax closure, we induced an RNA interference (RNAi) knockdown of D-Jun in the dorsal epithelium using the pnr-Gal4 driver (Heitzler et al, 1996). This resulted in mild to severe defects of thorax fusion in 23% of the UAS-D-JunRNAi/+; pnr-Gal4/+ flies (Figure 9C).
Figure 9.
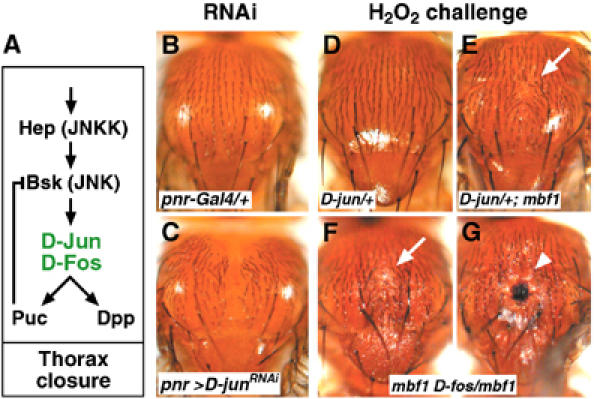
Genetic interaction between MBF1 and AP-1 during thorax closure. (A) The JNK cascade is required for Drosophila thorax closure (after Kockel et al, 2001). Hemipterous (Hep) phosphorylates a Jun N-terminal kinase Basket (Bsk). The redox-sensitive JNK substrates (AP-1) are in green. A JNK phosphatase Puckered (Puc) and a TGFβ signal Dpp (Decapentaplegic) are putative targets of AP-1. Incomplete function of Hep or D-Fos causes cleft adult thorax; Puckered is a negative regulator of the thorax closure. (B, C) RNAi knockdown of D-Jun, targeted to the dorsal epidermis using pnr-Gal4, prevents complete fusion of the thorax; a control expressing the driver alone has normal thorax (B). (D–G) Animals of indicated genotypes were challenged with H2O2 at the onset of metamorphosis. Emerging adults doubly mutant for mbf1 and either D-jun (E) or D-fos (F,G) occasionally displayed partially cleft thorax with naked cuticle (arrows), sometimes accompanied with necrosis (arrowhead).
The AP-1 proteins (Zeitlinger and Bohmann, 1999) and MBF1 (Figure 5D) are coexpressed in the wing imaginal discs. To examine whether the ability of MBF1 to protect D-Jun from oxidation is important for development, we tested whether mbf1 interacts genetically with D-jun/D-fos under an H2O2 challenge (Figure 9D–G). Animals that were heterozygous for the recessive lethal mutations in D-jun or D-fos did not show any defects when treated with H2O2 at the onset of metamorphosis, indicating that a single dose of D-jun or D-fos is sufficient to support normal development under the oxidative challenge. However, in mbf1 mutant background, D-jun/+ and D-fos/+ animals often produced adults with defects in the thorax, manifest as a depressed patch of naked cuticle at the dorsal midline (Figure 9E and F). In some mbf1 D-fos/mbf1 flies, a necrosis occurred at the site of the wound (Figure 9G). The thorax defects in mbf1− mutants were similar to the phenotypes resulting from compromised function of several components of the D-JNK cascade (Figure 9A) including D-Jun (Figure 9C) and D-Fos. These results thus suggest that as in vitro, the lack of MBF1 destabilizes AP-1 under oxidative conditions in vivo.
Discussion
MBF1 in the redox regulation of AP-1
Sensitivity of AP-1 to oxidation requires a mechanism to protect AP-1 activity. We introduce MBF1 as a new player that allows cells to maintain adequate AP-1 activity under oxidative stress. Drosophila AP-1 components D-Jun and D-Fos undergo oxidative inactivation via the same cysteine residues as the human orthologs. MBF1 prevents this oxidation and preserves the DNA-binding activity. In mbf1 mutants, an AP-1-dependent developmental process becomes hypersensitive to oxidative stress, suggesting that MBF1 also protects D-Jun from an oxidative modification in vivo. The protection is unlikely to be complete because it relies on the binding of MBF1 to Jun. Thus, the AP-1 action may be in an equilibrium between acceleration by the MBF1 protection of Jun and brake by the oxidative inactivation of Jun.
The mechanism by which MBF1 ensures the activity of AP-1 is different from that of the nuclear protein Ref-1, which reactivates oxidized AP-1 by reduction (Xanthoudakis and Curran, 1992). MBF1 was a much stronger enhancer of AP-1 activity when coexpressed and copurified with D-Jun from E. coli than when it was added to the DNA-binding assay separately. Unlike Ref-1, MBF1 was unable to restore AP-1 activity once lost. Thus, rather than reactivating AP-1, MBF1 protects it from oxidation in a preventive manner. Protection from oxidation is however one of several stabilizing effects that MBF1 exerts on AP-1, because MBF1 can stimulate DNA binding even of mutant AP-1 proteins, possessing serine instead of the redox-sensitive cysteine residues.
MBF1 enhanced the DNA-binding activity of AP-1 selectively through D-Jun. Since MBF1 bound D-Jun but not D-Fos in a direct interaction assay, we propose that the selectivity is based on an exclusive contact between MBF1 and D-Jun. This was unexpected as human MBF1 was shown to bind a GST-c-Fos fusion (Kabe et al, 1999). On the other hand, D-Jun and c-Jun share more similarity than the Fos orthologs; in particular, the critical cysteine context KCR reads RCR in D-Fos.
Although AP-1 regulation via the redox-sensitive cysteine residues was postulated more than a decade ago (Abate et al, 1990), the nature of the cysteine modification remained unknown. The prediction is that a regulatory oxidation may involve a reversible formation of sulfenic acid or a disulfide bond (Marshall et al, 2000). To examine how the critical cysteine is modified, we determined the molecular mass of the bacterially expressed D-Jun used in DNA-binding assays. The E. coli system allowed us to express D-Jun in the absence of endogenous MBF1. Surprisingly, we identified a previously undescribed modification of the critical cysteine, S-cystenyl cystenylation. In a striking contrast, no such modification occurred in D-Jun coexpressed with MBF1 or in D-Jun lacking the critical cysteine residue. S-glutathiolation of the cysteine, a similar modification that was known to prevent binding of c-Jun to an AP-1 site (Klatt et al, 1999), was not observed in D-Jun despite GSH:GSSG is an abundant redox system in E.coli (Sundquist and Fahey, 1989). Whether S-cystenyl cystenylation is only a product of the prokaryotic expression system or whether it represents true physiological regulation of AP-1 activity remains to be tested. However, our aim was to disclose the role for MBF1, and the ability of MBF1 to avert S-cystenyl cystenylation shows that this role is to protect D-Jun.
While our data illuminate the role of MBF1 in the protection of the redox-sensitive cysteine in D-Jun, MBF1 also stimulated DNA binding of the serine mutant (Figure 2). Thus the effect of MBF1 on D-Jun is not limited to protecting the critical cysteine but includes a more general stabilizing effect on the basic region. This is consistent with the observation that yeast MBF1 enhanced DNA binding of GCN4, which harbors a serine in the position of the oxidation-sensitive cysteine (Takemaru et al, 1998). Analysis of yeast MBF1 and GCN4 indicates that this serine resides within the region contacted by MBF1 (Takemaru et al, 1998). We speculate that it is this evolutionarily ancient function of MBF1 to support the activity of bZIP proteins that permitted the acquisition of the redox regulation of AP-1 by oxidation of the critical cysteine; in the absence of MBF1, such mutation (serine to oxidation-sensitive cysteine) would be prone to the total destruction of the AP-1 activity even under mild oxidative conditions. Interestingly, the yeast counterpart of AP-1 (yAP-1) is also required for antioxidant defense and is accordingly regulated by the redox state, albeit at the level of nuclear export (Kuge et al, 1997; Yan et al, 1998; Toone et al, 2001). The metazoan AP-1 may have introduced redox sensing at the DNA-binding step since it is directly involved in transcriptional regulation compared with the nuclear export.
MBF1 supports AP-1 functioning during oxidative stress in Drosophila
Despite the fact that evolutionary conservation of MBF1 suggests an essential role for the protein, null mutants lacking MBF1 proved to be viable in Drosophila (this study) and yeast (Takemaru et al, 1998) under laboratory conditions. Strikingly, however, in both organisms, MBF1 is essential during stress situations encountered in the real world: Drosophila mbf1 mutants are sensitive to oxidative stress induced by H2O2, and yeast MBF1 mutants are unable to overcome nutritional stress due to their inability to maintain the activity of GCN4, a regulator of amino-acid synthesis (Takemaru et al, 1998). A comparative advantage provided by MBF1 under stress conditions is thus the likely cause of its evolutionary conservation. We propose that in both yeast and Drosophila, MBF1 achieves these functions via the same mechanism, through binding a bZIP transcription factor.
The interaction between MBF1 and D-Jun, documented in this study, provides a molecular basis of the H2O2 sensitivity of mbf1 mutants. This is supported by the recently published evidence that JNK signaling is indeed required for oxidant resistance in Drosophila (Wang et al, 2003). A developmental defect that can occur in mbf1 mutants under oxidative stress is the failure to form a continuous cuticle at the dorsal midline. The cell shape changes of epithelia that occur at the dorsal closure during embryogenesis and adult morphogenesis are regulated by the JNK signaling pathway, culminating in the phosphorylation of D-Jun (Glise et al, 1995; Sluss et al, 1996; Glise and Noselli, 1997; Hou et al, 1997; Kockel et al, 1997; Riesgo-Escovar and Hafen, 1997a, 1997b; Zeitlinger et al, 1997; Agnes et al, 1999; Zeitlinger and Bohmann, 1999; Martin-Blanco et al, 2000). Using a knockdown experiment, we show here that also D-Jun is directly involved in the adult thorax closure. Because MBF1 exhibits a genetic interaction with AP-1 subunits under H2O2 challenge, it is likely that D-Jun requires its partner MBF1 to be protected from oxidation during its function in thorax closure. Necrotic wounds in mbf1 D-fos/mbf1 flies are a newly observed phenomenon, which may be connected with the exposure to H2O2 and may reflect a specific requirement for Fos in wound healing (Martin and Nobes, 1992). Another phenotype that mbf1 mutants display is the reduced longevity when challenged with H2O2. Since AP-1 is known to trigger antioxidant defense, we favor the idea that H2O2 hypersensitivity of mbf1 mutants is also due to their failure to protect Drosophila AP-1 activity during oxidative condition. For either phenotype function, the possibility remains that MBF1 also supports functions of other transcription factors.
Dual mode of coactivator action
MBF1 was first described as a coactivator that bridges bZIP transcription factors and the basal transcriptional machinery. Yeast MBF1 supports GCN4-dependent activation of the HIS3 gene (Takemaru et al, 1998) and Drosophila MBF1 serves as a coactivator of a bZIP protein Tracheae defective/Apontic during morphogenesis of the tracheal and nervous systems (Liu et al, 2003). In either case, MBF1 facilitates the formation of a ternary complex consisting of the bZIP protein, MBF1 and the general transcription factor TBP. MBF1 has been recently shown to interact also with human AP-1 proteins (Kabe et al, 1999) and function as a novel transcriptional coactivator of c-Jun in a human cell line (Busk et al, 2003).
Results presented here suggest a new function for coactivators. We demonstrate that MBF1 can prevent an oxidative modification of D-Jun produced in bacteria, and that MBF1 activity becomes important under oxidative environmental conditions in vivo. Association of MBF1 with D-Jun in Drosophila cells and the D-Jun-dependent nuclear localization of MBF1 suggest that endogenous Jun, once synthesized, is quickly bound by MBF1. Thus it is possible that transcriptional coactivators may exert a stabilizing or protective effect on their partner transcription factors even before they engage in transcription, and that the formation of the ternary complex is a two-step phenomenon involving a preformed complex and TBP.
Materials and methods
Expression and purification of recombinant proteins
DNA regions encoding amino acids from Met253 to Thr324 in D-Fos and from Met207 to Gly279 in D-Jun (Perkins et al, 1990) were N-terminally fused with a hexahistidine tag (6H) in the pET-28a vector (Novagen). Both proteins were terminated by converting the TGC codons of Cys325 in D-Fos and Cys280 in D-Jun to TGA. To produce redox-insensitive AP-1 forms, the single remaining Cys275 in D-Fos and Cys229 in D-Jun were substituted with serine (AGC) using PCR-based mutagenesis (Abate et al, 1990). The entire coding region of Drosophila MBF1 (Liu et al, 2003) was also cloned behind the 6H tag in pET-28a. For coexpression, bicistronic pET-28a vectors with tandemly cloned 6H-tagged MBF1 and D-Jun (or MBF1 and D-Fos) sequences (Figure 1) were constructed according to Li et al (1997). All proteins were expressed in E. coli strain BL21-CodonPlus (Stratagene) and recovered under native conditions. Bacteria at OD600=0.5 were induced with 1 mM IPTG and grown for 4–6 h at 25°C. Harvested cells were sonicated in buffer A (20 mM Tris–HCl (pH 7.2), 500 mM KCl, 5 mM imidazole, 1 mM phenylmethylsulfonyl fluoride and 0.1% Triton X-100) on ice and centrifuged (38 000 g, 20 min at 4°C). The supernatant was loaded onto an Ni-NTA agarose affinity column (Qiagen), and proteins were eluted with 250 mM imidazole in buffer A and stored in this solution at 4°C or frozen at −80°C.
Electrophoresis mobility shift assay
DNA-binding assays were carried out in 12.5. mM Hepes–KOH (pH 7.9), 50 mM KCl, 5 mM MgCl2, 10% glycerol, 4 mg/ml bovine serum albumin (BSA) and DTT (1 mM unless specified otherwise). Combinations of recombinant Jun, Fos and MBF1 proteins (each approximately 100 ng/20 μl reaction) were preincubated in this buffer for 30 min at 30°C, then 20 μg of poly(dI-dC) and 25 fmol of the 32P-labeled probe were added and the reaction was incubated for another 30 min at 30°C. The probe AP-1 site has been described previously (Perkins et al, 1990; Eresh et al, 1997). Complexes were resolved on 6% polyacrylamide gels in Tris–borate–EDTA (pH 8.3) at room temperature. Contrary to findings of others (Perkins et al, 1990; Eresh et al, 1997), we have not observed binding of D-Jun or D-Fos homodimers to the same AP-1 site under any conditions. This could be due to truncations, designed to eliminate a second cysteine C-terminal to the leucine zipper in both proteins.
GST pull-down assay
The entire MBF1 protein, N-terminally fused with GST, was expressed in E. coli using the pGEX-4T-3 vector and purified from a soluble fraction on Glutathione Sepharose 4B beads (Pharmacia). Interaction assay with the purified His-tagged D-Jun and D-Fos bZIP domains was performed as described (Takemaru et al, 1998) and the bound proteins were detected on Western blots with an anti-polyhistidine monoclonal antibody (Sigma) diluted 1:5000.
MALDI-TOF analysis
The bZIP region of D-Jun was expressed alone or coexpressed with MBF1 in E. coli and the 6H-tagged proteins were affinity purified as described above. The purified proteins were desalted on C18ZipTip and analyzed on a MALDI-TOF mass spectrometer Axima-CFR (Shimazu, Kyoto, Japan) using alpha-cyano-4-hydroxycinnamic acid as matrix.
MBF1 antibody, immunoblot and tissue staining
The entire 6H-tagged MBF1 protein was prepared as described above, then purified on Mono S column (Pharmacia) and used for immunization of rabbit. The polyclonal serum was diluted 1:50 000 for immunoblots of total Drosophila extracts, and 1:10 000 for tissue staining. For Western blots, embryos, larvae, pupae or flies were homogenized directly in a denaturing sodium dodecylsulfate (SDS) buffer, and proteins (15 μg per lane) were separated on an SDS–15% polyacrylamide gel. Detection was with a goat HRP-conjugated anti-rabbit antibody (1:3000) and a chemiluminescent substrate. For tissue staining, larvae were dissected in a phosphate-buffered saline (PBS) and fixed with 3.7% formaldehyde in PBS for 1 h. After permeabilization with 0.3% Triton X-100 in PBS and blocking in 5% normal goat serum, the tissues were incubated for 24 h with anti-MBF1, washed and then stained overnight with a Cy3-conjugated goat anti-rabbit antibody (Amersham) diluted 1:2000. DNA counterstaining was with DAPI (200 ng/ml).
Transfection, immunostaining and immunoprecipitation in Drosophila cultured cells
S2 cells were grown in Shields and Sang medium (Sigma) with 10% FBS (Invitrogen). Cl.8+ cells were kept in Shields and Sang medium supplemented with 2% FBS, 2.5% fly extract and 0.125 IU/ml insulin. Cells (106/ml) were transfected using calcium phosphate precipitation with 0.5 μg of the pIE1hr/PA plasmid (a gift from Dr Bock), expressing either MBF1, the entire D-Jun protein with a C-terminal hexahistidine tag (D-Jun-His), the 6H-tagged bZIP domain of D-Jun or the green fluorescent protein (GFP) under the IE baculoviral promoter. For immunoprecipitation, cells were lysed in 25 mM Hepes (pH 7.4), 60 mM NaCl, 1% Triton X-100, 1 mM EDTA, 1.5 mM MgCl2, 25 mM NaF, 1 mM Na3VO4 and mixed protease inhibitors (Sigma). Dynabeads Protein G (Dynal) magnetic beads bound with a monoclonal anti-polyhistidine antibody (Sigma) were incubated with 100 μl of the cell extract and washed as described (Liu et al, 2003). Co-immunoprecipitated D-Jun-His and MBF1 proteins were detected with anti-MBF1 and anti-D-Jun (Bohmann et al, 1994) rabbit polyclonal sera. For immunostaining, cells were fixed for 10 min with 3.7% formaldehyde and permeabilized for 10 min in 0.2% Triton X-100 in PBS. After blocking with 2.5% skim milk and 2.5% BSA in TBST (25 mM Tris–HCl (pH 7.5), 136 mM NaCl, 2.68 mM KCl, 0.1% Triton X-100), cells were incubated with the anti-MBF1 (1:10 000) and the anti-polyhistidine (1:800) antibodies in blocking solution for 2 h. After washing in TBST, MBF1 was visualized with anti-rabbit Cy3 (Amersham) and 6H-tagged D-Jun with anti-mouse DTAF (Jackson Immunoresearch).
Preparation and genetic rescue of mbf1 mutants
The P[lacW+] element sited in the first exon of the argos gene (styp2; Okano et al, 1992), about 100 kb to the left of mbf1 on the 3L chromosome, was mobilized in the presence of the P transposase Δ2-3. Close to 4500 males representing new insertion lines were individually mated with TM3-balanced virgins. The males were captured 4 days later, and in pools of 10 were prepared for PCR analysis (Gloor et al, 1993). P[lacW+] insertions near mbf1 were screened using PCR with one primer identical to the P-element 31-bp terminal repeat and two antisense mbf1-specific primers. All three primers were combined in standard reactions (1 min at 94°C, 1 min at 68°C, 3 min at 72°C, 30 cycles) with 1 μl of the fly extract and the ExTaq DNA polymerase (Takara). A single line with P[lacW+] around 20 bp upstream of the first mbf1 exon was isolated. Genomic DNA from homozygous flies was sequenced to determine the P[lacW+] position and to confirm that its original insertion site in argos was now wild type. Expression of mbf1 mRNA and protein products was unaffected by the P-element insertion. mbf1-null mutants were generated by remobilizing P[lacW+]. Imprecise excisions were detected among 500 males using PCR as described above, except that two primers flanking the mbf1 gene were used. Sequencing of the PCR products determined the extent of deletions in the resulting alleles. The absence of the genomic DNA, mRNA and protein in homozygous mbf1 mutants was confirmed with standard hybridization methods using the coding region of mbf1 as a probe, and by Western blot. Genetic rescue, which fully restored MBF1 expression, was carried out as described previously (Liu et al, 2003), except that the genomic fragment containing mbf1+ was placed on the second chromosome.
Fly stocks and RNAi
The mbf1-null mutants were used either as homozygotes or as hemizygotes over a Df(3L)st7P deficiency including mbf1. Loss of mbf1 was combined with recessive embryonic lethal alleles of D-jun (cn1JraIA109bw1sp1) and DJNK (bsk2cn1bw1sp1) on the second chromosome (Nüsslein-Volhard et al, 1984). The recessive lethal allele of D-fos (ru1h1th1st1cu1sr1eskay1ca1) (Jürgens et al, 1984) was recombined with mbf12 to produce mbf12sr1eskay1ca1, referred to in the text as mbf1 D-fos. RNAi silencing of D-jun was performed using the UAS/Gal4 system (Kennerdell and Carthew, 2000). The UAS transgenic line expressing a hairpin-loop D-jun RNA was a kind gift from Dr Yanicostas (Inst. Monod, Paris); the pannier (pnr) Gal4 driver was described previously (Calleja et al, 1996).
Oxidant resistance tests
H2O2 (0.5%) was added to 1.3% sucrose in 1% low melting point agarose at 40°C. The medium was dispensed into glass vials and allowed to solidify. Catalase (3-amino-1,2,4-triazole; Sigma) and GSH synthase (L-buthionine-[S,R]-sulfoximine; Sigma) inhibitors in aqueous solutions were applied the same way. Adult males 3–5 days old were placed in vials (30 per vial) and their survival was monitored. At least 120 flies were tested per genotype, over 500 for the mbf1− and rescued lines. Resistance of larvae was tested by placing first instar larvae on a sucrose–yeast medium containing 0.1 or 0.3% H2O2. Animals at the larval–pupal transition were challenged with H2O2 vapors using a tissue paper soaked with 30% H2O2 and placed into vials with wandering and freshly pupariating larvae.
Acknowledgments
We thank Shigeo Hayashi, Keith Yamamoto and Jean-Antoine Lepesant for discussions, Dino Yanicostas for D-junRNAi flies and Dirk Bohmann for anti-D-Jun antibody and D-Jun cDNA. Advice provided on oxidant sensitivity tests by Herve Tricoire and on the use of aminotriazole by Pedro Oliveira is appreciated. We acknowledge receiving fly stocks from the Bloomington center. This work was supported by 204/04/1371 from the Czech Grant Agency to MJ, who was also supported by a Center of Excellence Program of Japan, and by a Grant-in-aid for Scientific Research from MEXT of Japan to SH.
References
- Abate C, Patel L, Rauscher FJ, Curran T (1990) Redox regulation of Fos and Jun DNA-binding activity in vitro. Science 249: 1157–1161 [DOI] [PubMed] [Google Scholar]
- Agnes F, Suzanne M, Noselli S (1999) The Drosophila JNK pathway controls the morphogenesis of imaginal discs during metamorphosis. Development 126: 5453–5462 [DOI] [PubMed] [Google Scholar]
- Bohmann D, Bos TJ, Admon A, Nishimura T, Vogt PK, Tjian R (1987) Human protooncogene c-Jun encodes a DNA-binding protein with structural and functional properties of transcription factor AP-1. Science 238: 1386–1392 [DOI] [PubMed] [Google Scholar]
- Bohmann D, Ellis MC, Staszewski LM, Mlodzik M (1994) Drosophila Jun mediates Ras-dependent photoreceptor determination. Cell 78: 973–986 [DOI] [PubMed] [Google Scholar]
- Busk PK, Wulf-Andersen L, Strom CC, Enevoldsen M, Thirstrup K, Haunso S, Sheikh SP (2003) Multiprotein bridging factor 1 cooperates with c-Jun and is necessary for cardiac hypertrophy in vitro. Exp Cell Res 286: 102–114 [DOI] [PubMed] [Google Scholar]
- Calleja M, Moreno E, Pelaz S, Morata G (1996) Visualization of gene expression in living adult Drosophila. Science 274: 252–255 [DOI] [PubMed] [Google Scholar]
- Davis RJ (2000) Signal transduction by the JNK group of MAP kinases. Cell 103: 239–252 [DOI] [PubMed] [Google Scholar]
- Eresh S, Riese J, Jackson DB, Bohmann D, Bienz M (1997) A CREB-binding site as a target for decapentaplegic signalling during Drosophila endoderm induction. EMBO J 16: 2014–2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glise B, Bourbon H, Noselli S (1995) hemipterous encodes a novel Drosophila MAP kinase kinase, required for epithelial-cell sheet movement. Cell 83: 451–461 [DOI] [PubMed] [Google Scholar]
- Glise B, Noselli S (1997) Coupling of Jun amino-terminal kinase and decapentaplegic in Drosophila morphogenesis. Genes Dev 11: 1738–1747 [DOI] [PubMed] [Google Scholar]
- Gloor GB, Preston CR, Johnsonschlitz DM, Nassif NA, Phillis RW, Benz WK, Robertson HM, Engels WR (1993) Type-i repressors of P-element mobility. Genetics 135: 81–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heitzler P, Haenlin M, Ramain P, Calleja M, Simpson P (1996) A genetic analysis of pannier, a gene necessary for viability of dorsal tissues and bristle positioning in Drosophila. Genetics 143: 1271–1286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota K, Matsui M, Iwata S, Nishiyama A, Mori K, Yodoi J (1997) AP-1 transcriptional activity is regulated by a direct association between thioredoxin and Ref-1. Proc Natl Acad Sci USA 94: 3633–3638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou XS, Goldstein ES, Perrimon N (1997) Drosophila Jun relays the Jun amino-terminal kinase signal transduction pathway to the decapentaplegic signal transduction pathway in regulating epithelial cell sheet movement. Genes Dev 11: 1728–1737 [DOI] [PubMed] [Google Scholar]
- Jürgens G, Wieschaus E, Nüsslein-Volhard C, Kluding H (1984) Mutations affecting the pattern of the larval cuticle in Drosophila melanogaster 2. Zygotic loci on the 3rd chromosome. Wilhelm Roux's Arch Dev Biol 193: 283–295 [DOI] [PubMed] [Google Scholar]
- Kabe Y, Goto M, Shima D, Imai T, Wada T, Morohashi K, Shirakawa M, Hirose S, Handa H (1999) The role of human MBF1 as a transcriptional coactivator. J Biol Chem 274: 34196–34202 [DOI] [PubMed] [Google Scholar]
- Karin M (1995) The regulation of AP-1 activity by mitogen-activated protein- kinases. J Biol Chem 270: 16483–16486 [DOI] [PubMed] [Google Scholar]
- Kennerdell JR, Carthew RW (2000) Heritable gene silencing in Drosophila using double-stranded RNA. Nat Biotechnol 18: 896–898 [DOI] [PubMed] [Google Scholar]
- Klatt P, Molina EP, De Lacoba MG, Padilla CA, Martinez-Galisteo E, Barcena JA, Lamas S (1999) Redox regulation of c-Jun DNA binding by reversible S-glutathiolation. FASEB J 13: 1481–1490 [DOI] [PubMed] [Google Scholar]
- Kockel L, Homsy JG, Bohmann D (2001) Drosophila AP-1: lessons from an invertebrate. Oncogene 20: 2347–2364 [DOI] [PubMed] [Google Scholar]
- Kockel L, Zeitlinger J, Staszewski LM, Mlodzik M, Bohmann D (1997) Jun in Drosophila development: redundant and nonredundant functions and regulation by two MAPK signal transduction pathways. Genes Dev 11: 1748–1758 [DOI] [PubMed] [Google Scholar]
- Kuge S, Jones N, Nomoto A (1997) Regulation of yAP-1 nuclear localization in response to oxidative stress. EMBO J 16: 1710–1720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Schwabe JWR, Banayo E, Evans RM (1997) Coexpression of nuclear receptor partners increases their solubility and biological activities. Proc Natl Acad Sci USA 94: 2278–2283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu QX, Jindra M, Ueda H, Hiromi Y, Hirose S (2003) Drosophila MBF1 is a co-activator for Tracheae defective and contributes to the formation of tracheal and nervous systems. Development 130: 719–728 [DOI] [PubMed] [Google Scholar]
- Maki Y, Bos TJ, Davis C, Starbuck M, Vogt PK (1987) Avian-sarcoma virus-17 carries the jun oncogene. Proc Natl Acad Sci USA 84: 2848–2852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariotti M, De Benedictis L, Avon E, Maier JAM (2000) Interaction between endothelial differentiation-related factor-1 and calmodulin in vitro and in vivo. J Biol Chem 275: 24047–24051 [DOI] [PubMed] [Google Scholar]
- Marshall HE, Merchant K, Stamler JS (2000) Nitrosation and oxidation in the regulation of gene expression. FASEB J 14: 1889–1900 [DOI] [PubMed] [Google Scholar]
- Martin P, Nobes CD (1992) An early molecular-component of the wound-healing response in rat embryos—induction of c-Fos protein in cells at the epidermal wound margin. Mech Dev 38: 209–216 [DOI] [PubMed] [Google Scholar]
- Martin-Blanco E, Pastor-Pareja JC, Garcia-Bellido A (2000) JNK and decapentaplegic signaling control adhesiveness and cytoskeleton dynamics during thorax closure in Drosophila. Proc Natl Acad Sci USA 97: 7888–7893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan IM, Havarstein LS, Wong WY, Luu P, Vogt PK (1994) Efficient induction of fibrosarcomas by v-Jun requires mutations in the DNA-binding region and the transactivation domain. Oncogene 9: 2793–2797 [PubMed] [Google Scholar]
- Nüsslein-Volhard C, Wieschaus E, Kluding H (1984) Mutations affecting the pattern of the larval cuticle in Drosophila melanogaster 1. Zygotic loci on the 2nd chromosome. Wilhelm Roux's Arch Dev Biol 193: 267–282 [DOI] [PubMed] [Google Scholar]
- Okano H, Hayashi S, Tanimura T, Sawamoto K, Yoshikawa S, Watanabe J, Iwasaki M, Hirose S, Mikoshiba K, Montell C (1992) Regulation of Drosophila neural development by a putative secreted protein. Differentiation 52: 1–11 [DOI] [PubMed] [Google Scholar]
- Okuno H, Akahori A, Sato H, Xanthoudakis S, Curran T, Iba H (1993) Escape from redox regulation enhances the transforming activity of Fos. Oncogene 8: 695–701 [PubMed] [Google Scholar]
- Ordway JM, Eberhart D, Curran T (2003) Cysteine 64 of Ref-1 is not essential for redox regulation of AP-1 DNA binding. Mol Cell Biol 23: 4257–4266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins KK, Admon A, Patel N, Tjian R (1990) The Drosophila Fos-related AP-1 protein is a developmentally regulated transcription factor. Genes Dev 4: 822–834 [DOI] [PubMed] [Google Scholar]
- Riesgo-Escovar JR, Hafen E (1997a) Common and distinct roles of DFos and DJun during Drosophila development. Science 278: 669–672 [DOI] [PubMed] [Google Scholar]
- Riesgo-Escovar JR, Hafen E (1997b) Drosophila Jun kinase regulates expression of decapentaplegic via the ETS-domain protein Aop and AP-1 transcription factor DJun during dorsal closure. Genes Dev 11: 1717–1727 [DOI] [PubMed] [Google Scholar]
- Shaulian E, Karin M (2002) AP-1 as a regulator of cell life and death. Nat Cell Biol 4: E131–E136 [DOI] [PubMed] [Google Scholar]
- Sluss HK, Han ZQ, Barrett T, Davis RJ, Ip YT (1996) A JNK signal transduction pathway that mediates morphogenesis and an immune response in Drosophila. Genes Dev 10: 2745–2758 [DOI] [PubMed] [Google Scholar]
- Stronach BE, Perrimon N (1999) Stress signaling in Drosophila. Oncogene 18: 6172–6182 [DOI] [PubMed] [Google Scholar]
- Sundquist AR, Fahey RC (1989) Evolution of antioxidant mechanisms: thiol-dependent peroxidases and thioltransferases among procaryotes. J Mol Evol 29: 429–435 [DOI] [PubMed] [Google Scholar]
- Takemaru K, Harashima S, Ueda H, Hirose S (1998) Yeast coactivator MBF1 mediates GCN4-dependent transcriptional activation. Mol Cell Biol 18: 4971–4976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takemaru K, Li FQ, Ueda H, Hirose S (1997) Multiprotein bridging factor 1 MBF1 is an evolutionarily conserved transcriptional coactivator that connects a regulatory factor and TATA element-binding protein. Proc Natl Acad Sci USA 94: 7251–7256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toone WM, Morgan BA, Jones N (2001) Redox control of AP-1-like factors in yeast and beyond. Oncogene 20: 2336–2346 [DOI] [PubMed] [Google Scholar]
- Wang MC, Bohmann D, Jasper H (2003) JNK signaling confers tolerance to oxidative stress and extends lifespan in Drosophila. Dev Cell 5: 811–816 [DOI] [PubMed] [Google Scholar]
- Xanthoudakis S, Curran T (1992) Identification and characterization of Ref-1, a nuclear-protein that facilitates AP-1 DNA-binding activity. EMBO J 11: 653–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xanthoudakis S, Miao G, Wang F, Pan Y-CE, Curran T (1992) Redox activation of Fos–Jun DNA binding activity is mediated by a DNA repair enzyme. EMBO J 11: 3323–3335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan C, Lee LH, Davis LI (1998) Crm1p mediates regulated nuclear export of a yeast AP-1-life transcription factor. EMBO J 17: 7416–7429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeitlinger J, Bohmann D (1999) Thorax closure in Drosophila: involvement of Fos and the JNK pathway. Development 126: 3947–3956 [DOI] [PubMed] [Google Scholar]
- Zeitlinger J, Kockel L, Peverali FA, Jackson DB, Mlodzik M, Bohmann D (1997) Defective dorsal closure and loss of epidermal decapentaplegic expression in Drosophila fos mutants. EMBO J 16: 7393–7401 [DOI] [PMC free article] [PubMed] [Google Scholar]