

Abstract
Mutations in SURF1, the human homologue of yeast SHY1, are responsible for Leigh's syndrome, a neuropathy associated with cytochrome oxidase (COX) deficiency. Previous studies of the yeast model of this disease showed that mutant forms of Mss51p, a translational activator of COX1 mRNA, partially rescue the COX deficiency of shy1 mutants by restoring normal synthesis of the mitochondrially encoded Cox1p subunit of COX. Here we present evidence showing that Cox1p synthesis is reduced in most COX mutants but is restored to that of wild type by the same mss51 mutation that suppresses shy1 mutants. An important exception is a null mutation in COX14, which by itself or in combination with other COX mutations does not affect Cox1p synthesis. Cox14p and Mss51p are shown to interact with newly synthesized Cox1p and with each other. We propose that the interaction of Mss51p and Cox14p with Cox1p to form a transient Cox14p–Cox1p–Mss51p complex functions to downregulate Cox1p synthesis. The release of Mss51p from the complex occurs at a downstream step in the assembly pathway, probably catalyzed by Shy1p.
Keywords: COX assembly, COX14, mitochondria, MSS51, SHY1
Introduction
Assembly of yeast cytochrome oxidase (COX) requires the assistance of at least 20 nuclear gene products (McEwen et al, 1986; Tzagoloff and Dieckmann, 1990). These proteins act at all stages of the assembly process, including processing (Seraphin et al, 1989) and translation (Costanzo and Fox, 1993) of the mitochondrially encoded mRNAs for subunits 1, 2, and 3, membrane insertion of the hydrophobic subunits (Hell et al, 2001), and maturation of the heme and copper centers (Glerum et al, 1996a; 1996b; Barros et al, 2001).
Although much progress has been made in understanding some of the events leading to the assembly of COX, the functions of a number of gene products essential for this process have yet to be clarified (Barrientos et al, 2002a; 2002b). A case in point is Shy1p, a yeast mitochondrial protein needed for full expression of COX (Mashkevich et al, 1997; Barrientos et al, 2002b). The function of this protein is of considerable interest because mutations in its human homologue, Surf1p, have been shown to be responsible for most diagnosed cases of Leigh's syndrome (Tiranti et al, 1998; Zhu et al, 1998), a neuromuscular disease presenting a COX deficiency (DiMauro and De Vivo, 1996).
Extragenic suppressors of shy1 null mutants have been mapped to MSS51, a nuclear gene coding for a Cox1p-specific translational activator (Barrientos et al, 2002b), suggesting that unassembled Cox1p may downregulate its own translation by competitively trapping a translational activator complex in which Mss51p is one of the components. According to this model, the translational block is relieved by Shy1p-dependent assembly of Cox1p making Mss51p available for Cox1p synthesis (Barrientos et al, 2002b).
In the present study, we show that Cox1p synthesis is reduced in most COX assembly mutants, cox14 mutants being an exception. Mutant forms of Mss51p are able to restore Cox1p expression in strains carrying null alleles of either COX structural genes or genes coding for COX assembly factors. We also present evidence for an interaction of newly synthesized Cox1p with Mss51p and Cox14p, but not Shy1p. We propose that the formation and turnover of Cox1p/Mss51p/Cox14p couple Mss51p-dependent translation of Cox1p to its utilization for COX assembly.
Results
Synthesis of Cox1p is repressed in assembly-arrested mutants
To ascertain if the Cox1p labeling defect previously noted in shy1 mutants (Barrientos et al, 2002b) is general to all mutants blocked in COX assembly, a wide range of strains with lesions in COX subunits or assembly factors were pulsed with [35S]methionine in vivo in the presence of cycloheximide. Labeling of Cox1p, but not Cox2p or Cox3p, was visibly reduced in most mutants (Figures 1, 2 and 3). The only exception was the cox14 mutant (Glerum et al, 1995). The precise function of Cox14p is not known at present.
Figure 1.
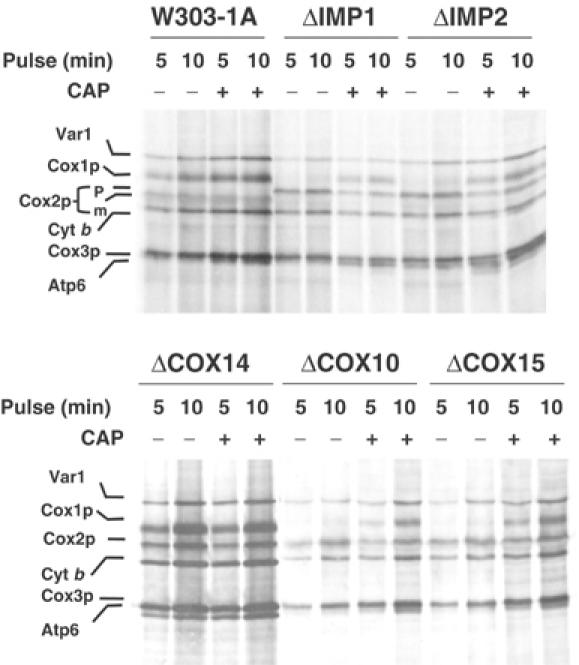
In vivo labeling of mitochondrial gene products in COX mutants. Wild-type (W303-1A) and mutant cells (described in Table I) were labeled with [35S]methionine at 30°C for the indicated times in the presence of cycloheximide. One-half of each culture was incubated in the presence of 2 mg/ml chloramphenicol during the last 2 h of growth prior to labeling (+CAP). Samples were removed after the indicated times of labeling and processed as detailed in Materials and methods. The mitochondrial translation products are identified in the margin. Cox2p is not processed in ΔIMP1 and ΔIMP2 mutants. The Cox2p precursor (p) in these strains migrates slower that the mature Cox2p (m).
Figure 2.
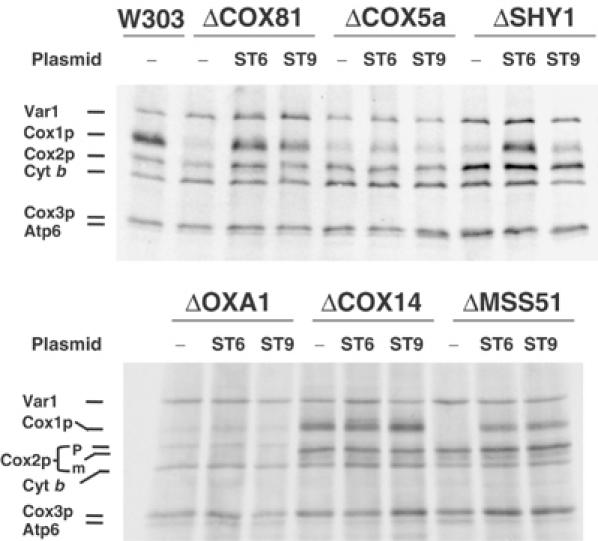
In vivo labeling of mitochondrial gene products in COX mutants expressing different alleles of MSS51. MSS51 and the suppressor mss51T167R, which partially suppress the respiratory defect of shy1 mutants, were cloned in YIp351. The resultant constructs pSG91/ST9 and pSG91/ST6, respectively, were integrated at the chromosomal LEU2 locus of the indicated mutants (see Table I for description of mutants). The mutants (−) and transformants were labeled with [35S]methionine at 30°C for 15 min in the presence of cycloheximide.
Figure 3.
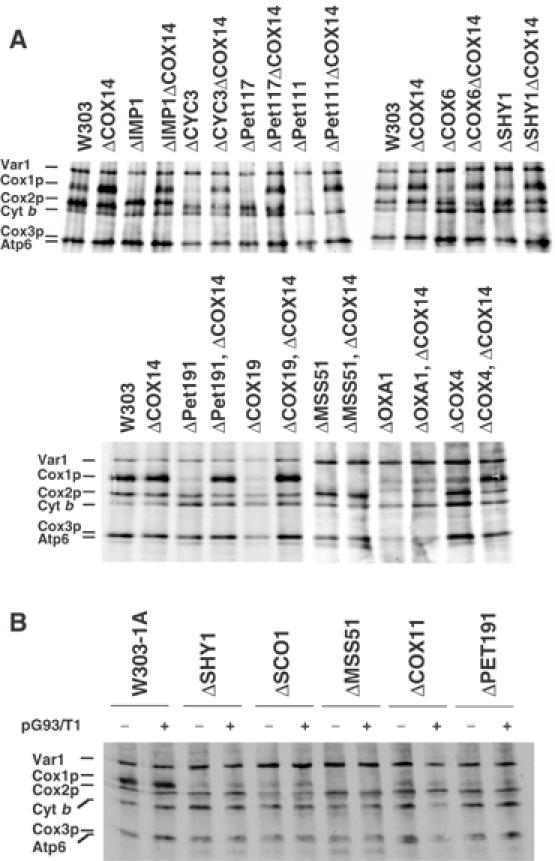
In vivo labeling of mitochondrial gene products in COX14 mutants and effect of overexpression of COX14p on Cox1p labeling. (A) Wild type (W303), different COX mutants, and the same mutants carrying an additional null mutation in COX14 were labeled with [35S]methionine at 30°C for 15 min in the presence of cycloheximide. With the exception of CYC3, which codes for a cytochrome c-specific heme lyase, the functions affected in the different strains are described in Table I. (B) The wild-type W303-1A, the indicated COX mutants, and the same strains harboring COX14 on a multicopy plasmid (pG93/T1) were pulse-labeled with [35S]methionine and equivalent amounts of protein were separated by SDS–PAGE on a 17.5% polyacrylamide gel.
The reduced Cox1p labeling in the mutants was seen even with 5 min pulses, the shortest time at which incorporation of [35S]methionine into the mitochondrial translation products could be consistently detected in the wild type. This phenotype is easy to explain in the case of the mss51 and pet309 mutants, both of which have mutations in Cox1p-specific translational factors (Decoster et al, 1990; Manthey and McEwen, 1995). Similarly, the poor labeling of Cox1p in the oxa1 mutant (Figure 3) may be the result of rapid turnover when Oxa1p-dependent membrane insertion of Cox1p is blocked (Hell et al, 2001). The Cox1p deficit in the other strains, however, is difficult to rationalize since the functions affected are unrelated to translation of this protein.
With the exception of the mss51, pet309, and oxa1 mutants, Cox1p labeling was increased in mutants and in wild type when cells were preincubated in chloramphenicol prior to the pulse (Figure 1A and B). The improved translation of mitochondrial products following chloramphenicol treatment is presumed to occur as a result of the larger pools of nuclear-encoded subunits available for assembly of intermediates and/or because of the accumulation of nuclear-encoded factors required for mitochondrial gene expression. Methionine incorporation into Cox1p in the mutants after the chloramphenicol incubation was comparable to that seen in wild type under normal pulse-labeling conditions, indicating that the translation apparatus is fully functional in the mutants and that the phenotype stems from a decreased rate of synthesis and/or increased rate of turnover of Cox1p. Turnover seemed less likely in view of the ability of cox14 mutants to synthesize Cox1p at rates similar to wild type even though they are also blocked in COX assembly and displays low steady-state concentrations of Cox1p and Cox2p (Glerum et al, 1995). This and other observations discussed below favor decreased translation as the more likely explanation for the observed deficit of newly synthesized Cox1p in the various COX-deficient mutants.
Cox1p synthesis defect in COX assembly mutants in suppressed by the mss51T167R allele
A single copy of the mss51T167R allele or an extra copy of wild-type MSS51 was shown to suppress partially the respiratory defect of shy1 mutants by increasing Cox1p translation (Barrientos et al, 2002b). This suggested that the shy1 mutation may inactivate or reduce the effective concentration of Mss51p as a translational activator of the COX1 mRNA. These observations have been extended to other COX-deficient mutants. Mitochondrial translation products were labeled in vivo with [35S]methionine in an assortment of COX mutants, with and without an extra copy of the wild-type MSS51 or the mss51T167R suppressor integrated at the leu2 locus of nuclear DNA. In all the strains except the cox14 mutant examined, synthesis of Cox1p, but not of the other COX subunits, was markedly increased by the suppressor and to a lesser extent also by the extra copy of wild-type MSS51 (Figure 2).
It is significant that the higher expression of Cox1p leads to a partial rescue of respiration and COX activity in the shy1 mutant (Barrientos et al, 2002b), but not in any of the other mutants examined. This suggests that the functions of Shy1p and Mss51p are related. In other COX mutants, however, restoration of normal rates of Cox1p synthesis by the mss51T167R suppressor (see ΔCOX18 in Figure 2) is not a sufficient condition to compensate for the assembly defect because the impaired function is unrelated to expression of this subunit.
Increased Cox1p synthesis in cox14 mutants is epistatic in mutants with lesions in other COX assembly factors
The lack of effect of the cox14 mutation on Cox1p synthesis (Figures 1, 2, 3 and 4) suggested that Cox14p might negatively regulate translation of this subunit. This was tested by measuring Cox1p synthesis in strains carrying mutations in COX14 and other COX-specific genes. The cox14 mutation restored normal Cox1p synthesis in all the COX mutants except the mss51, pet309 (not shown), and oxa1 mutants (Figure 3A). Despite being epistatic with respect to Cox1p expression, the cox14 mutation did not rescue either respiration or the ability of the mutants, including the of shy1 mutant, to assemble COX (not shown).
Figure 4.
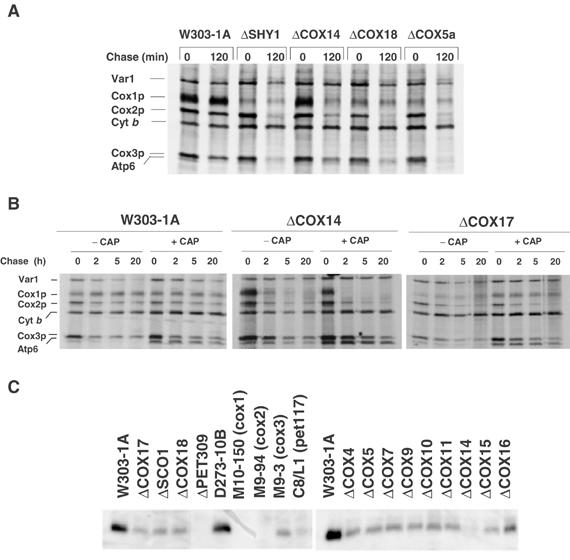
Turnover of in vivo-labeled mitochondrial translation products and steady-state concentration of Cox1p in wild type and COX mutants. (A) Wild type (W303-1A) and mutants (described in Table I) were grown and labeled for 20 min at 30°C with [35S]methionine. Labeling was terminated by addition of 80 μmol cold methionine and 12 μg/ml puromycin (0 time). Samples of the cultures were collected after the indicated times of incubation at 30°C and processed as in Figure 1. (B) The wild-type strain W303-1A and the cox14 and cox17 mutants were labeled and chased for the indicated times as in panel A. One-half of each culture was incubated in the presence of chloramphenicol as in Figure 1 prior to labeling. (C) The wild-type W303-1A and D273-10B and mutant strains were grown in 2% galactose, 1% yeast extract, and 2% peptone to stationary phase. Mitochondria were prepared and 10 μg of protein was separated by SDS–PAGE on a 12% polyacrylamide gel. The proteins were transferred to nitrocellulose and probed with a polyclonal antibody against yeast Cox1p. The antibody–antigen complexes were visualized by a secondary reaction with [125I]protein A.
Overexpression of Cox14p does not alter synthesis of Cox1p
Normal labeling of Cox1p in cox14 single and double mutants could indicate that Cox14p acts to increase degradation of unassembled or incompletely assembled Cox1p. If this were the case, overexpression of Cox14p in a wild-type or mutant background might be expected to affect the amount of newly synthesized Cox1p. This was examined by transforming the wild-type strain and several COX assembly-deficient mutants with the episomal plasmid pG93/T1 (Glerum et al, 1995), which contains a wild-type COX14 gene. Overexpression of Cox14p in these strains did not affect in vivo labeling of Cox1p (Figure 3B) or restore respiration (data not shown).
Stability of newly synthesized Cox1p in wild type and COX mutants
The stability of unassembled Cox1p was assessed in wild type and different COX mutants by pulse-chase. Most of the translation products, including the three COX subunits, were stable during 2 h of chase in wild type but not in the mutants in which a significant fraction of Cox2p and Cox3p were degraded (Figure 4A). In contrast, the small amount of labeled Cox1p detected in the mutants was stable during the chase (Figure 4A). The exception was the cox14 mutants, in which most of the Cox1p was degraded (Figure 4A and B). The kinetics of Cox1p turnover was also examined during longer periods of chase of cells pulse-labeled with and without a prior incubation in the presence of chloramphenicol (Figure 4B). Cox1p was stable even after 20 h of chase, but under the same conditions most of Cox1p in the cox14 mutant was degraded after 2 h of chase. The small amount of Cox1p detected in the cox17 mutant appears to be as stable as in wild type. The greater lability of Cox1p in the cox14 mutant is also supported by the results of Western analysis of the steady-state concentrations of this subunit in different mutants (Figure 4C).
Cox1p, Cox2p, and Cox3p synthesized in cycloheximide-inhibited wild-type cells are not incorporated into the holoenzyme, even following a 2 h period of chase (data not shown). The relatively high stability of these COX subunits, however, suggests that they are in a protease-protected environment either as monomers or partially assembled intermediates, or that they are complexed to a ‘stabilizing' factor(s).
Do Shy1p and Cox14p act post-translationally?
Manthey and McEwen (1995) have shown that ρ− genomes in which COX1 is fused to the 5′ leader of the mitochondrial COB (SUP2; see Figure 5A) or COX3 (SUP1) genes are able to suppress pet309 mutants. This argues strongly against a post-translational role of Pet309p in expression of COX. In this study, we have constructed strains heteroplasmic for wild type and the SUP2 ρ− suppressor in the context of shy1, mss51, or cox14 null mutations.
Figure 5.
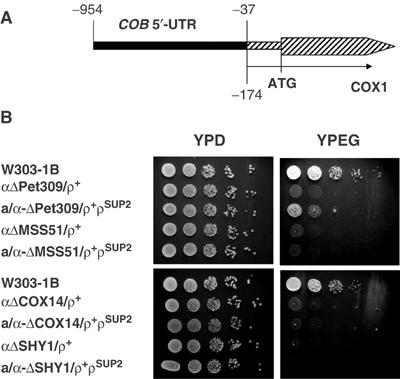
The cox14 defect is not rescued by a ρ− genome with COX1 fused to the 5′-UTR of COB. (A) Map of the mitochondrial bypass suppressor ρSUP2. The rearrangement in the suppressor leads to a fusion of the 5′-UTR of COB to nucleotide −174 of COX1 (Manthey and McEwen, 1995). (B) ρSUP2 was transferred by cytoduction to a kar1 mutant (Conde and Fink, 1976) lacking mitochondrial DNA (ρ°). The SUP2 suppressor was transferred from the kar1 donor to ρ° derivatives of null mutants of PET309, MSS51, COX14, and SHY1. The different mutants with the SUP2 genome (ρSUP2) were then crossed to the isogenic mutants with wild-type mitochondrial DNA (ρ+) to obtain the heteroplasmic diploid mutants of PET309 (a/α-ΔPET309/ρ+ρSUP2), MSS51 (a/α-ΔMSS51/ρ+ρSUP2), COX14 (a/α-ΔCOX14309/ρ+ρSUP2), and SHY1 (a/α-ΔSHY1/ρ+ρSUP2). Serial dilutions of the haploid mutants with wild-type mitochondrial DNA and of the diploid strains with the wild-type and suppressor genomes were spotted on YPD and YPEG plates and incubated at 30°C for 2.5 days.
The failure of the SUP2 suppressor to rescue cox14 and shy1 mutants (Figure 5B) indicates that Cox14p and Shy1p are required at a post-translational stage of COX assembly, although it does not exclude a role in translation as well. In agreement with the findings of Perez-Martinez et al (2003) who used SUP1 in their studies, SUP2 does not restore the COX deficiency of mss51 mutants (Figure 5B).
Cox14p interacts with newly synthesized Cox1p
Restoration of normal Cox1p translation in mutants that have a second mutation in COX14 suggested that Cox14p might be regulating Cox1p synthesis. A physical interaction of Cox1p with Cox14p was tested by expressing the latter as a GST fusion protein from a chromosomally integrated gene. The GST-tagged Cox14p was able to fully complement the respiratory defect of the cox14 mutant (data not shown). Mitochondria from aW303ΔCOX14/ST32 expressing the Cox14p-GST fusion protein were labeled in organello with [35S]methionine, extracted with lauryl maltoside, and adsorbed onto glutathione–Sepharose beads. The proteins that were recovered from the beads indicated a selective and virtually quantitative enrichment of labeled Cox1p (Figure 6A). A similar enrichment of Cox1p was seen when mitochondria were isolated from a strain expressing Mss51p-GST but not from wild type or from a strain expressing a Shy1p-GST fusion protein. Like Cox14p-GST, the latter two fusions also complemented the respective null mutants. An association of HA-tagged Mss51p with newly synthesized Cox1p was also reported by Perez-Martinez et al (2003). Trace amounts of cytochrome b, Cox2p, and Cox3p were also adsorbed to the beads but the signals varied in different experiments. The enrichment of the Cox2p precursor seen in the pull-down of the strain expressing Cox14p-GST was consistent but was investigated further.
Figure 6.
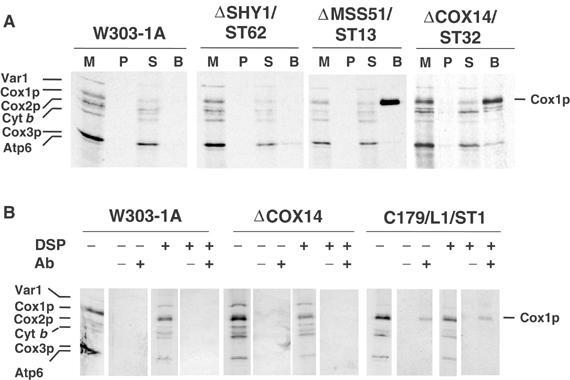
Cox14p and Mss51p interact with Cox1p. (A) Mitochondria were prepared from the wild-type W303-1A, a shy1 null mutant (ΔSHY1/ST62) with a chromosomally integrated plasmid expressing the Shy1p-GST fusion protein, an mss51 null mutant (ΔMSS51/ST13) with a chromosomally integrated plasmid expressing Mss51p-GST, and a cox14 null mutant (ΔCOX14/ST32) with a chromosomally integrated plasmid expressing Cox14p-GST. Mitochondria were labeled with [35S]methionine for 30 min and extracted with 1% lauryl maltoside, 1 M KCl, and 1 mM PMSF. The extract was clarified by centrifugation at 50 000 gav for 30 min and incubated with glutathione–Sepharose beads for 4 h at 4°C. After centrifugation at 1500 rpm for 5 min, the supernatant was collected and the beads were washed three times with PBS. Mitochondria (M) corresponding to 2 μg protein, equivalent volumes of the membrane pellet (P) after lauryl maltoside extraction and of the supernatant from the glutathione–Sepharose beads (S) were separated on a 17.5% polyacrylamide gel by SDS–PAGE. The amount of washed beads (B), however, corresponded to ∼500 μg of the starting mitochondria. (B) Mitochondria from W303-1A, the cox14 null mutant (ΔCOX14), and a cox14 point mutant transformed with a high-copy plasmid containing COX14 (C179/L1/ST1) were labeled for 30 min at 30°C in the presence of [35S]methionine. After a 5 min pulse, the samples were treated with the crosslinker DSP (+) or were mock-treated (−) as described (Hell et al, 2000). Immunoprecipitation of crosslinked adducts was performed using antiserum specific for Cox14p (+) and preimmune serum (−). Immunoprecipitates were analyzed by SDS–PAGE and autoradiography as in Figure 1.
To test if the binding of Mss51p to newly synthesized Cox1p requires the presence of Cox14p, an mss51 and cox14 double mutant was transformed with an integrative plasmid expressing Mss51p-GST. Co-precipitation of newly synthesized Cox1p with Mss51p-GST in the cox14 null background (data not shown) indicates that the interaction of Mss51p and Cox1p is not Cox14p dependent.
The interaction of Cox14p with newly synthesized Cox1p was also studied by labeling mitochondria in organello with [35S]methionine in the absence or presence of the cleavable crosslinker dithio-bis-succinimidyl propionate (DSP) to trap transient complexes that might be formed early after completion of Cox1p synthesis. Detergent extracts containing the labeled translation products were treated with antibody to Cox14p and analyzed by SDS–PAGE under conditions causing cleavage of the crosslinker. A small fraction of newly synthesized Cox1p was present in the immunoprecipitate obtained with the Cox14p antibody in a strain overexpressing Cox14p but not in a wild-type strain (Figure 6B). The co-immunoprecipitation of Cox1p did not depend on the inclusion of DSP during translation. The poor recovery of Cox1p in this procedure is probably due to the low efficiency of immunoprecipitation with the Cox14p antibody.
Cox14p interacts with Mss51p
The interaction of Cox14p with Mss51p was examined in strains of yeast, expressing either Cox14p-GST or Mss51p-GST in cox14 and mss51 null backgrounds, respectively. Pull-down assays of mitochondria extracted with lauryl maltoside indicated that approximately 61% of Mss51p-GST and 48% of Cox14p were adsorbed onto the beads (Figure 7A). Likewise, when crude mitochondrial extracts containing Cox14p-GST were adsorbed onto glutathione–Sepharose beads, more than 75% of Cox14p-GST and 50% of Mss51p were pulled down (Figure 7B).
Figure 7.
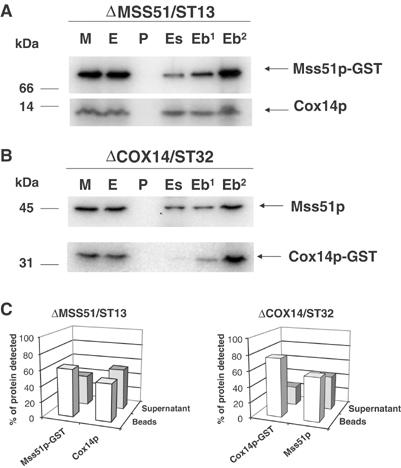
Cox14p interacts with Mss51p. (A) Mitochondria (M) from an mss51 null mutant with a chromosomally integrated plasmid expressing Mss51p-GST fusion protein (ΔMSS51/ST13) were extracted with 1% lauryl maltoside, 1 M KCl, and 1 mM PMSF. The pellet (P) after centrifugation at 50 000 gav for 30 min was suspended in the starting volume of buffer and the extract (E) was mixed and incubated for 4 h at 4°C with glutathione–Sepharose. The supernatants (Es) from the beads were collected and the beads (Eb) were washed three times with PBS. The different fractions adjusted for volume were separated by SDS–PAGE. The lane labeled (Eb2) was loaded with two times the amount of beads. Cox14p and Mss51p-GST were detected by Western blot analysis using specific antibodies against each protein. The proteins were visualized by a secondary reaction with [125I]protein A and the radiolabeled bands were detected with a PhosphorImager (Molecular Dynamics). (B) Same as (A) except that the mitochondria were prepared from a cox14 null mutant with an integrated plasmid expressing a Cox14p-GST fusion protein (ΔCOX14/ST32). (C) The bands shown in (A, B) were quantified with the PhosphorImager. The open bars represent the percentage of the corresponding protein bound to the beads, and the filled bars represent the percentage of unbound protein recovered in the supernatant fraction.
Part of the lauryl maltoside extracts was centrifuged on a linear 7.5–25% sucrose gradient. Gradient fractions were analyzed for the distributions of Mss51p and Cox14p and the peak fractions with Mss51p-GST and Cox14p or Cox14p-GST and Mss51p were treated with glutathione–Sepharose beads. These pull-down assays confirmed that the Cox14p cosedimenting with Mss51p-GST and vice versa were complexed to each other (data not shown). Our data, however, do not discriminate between a direct interaction of the two proteins and an interaction mediated by other proteins that may constitute the complex.
The GST pull-down assays suggested that only a fraction of Mss51p is complexed to Cox14p. This was supported by the results of sucrose gradient sedimentations of mitochondrial detergent extracts. Mss51p and Cox14p sedimented similarly in a gradient loaded with a lauryl maltoside extract of wild-type mitochondria. Both proteins peaked only a fraction behind lactate dehydrogenase with estimated masses of 130 kDa (Figure 8A). The distribution of Mss51p and Cox14p in this gradient, however, was not symmetrical, indicating the presence of higher molecular weight specie(s) (Figure 8A). The gradient of the lauryl maltoside extract of mitochondria from the mss51 deletion mutant showed a symmetrical distribution of Cox14p with a peak at approximately the same position as Cox14p in the wild-type extract (Figure 8B). The symmetrical Cox14p peak in the gradient of the mutant extract suggests that the faster sedimenting component(s) in the wild-type extract may represent that fraction of Cox14p complexed to Mss51p. All of the Cox14p in the mutant and most of Cox14p in wild type were estimated to have a mass at least 10 times larger than the monomer. This indicates that in addition to interacting with Mss51p, Cox14p also exists as part of a larger homo or hetero-oligomeric complex.
Figure 8.
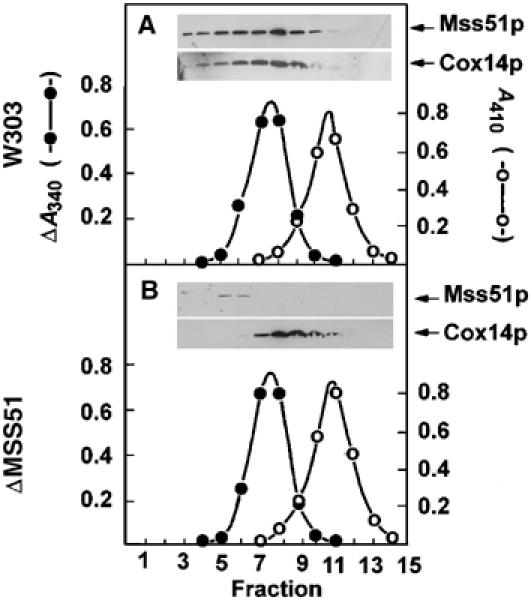
Sedimentation of Mss51p and Cox14p in sucrose gradients. (A) Mitochondria of the wild-type strain W303-1A were extracted at a protein concentration of 10 mg/ml with 1% lauryl maltoside, 20 mM Tris–HCl (pH 7.5), and 0.5 M KCl. The extract (0.4 ml) was mixed with 2.5 mg of hemoglobin and 60 μg lactate dehydrogenase and applied to 4.6 ml of a linear 7–25% sucrose gradient containing 10 mM Tris–HCl and 0.1% Triton X-100. Following centrifugation at 65 000 rpm in a Beckman SW65Ti rotor for 6 h, 14.5 fractions were collected, separated on a 12% polyacrylamide gel, transferred to nitrocellulose and probed with rabbit antiserum against Mss51p or Cox14p followed by a secondary goat peroxidase-conjugated antibody against rabbit IgG. Antibody–antigen complexes were visualized with the Super Signal reagent (Pierce Chemical Co., Rockford, IL). Hemoglobin (○- - -○) was estimated from absorbance at 410 nm and lactate dehydrogenase (⧫- -⧫) was assayed by measuring oxidation of NADH at 340 nm with pyruvate as the substrate. (B) Same as (A) except that the mitochondria were isolated from the mss51 null mutant aW303ΔMSS51 and the gradient was collected in 15 fractions.
Unlike Cox14p and Mss51p, Shy1p was not adsorbed onto the glutathione beads from mitochondrial extracts containing either Mss51p or Cox14p fused to GST (data not shown).
COX14p is a mitochondrial inner membrane protein facing the matrix
Cox14p and Mss51p were previously shown to be associated with the inner membrane of mitochondria (Glerum et al, 1995; Siep et al, 2000). To see if the localization of the two proteins is consistent with their proposed functions, we have determined their topology and solubility properties. Sonic irradiation of wild-type mitochondria solubilized cytochrome b2, a soluble protein of the intermembrane space, but not Cox14p or Mss51p (Figure 9A). Cox14p and Mss51p, however, were solubilized with alkaline carbonate, suggesting that they are peripheral proteins (Figure 9A). In this experiment, Shy1p was recovered in the membrane fraction, confirming earlier evidence that it is an intrinsic protein of the inner membrane (Barrientos et al, 2002b).
Figure 9.
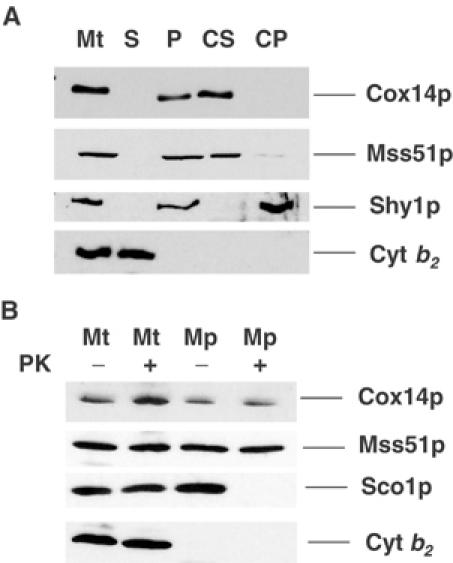
Localization and topology of Cox14p and Mss51p. (A) Mitochondria and the post-mitochondrial supernatant fractions were prepared from the wild-type strain W303-1A. A sample of mitochondria at 4 mg/ml was sonically irradiated and centrifuged at 50 000 gav for 30 min. The membrane pellet was suspended in the starting volume of buffer. To 500 μl of the membranes at a protein concentration of 1 mg/ml was added 50 μl of 1 M Na2CO3 (pH 11.3) and 50 mM EDTA. After 30 min on ice, the sample was centrifuged at 100 000 gav for 15 min at 4°C to separate the soluble from the insoluble intrinsic membrane proteins. Equivalent volumes of mitochondria (Mt), membranes (P), the supernatant obtained after centrifugation of the sonicated mitochondria (S), the carbonate supernatant (CS), and pellet (CP) were separated on a 12% polyacrylamide gel, transferred to nitrocellulose, and treated with antiserum against Cox14p as in Figure 8. Antibodies against Mss51p, Shy1p, and cytochrome b2 (Cyt b2) were used to monitor the conversion of mitochondria to mitoplasts and the intactness of the latter. (B) Mitochondria from W303-1A (Glick and Pon, 1995) at a protein concentration of 8 mg/ml in 0.6 M sorbitol and 20 mM Hepes (pH 7.5) (SH) were converted to mitoplasts (Mp) by dilution with eight volumes of 20 mM Hepes (pH 7.5). For controls, mitochondria (Mt) were diluted with eight volumes of SH. Proteinase K (prot K) was added to one-half of each sample at a final concentration of 100 μg/ml. After 60 min on ice, the reaction was stopped by addition of PMSF to a final concentration of 2 mM and the mitochondria and mitoplasts were recovered by centrifugation at 100 000 gav. The pellets were suspended in SH, and proteins were precipitated by addition of 0.1 volume of 50% trichloroacetic acid and heated for 10 min at 65°C. Mitochondrial and mitoplast proteins were separated by SDS–PAGE on a 12% polyacrylamide gel, transferred to nitrocellulose, and probed with antibody against Cox14p, Sco1p, Mss51p, and cytochrome b2. Antibody–antigen complexes were visualized as in (A).
Both Cox14p and Mss51p are located on the matrix side of the inner membrane as evidenced by their resistance to proteinase K in mitochondria and in mitoplasts (Figure 9B). Sco1p, an inner membrane protein previously shown to face the intermembrane space (Glerum et al, 1996b), was digested in the mitoplasts but not in mitochondria (Figure 9B). Disruption of the outer membrane under the hypotonic conditions used to obtain mitoplasts was confirmed by the loss of cytochrome b2, a soluble protein marker of the intermembrane space.
Discussion
Mitochondrial translation of Cox1p, the heme-bearing subunit of COX, was previously proposed to be negatively regulated by means of its interaction with the Cox1p-specific translation factor Mss51p (Barrientos et al, 2002b). This is supported by the recent demonstration of an interaction between Mss51p and newly synthesized Cox1p (Perez-Martinez et al, 2003). In this study, we attempted to learn more about the relationship of COX assembly to Mss51-dependent synthesis of Cox1p by analyzing a wide range of different COX-deficient mutants.
In vivo assays of mitochondrial translation revealed that, with a few exceptions, labeling of Cox1p but not of the other mitochondrial gene products is 10 times less efficient in COX mutants than in wild type. Decreased labeling of Cox1p relative to other mitochondrial translation products has also been observed in pet111 mutants that have a lesion in a translational activator of COX2 mRNA (Poutre and Fox, 1987) and in cox7 mutants, lacking the nuclear-encoded subunit 7 (Calder and McEwen, 1991). This phenotype cannot be accounted for by a defect in the translation system since the mutations are not in proteins (except those in mss51, pet309) related to this mitochondrial activity. Several lines of evidence also argue against a rapid turnover of Cox1p (except in the oxa1 mutant) as an explanation for the phenotype. The decreased labeling of Cox1p is manifest even with very short pulse times. Additionally, turnover seems unlikely since cox14 mutants, which are also compromised in COX assembly, do not display the Cox1p labeling defect. A more reasonable explanation is that expression of Cox1p is downregulated in most mutants at the translational level.
The ability of the cox14 null mutation to suppress the Cox1p translational defect in all the mutants except the mss51, pet309, and oxa1 mutants suggested that Cox14p either represses translation of the COX1 mRNA or decreases the effective pool of translational activators of Cox1p such as Mss51p or Pet309p. The failure of a bypass ρ− genome in which COX1 is fused to the 5′-UTR of the cytochrome b gene (Manthey and McEwen, 1995) to restore respiration in the cox14 mutant makes it unlikely that Cox14p acts as a translational repressor. Furthermore, overexpression of Cox14p does not diminish Cox1p synthesis.
A more plausible explanation for the effect of the cox14 mutation on Cox1p expression is that Cox14p prevents Mss51p and/or Pet309p from promoting translation of the COX1 mRNA in mutants unable to complete assembly of functional COX. An essential feature of the mechanism proposed here (Figure 10) is that the entry of newly translated Cox1p into the COX assembly pathway depends on the interaction of Cox1p with Cox14p and Mss51p and that when bound to Cox1p and Cox14p, Mss51p is forestalled from acting as a translational activator. Recently, Perez-Martinez et al (2003) provided evidence that in addition to enhancing initiation of translation of COXI mRNA by interacting with the 5′-UTR, Mss51p also acts on target(s) within the COX1 coding sequence to promote translation elongation. The effect of Mss51p on elongation could be mediated either by an interaction with the mRNA or the nascent protein (Perez-Martinez et al, 2003). Since translational regulation by nascent chains has been reported previously (reviewed in Tenson and Ehrenberg, 2002), the latter seems to be the more plausible hypothesis. For reasons of simplicity, the scheme in Figure 10 does not distinguish between these two possibilities. Our results indicate that the interaction of Mss51p with Cox1p does not depend on Cox14p. However, the absence of Cox14p may decrease either the stability of Cox1p itself or the Cox1p/Mss51p complex, thereby raising the effective concentration of Mss51p available for translation. This is consistent with the phenotype of cox14 mutants and with their increased turnover of Cox1p.
Figure 10.
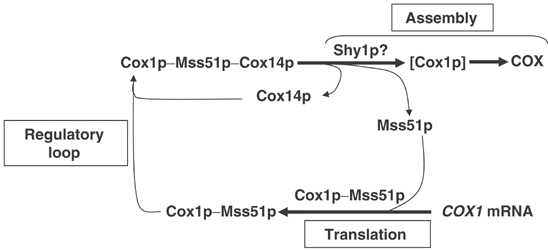
Model depicting Cox1p expression coupled to Shy1p-dependent cytochrome oxidase assembly. Mss51p promotes initiation and elongation of Cox1p translation (see Discussion). Cox14p and Mss51p form a ternary complex with newly synthesized Cox1p. The release of Mss51p from the ternary complex depends on a downstream Cox1p assembly step(s) perhaps involving Shy1p. According to this scheme, mutations blocking assembly trap Mss51p in the ternary complex, thereby limiting its availability for translation. In the cox14 mutant, Mss51p is still able to complex with Cox1p. The resultant binary complex is not assembly-competent causing Cox1p to be diverted to degradation with a concomitant release of Mss51p for additional rounds of Cox1p synthesis.
According to the model proposed here, the dissociation of Mss51p from Cox1p occurs during assembly and could be catalyzed by Shy1p or other COX assembly factors. The enhancement of Cox1p translation in the presence of the mss51 suppressor or an extra copy of wild-type MSS51 may circumvent the Shy1p requirement and account for the partial rescue of shy1 mutants (Barrientos et al, 2002b). The failure of the cox14 null mutant to form COX and to suppress shy1 mutants, despite normal synthesis of Cox1p, indicates that the interaction of Cox14p with Cox1p is essential for a later step in the assembly pathway.
The coupling of Cox1p synthesis to COX assembly, implicit in this regulatory scheme, is similar to the ‘control by epistasy of synthesis' (CES) mechanism that has been proposed to regulate biogenesis of the photosystem complex in chloroplasts of Chlamydomonas reinhardtii (Wollman et al, 1999), and to act in translational regulation of flagellar assembly in bacteria (Aldridge and Hughes, 2002). In these systems, translation of certain subunits is contingent on the availability of their assembly partners, thereby acting as a negative feedback loop that paces translation of a subunit to its utilization during assembly of the complex (Choquet and Vallon, 2000). In the case of membrane complexes, this form of negative regulation may prevent nonproductive aggregation of unassembled hydrophobic membrane proteins by restricting their steady-state concentration.
The involvement of Mss51p and Cox14p in the regulation of Cox1p expression is supported not only by the results of the in vivo mitochondrial translation assays discussed above but also by the interactions of Mss51p and Cox14p with each other and with Cox1p. Reciprocal GST pull-down experiments indicate a transient (or weak) interaction (direct or indirect) of Cox14p with Mss51p. In these experiments, approximately 50% of the untagged Cox14p or Mss51p was co-adsorbed onto the glutathione–sepharose beads. Similar experiments demonstrated that nearly all of newly synthesized Cox1p is complexed to Cox14p but not to Shy1p. An interaction of Cox1p with wild-type Cox14p was also detected by co-immunoprecipitation with a Cox14p-specific antibody. The pull-down assays have also confirmed a complex of newly synthesized Cox1p with Mss51p as reported by Perez-Martinez et al (2003). Finally, Cox14p and Mss51p are extrinsic proteins facing the matrix side of the inner mitochondrial membrane where synthesis of Cox1p is presumed to occur. This localization is consistent with the role of Mss51p as a translational activator of COX1 mRNA (Decoster et al, 1990; Siep et al, 2000) and with the secondary role of Mss51p and Cox14p as sensors of unassembled Cox1p.
Since COX subunits translated on mitochondrial ribosomes in cycloheximide-poisoned cells do not assemble into a native size complex (unpublished), the stability of Cox1p, Cox2p, and Cox3p in wild type under these conditions was unexpected. In contrast to wild type, newly synthesized Cox2p and Cox3p in the mutants were unstable and degraded during the chase. This was not true of Cox1p, which, despite its low initial concentration, was not reduced further, even after a long period of chase. It may be significant that the three COX subunits reached an approximately stoichiometric ratio following 2 h of chase. These observations suggest that in the wild type, the mitochondrial translation products form a protease-resistant intermediate or are sequestered in a protease-protected compartment of the membrane. The stability of Cox1p in the mutants indicates that the small amount of the subunit synthesized in these strains, as well as a similar fraction of Cox2p and Cox3p, is able to attain a protected state similar to that seen in wild type. Most of Cox2p and Cox3p in the mutants, however, are susceptible to proteolytic degradation. This also applies to Cox1p in the single and double cox14 mutants. The extra Cox1p made in these strains, as a result of impaired translational regulation, is prevented from being converted to a protease-resistant form because of its failure to be integrated into an assembly intermediate or incorporated into a protected compartment.
At present, it is not clear if translation of Cox1p in other organisms is also subject to regulation by downstream events. Of the more than dozen yeast nuclear genes governing different post-translational events in COX assembly, half are currently known to have human homologues (Barrientos et al, 2002a). Shy1p is a conserved protein found in all eukaryotes and some prokaryotes (Poyau et al, 1999). Homologues of Cox14p, however, have only been found in Kluyveromyces lactis (Fiori et al, 2000), and of Mss51p in Schizosaccharomyces pombe and in Neurospora crassa. This does not necessarily exclude the possibility that they may exist in mammalian and other genomes but have not yet been recognized because of their smaller size and/or divergent sequences. For example, the products of yeast PET309 and human LRPPRC (LRP130), responsible for the Canadian form of Leigh's syndrome (Mootha et al, 2003), display very weak sequence similarity, even though they both bind to mitochondrial RNAs and are essential for COX expression. It is also possible that translational activators such as Pet309p and Mss51p have more than one function (i.e. mRNA metabolism, membrane insertion of the newly synthesized protein), only some of which are conserved among different organisms. Understanding the nature and players involved in the mechanism regulating COX assembly in yeast will help to identify their functional homologues in humans.
Materials and methods
Strains and media
The genotypes and sources of the Saccharomyces cerevisiae strains carrying null alleles of COX-related genes are listed in Table I.
Table 1.
Genotypes and sources of yeast strains carrying null alleles of COX-related genes
| Strain | Genotype | Source |
|---|---|---|
| Structural subunits | ||
| W303ΔCOX4 | ade2-1 his3-1,15 leu2-3,112 trp1-1 ura3-1 Δcox4∷URA3 | Glerum and Tzagoloff (1997) |
| W303ΔCOX5a | ade2-1 his3-1,15 leu2-3,112 trp1-1 ura3-1 Δcox5a∷HIS3 | Glerum and Tzagoloff (1997) |
| W303ΔCOX6 | ade2-1 his3-1,15 leu2-3,112 trp1-1 ura3-1 Δcox6∷URA3 | Glerum and Tzagoloff (1997) |
| W303ΔCOX7 | ade2-1 his3-1,15 leu2-3,112 trp1-1 ura3-1 Δcox7∷URA3 | This study |
| W303ΔCOX9 | ade2-1 his3-1,15 leu2-3,112 trp1-1 ura3-1 Δcox9∷URA3 | This study |
| M10-150 | cox1 | Tzagoloff et al (1975) |
| M9-94 | cox2 | Tzagoloff et al (1975) |
| M9-3 | cox3 | Tzagoloff et al (1975) |
| Cytochrome c maturation | ||
| W303ΔCYC3 | ade2-1 his3-1,15 leu2-3,112 trp1-1 ura3-1 Δcyc3∷URA3 | This study |
| COX1 expression | ||
| W303ΔPET309 | ade2-1 his3-1,15 leu2-3,112 trp1-1 ura3-1 Δpet309∷HIS3 | Glerum and Tzagoloff (1997) |
| W303ΔMSS51 | ade2-1 his3-1,15 leu2-3,112 trp1-1 ura3-1 Δmss51∷HIS3 | Barrientos et al (2002b) |
| Maturation of CuA or CuB centers | ||
| W303ΔCOX17 | ade2-1 his3-1,15 leu2-3,112 trp1-1 ura3-1 Δcox17∷TRP1 | Glerum et al (1996a) |
| W303ΔSCO1 | ade2-1 his3-1,15 leu2-3,112 trp1-1 ura3-1 Δsco1∷URA3 | Glerum et al (1996b) |
| W303ΔCOX11 | ade2-1 his3-1,15 leu2-3,112 trp1-1 ura3-1 Δcox11∷HIS3 | Tzagoloff et al (1990) |
| COX2 expression | ||
| W303ΔOXA1 | ade2-1 his3-1,15 leu2-3,112 trp1-1 ura3-1 Δoxa1∷HIS3 | Hell et al (2000) |
| W303ΔCOX18 | ade2-1 his3-1,15 leu2-3,112 trp1-1 ura3-1 Δcox18∷URA3 | Souza et al (2000) |
| W303ΔPET111 | ade2-1 his3-1,15 leu2-3,112 trp1-1 ura3-1 Δpet111∷HIS3 | Barros and Tzagoloff (2002) |
| W303ΔIMP1 | ade2-1 his3-1,15 leu2-3,112 trp1-1 ura3-1 Δimp1∷HIS3 | This study |
| W303ΔIMP2 | ade2-1 his3-1,15 leu2-3,112 trp1-1 ura3-1 Δimp2∷URA3 | Barros et al (2001) |
| Heme biosynthesis | ||
| W303ΔCOX10 | ade2-1 his3-1,15 leu2-3,112 trp1-1 ura3-1 Δcox10∷HIS3 | Nobrega et al (1990) |
| W303ΔCOX15 | ade2-1 his3-1,15 leu2-3,112 trp1-1 ura3-1 Δcox15∷HIS3 | Glerum et al (1997) |
| Assembly/unknown | ||
| W303ΔPET117 | ade2-1 his3-1,15 leu2-3,112 trp1-1 ura3-1 Δpet117∷HIS3 | Barros and Tzagoloff (2002) |
| C8/L1 | leu2 pet117 | Tzagoloff and Dieckmann (1990) |
| W303ΔSHY1/U2 | ade2-1 his3-1,15 leu2-3,112 trp1-1 ura3-1 Δshy1∷URA3 | Barrientos et al (2002b) |
| W303ΔPET191 | ade2-1 his3-1,15 leu2-3,112 trp1-1 ura3-1 Δpet191∷HIS3 | This study |
| W303ΔCOX14 | ade2-1 his3-1,15 leu2-3,112 trp1-1 ura3-1 Δcox14∷TRP1 | This study |
| W303ΔCOX16 | ade2-1 his3-1,15 leu2-3,112 trp1-1 ura3-1 Δcox14∷URA3 | Carlson et al (2003) |
| W303ΔCOX19 |
ade2-1 his3-1,15 leu2-3,112 trp1-1 ura3-1 Δcox19∷URA3 |
Nobrega et al (2002) |
| The part headings indicate the functional category of the deleted gene products. | ||
| All null mutations have been created or are available in both, a and α, mating types. | ||
In vivo and in organello mitochondrial protein synthesis
Mitochondrial gene products were labeled with [35S]methionine in whole cells at 30°C in the presence of cycloheximide (Barrientos et al, 2002b). For in organelle translation, mitochondria were prepared by the method of Herrmann et al (1994) and labeled with [35S]methionine as described (Hell et al, 2000). Equivalent amounts of total cellular or mitochondrial proteins were separated by SDS–PAGE on a 17.5% polyacrylamide gel, transferred to a nitrocellulose membrane, and exposed to an X-ray film.
Preparation of antibodies to Cox14p
Antibodies were obtained against Cox14p expressed from a trpE fusion gene. The full coding region of the COX14 gene was PCR amplified with the primers 5′-GGCGGATCCAT-GTCCAAATACGCTTGG and 5′-GGCAAGCTTGGAACCAGC-ACACTACGT. The amplified BamHI–HindIII fragment was fused in-frame to the amino-terminal half of trpE in pATH21. Escherichia coli transformed with this plasmid expresses a fusion protein of about 45 kDa, constituting most of the insoluble proteins of the cells. This fraction was dissolved in 2% SDS, 10 mM Tris–HCl (pH 7.5), 1 mM EDTA, 5 mM β-mercaptoethanol, and 20 μg/ml PMSF and was further purified on a Bio-Gel A 0.5 column developed with a buffer containing 0.1% SDS, 10 mM Tris–HCl (pH 7.5), and 0.1 M NaCl. Fractions enriched for the fusion protein were pooled, and precipitated with acetone precipitation. The precipitated protein was dissolved in 0.2% SDS and used to raise antibodies in rabbits.
Construction of GST chimeras
The construction of plasmids containing chimeric SHY1-GST, MSS51-GST, and COX14-GST genes was performed in two steps. First, the GST gene was amplified by PCR and, second, cloned in-frame to the corresponding genes into plasmids already containing them.
The GST gene plus a thrombin cleavage site at its N-terminus was amplified from pGEX-3X (Amersham Biosciences Corp., Piscataway, NJ) with primers 5′-GGCTGCAGCTGGTTCCGCGTGGATCCGGAGGAATG TCCCCTATACTAGGT and 5′-CCGGGAGCTCGGATCCACGCGGAACCAGATCC or with primers 5′-GGGGTACCTACTGGTTCCGCGTGGATCCGGAGGAA TGTCCCCTATACTAGGT and 5′-CCGGGAG-CTCGGATCCACGCGGAACCAGATCC. The ∼600-bp products were digested with PstI/SacI and KpnI/SacI respectively and kept until used for further cloning.
To prepare a SHY1-GST chimeric gene with a thrombin cleavage site, a portion of the SHY1 gene, starting at the BamHI site, was amplified with primers 5′-GGCGAGCTCCTGCAGATATTTCCTTGAATGCTTC and 5′-TGGGCGAAAA-AGGATCCAAATTC using the plasmid pG91/T1 (Mashkevich et al, 1997). The amplicon was cloned into the integrative plasmid pG91/ST17 (Mashkevich et al, 1997) from which the BamHI/SacI had been removed to yield the plasmid pG91/ST60. The previously amplified GST gene was cloned into this plasmid as PstI–SacI, in-frame with the 3′ end of SHY1 to obtain pG91/ST62.
To construct the COX14-GST chimeric gene with a thrombin cleavage site, the COX14 gene was amplified with the primers 5′-GGCGGAATTCAACTAATGATTGG and 5′-GGCAAGCTTGAGCTCCTGCAGCTCGGTAGGAGGAG GTGCAG using the plasmid pG93/T1 (Glerum et al, 1995) as the template, and cloned as an EcoRI/HindIII fragment into YIp352. The previously amplified GST gene was cloned into this plasmid as PstI–SacI, in-frame with the 3′ end of COX14 to obtain pG93/ST32.
The MSS51-GST chimeric gene was constructed by first cloning MSS51 as a KpnI/SphI fragment in YIp352. The sequence coding for GST was then cloned into this plasmid as a PstI–SacI fragment, in-frame with the 3′ end of MSS51 to obtain pG96/ST13.
Miscellaneous procedures
Standard procedures were used for the preparation and ligation of DNA fragments, and for transformation and recovery of plasmid DNA from E. coli (Maniatis et al, 1982). Yeast were transformed by the method of Schiestl and Gietz (1989). The one-step gene insertion method (Rothstein, 1983) was used to integrate linear plasmids at the URA3 or LEU2 locus of yeast chromosomal DNA.
Acknowledgments
This research was supported by an NIH research grant GM50187 (to AT) and a research grant from the MDA (to AB).
References
- Aldridge P, Hughes KT (2002) Regulation of flagellar assembly. Curr Opin Microbiol 5: 160–165 [DOI] [PubMed] [Google Scholar]
- Barrientos A, Barros MH, Valnot I, Rotig A, Rustin P, Tzagoloff A (2002a) Cytochrome oxidase in health and disease. Gene 286: 53–63 [DOI] [PubMed] [Google Scholar]
- Barrientos A, Korr D, Tzagoloff A (2002b) Shy1p is necessary for full expression of mitochondrial COX1 in the yeast model of Leigh's syndrome. EMBO J 21: 43–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barros MH, Carlson CG, Glerum DM, Tzagoloff A (2001) Involvement of mitochondrial ferredoxin and Cox15p in hydroxylation of heme O. FEBS Lett 492: 133–138 [DOI] [PubMed] [Google Scholar]
- Barros MH, Tzagoloff A (2002) Regulation of the heme A biosynthetic pathway in Saccharomyces cerevisiae. FEBS Lett 516: 119–123 [DOI] [PubMed] [Google Scholar]
- Calder KM, McEwen JE (1991) Deletion of the COX7 gene in Saccharomyces cerevisiae reveals a role for cytochrome c oxidase subunit VII in assembly of remaining subunits. Mol Microbiol 5: 1769–1777 [DOI] [PubMed] [Google Scholar]
- Carlson CG, Barrientos A, Tzagoloff A, Glerum DM (2003) COX16 encodes a novel protein required for the assembly of cytochrome oxidase in Saccharomyces cerevisiae. J Biol Chem 278: 3770–3775 [DOI] [PubMed] [Google Scholar]
- Choquet Y, Vallon O (2000) Synthesis, assembly and degradation of thylakoid membrane proteins. Biochimie 82: 615–634 [DOI] [PubMed] [Google Scholar]
- Conde J, Fink GR (1976) A mutant of Saccharomyces cerevisiae defective for nuclear fusion. Proc Natl Acad Sci USA 73: 3651–3655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo MC, Fox TD (1993) Suppression of a defect in the 5′ untranslated leader of mitochondrial COX3 mRNA by a mutation affecting an mRNA-specific translational activator protein. Mol Cell Biol 13: 4806–4813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decoster E, Simon M, Hatat D, Faye G (1990) The MSS51 gene product is required for the translation of the COX1 mRNA in yeast mitochondria. Mol Gen Genet 224: 111–118 [DOI] [PubMed] [Google Scholar]
- DiMauro S, De Vivo DC (1996) Genetic heterogeneity in Leigh syndrome. Ann Neurol 40: 5–7 [DOI] [PubMed] [Google Scholar]
- Fiori A, Saliola M, Goffrini P, Falcone C (2000) Isolation and molecular characterization of KlCOX14, a gene of Kluyveromyces lactis encoding a protein necessary for the assembly of the cytochrome oxidase complex. Yeast 16: 307–314 [DOI] [PubMed] [Google Scholar]
- Glerum DM, Koerner TJ, Tzagoloff A (1995) Cloning and characterization of COX14, whose product is required for assembly of yeast cytochrome oxidase. J Biol Chem 270: 15585–15590 [DOI] [PubMed] [Google Scholar]
- Glerum DM, Muroff I, Jin C, Tzagoloff A (1997) COX15 codes for a mitochondrial protein essential for the assembly of yeast cytochrome oxidase. J Biol Chem 272: 19088–19094 [DOI] [PubMed] [Google Scholar]
- Glerum DM, Shtanko A, Tzagoloff A (1996a) Characterization of COX17, a yeast gene involved in copper metabolism and assembly of cytochrome oxidase. J Biol Chem 271: 14504–14509 [DOI] [PubMed] [Google Scholar]
- Glerum DM, Shtanko A, Tzagoloff A (1996b) SCO1 and SCO2 act as high copy suppressors of a mitochondrial copper recruitment defect in Saccharomyces cerevisiae. J Biol Chem 271: 20531–20535 [DOI] [PubMed] [Google Scholar]
- Glerum DM, Tzagoloff A (1997) Submitochondrial distributions and stabilities of subunits 4, 5, and 6 of yeast cytochrome oxidase in assembly defective mutants. FEBS Lett 412: 410–414 [DOI] [PubMed] [Google Scholar]
- Glick BS, Pon LA (1995) Isolation of highly purified mitochondria from Saccharomyces cerevisiae. Methods Enzymol 260: 213–223 [DOI] [PubMed] [Google Scholar]
- Hell K, Neupert W, Stuart RA (2001) Oxa1p acts as a general membrane insertion machinery for proteins encoded by mitochondrial DNA. EMBO J 20: 1281–1288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hell K, Tzagoloff A, Neupert W, Stuart RA (2000) Identification of Cox20p, a novel protein involved in the maturation and assembly of cytochrome oxidase subunit 2. J Biol Chem 275: 4571–4578 [DOI] [PubMed] [Google Scholar]
- Herrmann JM, Foelsch H, Neupert W, Stuart RA (1994) Isolation of yeast mitochondria and study of mitochondrial protein translation. In Cell Biology: A Laboratory Handbook, Celis JE (ed) Vol. I, pp 538–544. San Diego, CA: Academic Press [Google Scholar]
- Maniatis T, Fritsch EF, Sambrook J (1982) Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press [Google Scholar]
- Manthey GM, McEwen JE (1995) The product of the nuclear gene PET309 is required for translation of mature mRNA and stability or production of intron-containing RNAs derived from the mitochondrial COX1 locus of Saccharomyces cerevisiae. EMBO J 14: 4031–4043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mashkevich G, Repetto B, Glerum DM, Jin C, Tzagoloff A (1997) SHY1, the yeast homolog of the mammalian SURF-1 gene, encodes a mitochondrial protein required for respiration. J Biol Chem 272: 14356–14364 [DOI] [PubMed] [Google Scholar]
- McEwen JE, Ko C, Kloeckner-Gruissem B, Poyton RO (1986) Nuclear functions required for cytochrome c oxidase biogenesis in Saccharomyces cerevisiae. Characterization of mutants in 34 complementation groups. J Biol Chem 261: 11872–11879 [PubMed] [Google Scholar]
- Mootha VK, Lepage P, Miller K, Bunkenborg J, Reich M, Hjerrild M, Delmonte T, Villeneuve A, Sladek R, Xu F, Mitchell GA, Morin C, Mann M, Hudson TJ, Robinson B, Rioux JD, Lander ES (2003) Identification of a gene causing human cytochrome c oxidase deficiency by integrative genomics. Proc Natl Acad Sci USA 100: 605–610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobrega MP, Bandeira SC, Beers J, Tzagoloff A (2002) Characterization of COX19, a widely distributed gene required for expression of mitochondrial cytochrome oxidase. J Biol Chem 277: 40206–40211 [DOI] [PubMed] [Google Scholar]
- Nobrega MP, Nobrega FG, Tzagoloff A (1990) COX10 codes for a protein homologous to the ORF1 product of Paracoccus denitrificans and is required for the synthesis of yeast cytochrome oxidase. J Biol Chem 265: 14220–14226 [PubMed] [Google Scholar]
- Perez-Martinez X, Broadley SA, Fox TD (2003) Mss51p promotes mitochondrial Cox1p synthesis and interacts with newly synthesized Cox1p. EMBO J 22: 5951–5961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poutre CG, Fox TD (1987) PET111, a Saccharomyces cerevisiae nuclear gene required for translation of the mitochondrial mRNA encoding cytochrome c oxidase subunit II. Genetics 115: 637–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poyau A, Buchet K, Godinot C (1999) Sequence conservation from human to prokaryotes of Surf1, a protein involved in cytochrome c oxidase assembly, deficient in Leigh syndrome. FEBS Lett 462: 416–420 [DOI] [PubMed] [Google Scholar]
- Rothstein RJ (1983) One-step gene disruption in yeast. Methods Enzymol 101: 202–211 [DOI] [PubMed] [Google Scholar]
- Seraphin B, Simon M, Boulet A, Faye G (1989) Mitochondrial splicing requires a protein from a novel helicase family. Nature 337: 84–87 [DOI] [PubMed] [Google Scholar]
- Schiestl RH, Gietz RD (1989) High efficiency transformation of intact yeast cells using single stranded nucleic acids as a carrier. Curr Genet 16: 339–346 [DOI] [PubMed] [Google Scholar]
- Siep M, van Oosterum K, Neufeglise H, van der Spek H, Grivell LA (2000) Mss51p, a putative translational activator of cytochrome c oxidase subunit-1 (COX1) mRNA, is required for synthesis of Cox1p in Saccharomyces cerevisiae. Curr Genet 37: 213–220 [DOI] [PubMed] [Google Scholar]
- Souza RL, Green-Willms NS, Fox TD, Tzagoloff A, Nobrega FG (2000) Cloning and characterization of COX18, a Saccharomyces cerevisiae PET gene required for the assembly of cytochrome oxidase. J Biol Chem 275: 14898–14902 [DOI] [PubMed] [Google Scholar]
- Tenson T, Ehrenberg M (2002) Regulatory nascent peptides in the ribosomal tunnel. Cell 108: 591–594 [DOI] [PubMed] [Google Scholar]
- Tiranti V, Hoertnagel K, Carrozzo R, Galimberti C, Munaro M, Granatiero M, Zelante L, Gasparini P, Marzella R, Rocchi M, Bayona-Bafaluy MP, Enriquez J A, Uziel G, Bertini E, Dionisi-Vici C, Franco B, Meitinger T, Zeviani M (1998) Mutations of SURF-1 in Leigh disease associated with cytochrome c oxidase. Am J Hum Genet 63: 1609–1621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzagoloff A, Akai A, Needleman RB, Zulch G (1975) Cytoplasmic mutants of S. cerevisiae with deficiencies in respiratory activities and in the ATPase. J Biol Chem 250: 8236–8242 [PubMed] [Google Scholar]
- Tzagoloff A, Capitanio N, Nobrega MP, Gatti D (1990) Cytochrome oxidase assembly in yeast requires the product of COX11, a homolog of the P. denitrificans protein encoded by ORF3. EMBO J 9: 2759–2764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzagoloff A, Dieckmann CL (1990) PET genes of Saccharomyces cerevisiae. Microbiol Rev 54: 211–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wollman FA, Minai L, Nechushtai R (1999) The biogenesis and assembly of photosynthetic proteins in thylakoid membranes. Biochim Biophys Acta 1411: 21–85 [DOI] [PubMed] [Google Scholar]
- Zhu Z, Yao J, Johns T, Fu K, De Bie I, Macmillan C, Cuthbert AP, Newbold RF, Wang J, Chevrette M, Brown GK, Brown RM, Shoubridge EA (1998) SURF1, encoding a factor involved in the biogenesis of cytochrome c oxidase, is mutated in Leigh syndrome. Nat Genet 20: 337–343 [DOI] [PubMed] [Google Scholar]