

Abstract
Enteropathogenic Escherichia coli delivers a subset of effectors into host cells via a type III secretion system, and this step is required for the progression of disease. Here, we show that the type III effectors, EspG and its homolog Orf3, trigger actin stress fiber formation and the destruction of the microtubule networks beneath adherent bacteria. Both effectors were shown to possess the ability to interact with tubulins, and to stimulate microtubule destabilization in vitro. A recent study showed that microtubule-bound GEF-H1, a RhoA-specific guanine nucleotide exchange factor, was converted to its active form by microtubule destabilization, and this sequence of events resulted in RhoA stimulation. Indeed, EspG- and Orf3-induced stress fiber formation was inhibited by the expression of dominant-negative forms of GEF-H1 and RhoA, but not of Rac1 and Cdc42, and by treatment with a ROCK inhibitor. These results indicate that the impact of EspG/Orf3 on microtubule networks triggers the activation of the RhoA–ROCK signaling pathway via GEF-H1 activity. This report reveals for the first time that a pathogen can exploit the host factor GEF-H1.
Keywords: enteropathogenic E. coli, EspG, GEF-H1, RhoA, type III secretion
Introduction
Enteropathogenic Escherichia coli (EPEC) remains a major cause of infantile diarrhea worldwide, mostly in the developing world (Donnenberg and Kaper, 1992). Related pathogens, which cause disease by similar mechanisms (DeVinney et al, 1999), include enterohemorrhagic E. coli O157:H7, rabbit EPEC, and Citrobacter rodentium, which is found in mice. These pathogens induce a characteristic histopathological lesion, referred to as an attaching/effacing (A/E) lesion (Moon et al, 1983), which is defined by the intimate attachment of bacteria to the intestinal epithelial surface and the effacement of host cell microvilli, which is involved in the development of diarrhea (Abe et al, 1998). Factors responsible for A/E lesion formation are encoded by a 35-kilobase pair region referred to as the locus of enterocyte effacement (LEE) (McDaniel et al, 1995), which includes the type III secretion system, the translocated intimin receptor (Tir) (Kenny et al, 1997), intimin (Donnenberg et al, 1993b), and EPEC-secreted proteins (Esps). We demonstrated the supermolecular structure of the type III secretion system (Sekiya et al, 2001), which includes an EscF-needle portion and an EspA-sheath-like structure at the tip of the needle. EscN, a cytoplasmic ATPase, is essential for the secretion of effectors (Jarvis et al, 1995), and the presumed pore-forming factors EspB and EspD are delivered to the host plasma membrane, and these factors are a prerequisite for the translocation of effectors into host cells (Ide et al, 2001). To date, five effectors, which are translocated into host cells via the type III secretion system, have been identified and characterized in EPEC; these effectors include Tir (Kenny et al, 1997), EspF (McNamara et al, 2001), EspG (Elliott et al, 2001), EspH (Tu et al, 2003), and Map (Kenny et al, 2002). In addition, the EspG homolog Orf3, which is encoded on the EspC pathogenicity islet within the EPEC genome (distinct from the LEE), is thought to be an effector (Elliott et al, 2001). Both EspG and Orf3 effectors share homology with the Shigella VirA effector. Although it has been demonstrated that Shigella VirA promotes the destabilization of host microtubules and Rac1 activation (Yoshida et al, 2002), the contributions of EspG/Orf3 effectors to EPEC virulence remain unclear. Map induces the transient formation of Cdc42-dependent filopodia, while EspH represses filopodium formation and enhances the actin-rich pedestal formation beneath adherent bacteria. A/E lesion formation is mediated by a Tir–intimin interaction (Kenny et al, 1997), which is required for the initiation of the disease process (Abe et al, 1998). However, an in vivo study with adult human volunteers suggested that additional EPEC factor(s) trigger diarrhea, since diarrhea still developed in four of 11 volunteers who ingested an intimin mutant strain that was incapable of A/E lesion formation (Donnenberg et al, 1993a). It has been reported that EPEC-induced diarrhea may be involved in a loss of intestinal barrier function, with ion and fluid movement from the intestinal submucosa into the lumen (Hecht, 2001). When polarized epithelial cells were infected with EPEC, redistribution of the tight junction-associated protein occludin was observed, and this redistribution was correlated with the disruption of the tight junction barrier (Yuhan et al, 1997; Simonovic et al, 2000). Although the precise function of EspF in host cells is not yet clear, EspF is known to be required for the disruption of epithelial cell junctions during EPEC infection, but EspF is not involved in the formation of the A/E lesion (McNamara et al, 2001). Thus, EPEC effectors have multiple functions in host cells, and their synergistic effects are thought to contribute to EPEC-mediated diarrhea.
In this study, we demonstrated that actin stress fiber formation was induced by EPEC infection in Swiss 3T3 cells. This effect was dependent on the type III secretion system, but not on Tir–intimin interactions. It has been reported that the formation of actin stress fibers is regulated by the RhoA–ROCK signaling pathway (Kimura et al, 1996; Leung et al, 1996; Hall, 1998). Although the Tir-induced accumulation of actin filaments has been observed during A/E lesion formation, this phenotype was not found to be affected by the addition of the RhoA inhibitor (Ben-Ami et al, 1998); moreover, this phenotype was required for the activity of N-WASP (Kalman et al, 1999), the Arp2/3 complex, and Nck (Gruenheid et al, 2001), indicating that EPEC may exploit different signal transduction pathways in the formation of A/E lesions and actin stress fibers.
To determine whether or not the type III effectors are involved in actin stress fiber formation, various effector mutants were constructed, and an infection assay was carried out. We found that EspG and Orf3 effectors are involved in the induction of actin stress fiber formation in a Tir–intimin interaction-independent manner. We further demonstrated that both effectors induce the disruption of microtubule networks beneath adherent bacteria by the direct association of EspG and Orf3 with tubulins. One of the aims of the present study was to determine how EspG/Orf3 governs the crosstalk between the actin cytoskeleton and microtubules. We revealed here that a newly identified guanine nucleotide exchange factor, GEF-H1 (Ren et al, 1998; Krendel et al, 2002), is exploited by EspG/Orf3 effectors to induce actin stress fiber formation during EPEC infection. A recent study also showed that endogenous GEF-H1 is localized in the microtubules in its inactive form and it switches to an active form when it is released from microtubules. Indeed, both EspG/Orf3 effectors were revealed to possess the ability to release GEF-H1 from the cytoskeleton to the cytosol as a result of the microtubule destabilization. We report here that the GEF-H1-mediated signaling pathway is triggered by EPEC infection via the activity of EspG/Orf3 effectors.
Results
EPEC infection of mammalian cells induces stress fiber formation
To investigate whether EPEC infection influences the host actin cytoskeleton, serum-starved Swiss 3T3 cells were infected with wild-type (WT) EPEC and the architecture of the actin networks was visualized by F-actin staining with rhodamine-labeled phalloidin. Although EPEC is a human pathogen, we used mouse Swiss 3T3 cells for this experiment, since the dynamics of the actin cytoskeleton are easily observed in these fibroblastic cells. EPEC has the ability to adhere to and form tight microcolonies on tissue culture cells and human intestinal cells (Rosenshine et al, 1992); these activities are mediated by a bundle-forming pilus (Giron et al, 1991). As expected, the accumulation of F-actin beneath microcolonies of adherent bacteria was also observed in the mouse fibroblastic cell line, Swiss 3T3 (Figure 1A, arrows). In addition to this accumulation, actin stress fibers with parallel bundles extending into projections of the cell periphery were prominently induced in the cytoplasm under conditions of EPEC infection (Figure 1A, arrowhead). Although the formation of a pedestal structure, represented by the accumulation of F-actin beneath the adherent bacteria, was not detected in all of the infected cells with the eae mutant (defective in intimin production) or the tir mutant (defective in Tir production), these cells still developed extensive actin stress fibers (Figure 1B and C). It was of note that stress fiber formation, observed here in EPEC WT-infected cells, was not detected with either the espA mutant (defective in the formation of the EspA-sheath in type III machinery) or the escF EPEC mutant (defective in the formation of the EscF-needle in type III machinery) (Figure 1B and C). The same was true for the espB and espD mutants (defective in pore formation on the host plasma membrane) (Figure 1B and C) and the escN mutant (defective in secretion of type III-secreted proteins) (Figure 2A and B). Therefore, the results of the present series of experiments strongly suggested that certain unknown type III-secreted effector(s) are involved in EPEC-induced actin stress fiber formation in infected cells.
Figure 1.
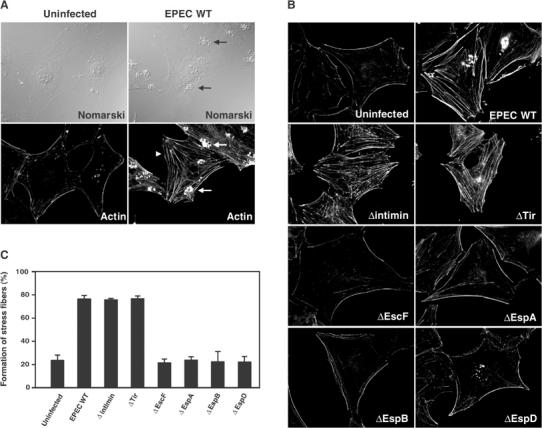
The host cytoskeletal rearrangement as a result of EPEC infection. (A) Swiss 3T3 cells were infected with EPEC WT for 3 h, and cells were fixed and stained with rhodamine-labeled phalloidin. The formation of pedestal structures beneath EPEC microcolonies (arrows) and actin stress fibers (arrowhead) was observed in the cells infected with EPEC. (B) Swiss 3T3 cells were infected with EPEC WT or its mutants for 3 h, and the cells were fixed and visualized as in (A). All fluorescence images were taken with the same exposure time using a CCD camera. (C) The percentage of Swiss 3T3 cells showing marked formation of actin stress fibers is indicated. Swiss 3T3 cells were infected with EPEC WT or its mutants for 3 h, and the cells were fixed and visualized as in (A). Percentages were based on counts of at least 150 cells, and the values are the means±standard deviation from three independent experiments.
Figure 2.
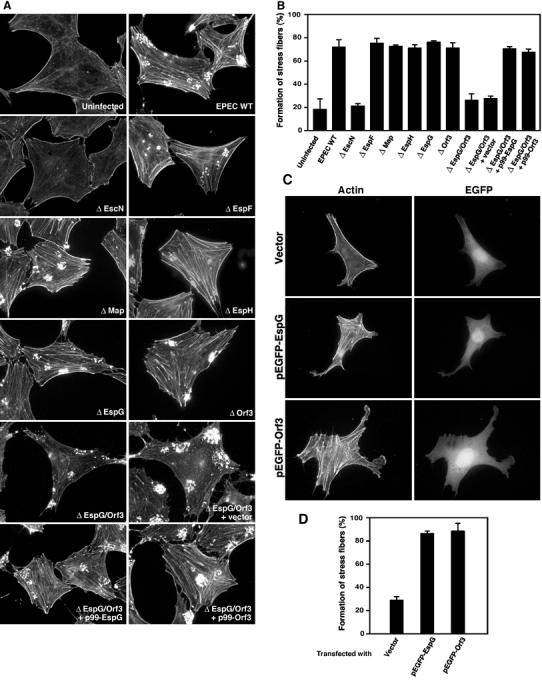
Determination of the EPEC type III effectors required for stress fiber formation. (A) Swiss 3T3 cells were infected with EPEC WT, various type III effector mutants, or an espG/orf3 mutant complemented with cloned espG (p99-EspG), orf3 (p99-Orf3), or vector alone for 3 h. Cells were fixed and stained with rhodamine-labeled phalloidin in order to detect F-actin. All fluorescence images were taken with the same exposure time. (B) The percentage of Swiss 3T3 cells showing marked formation of actin stress fibers is indicated. Swiss 3T3 cells were infected with EPEC WT or its mutants for 3 h, and the cells were fixed and visualized as in (A). Percentages were based on counts of at least 150 cells, and the values are the means±standard deviation from three independent experiments. (C) Swiss 3T3 cells were transfected with the plasmids encoding EGFP-tagged EspG, EGFP-tagged Orf3, and vector alone. Cells were fixed and stained as in (A), and the transfected cells were visualized by EGFP fluorescence. (D) EGFP fluorescence-positive Swiss 3T3 cells were randomly obtained from panel C, and the percentage of cells showing actin stress fiber formation is indicated. Percentages were based on a count of at least 100 cells, and the values are the means±standard deviation from three independent experiments.
EspG/Orf3 is involved in stress fiber formation in host cells infected with EPEC
To identify the secreted bacterial protein(s) involved in the formation of stress fibers in host cells, we constructed a series of nonpolar mutants with a defect in the genes espF, orf19 (defective in Map production), espG, espH, or orf3, and we also considered an espG/orf3 double-knockout mutant (due to the fact that EspG shares 43.5% identity with Orf3; Elliott et al, 2001). Swiss 3T3 cells were infected with each of the mutants in order to investigate their ability to elicit actin stress fiber formation. The results demonstrated that the ability of the espG/orf3 double-knockout mutant to induce stress fiber formation was greatly reduced, in comparison with that of the other type III effector mutants, such as the espF, orf19, espG, espH, and orf3 mutants (Figure 2A and B). Complementation of the espG/orf3 double-knockout mutant with a plasmid encoding EspG (p99-EspG) or Orf3 (p99-Orf3) restored the ability to form actin stress fibers (Figure 2A and B), indicating that both proteins participated in the formation of stress fibers. To confirm this possibility, pEGFP-EspG or pEGFP-Orf3 (enhanced green fluorescent protein (EGFP)-tagged EspG or Orf3, respectively) was transfected into Swiss 3T3 cells and the cells were observed for the formation of stress fibers. The Swiss 3T3 cells overexpressing EGFP-EspG or EGFP-Orf3, but not EGFP alone, were able to induce actin stress fibers (Figure 2C and D), thereby suggesting the following conclusions: (i) EspG and Orf3 are responsible for the formation of stress fibers, and they possess functional redundancy, and (ii) other effectors are not involved in this series of events.
Although EspG is secreted via the type III machinery (Elliott et al, 2001), it remains unclear as to how Orf3 is secreted. To address this issue, the secretion profile of Orf3 was analyzed by immunoblotting using anti-Orf3 antibodies. We detected Orf3 in the culture supernatant prepared from the EPEC WT, but not from the type III machinery mutant, indicating that Orf3 is also secreted via the type III machinery (data not shown).
EspG/Orf3 interaction with tubulins and stimulation of microtubule destruction
EspG and Orf3 shared a striking homology not only with each other but also with the Shigella VirA effector (Elliott et al, 2001). VirA is secreted via the type III machinery, and is required for promoting the uptake of bacteria by epithelial cells. Since VirA has the ability to interact with tubulins and trigger microtubule destabilization, this finally leads to Rac1 activation (Yoshida0 et al, 2002). Interestingly, cloned EPEC espG and orf3 were able to individually rescue the ability of the Shigella virA mutant to invade host cells, implying that both EspG and Orf3 shared functions similar to those of VirA (Elliott et al, 2001). To investigate the effects of both EspG and Orf3 on host microtubule networks, Swiss 3T3 cells were infected for 3 h with EPEC WT or the espG/orf3 double mutant, and the reorganization of both F-actin and the microtubule structures was observed by immunofluorescence microscopy. As shown in Figure 3A, the local microtubule structures beneath the EPEC microcolonies were destabilized by infection with EPEC WT (Figure 3A, arrows), but not with the espG/orf3 mutant. These events were reproducibly detected in EPEC WT-infected HeLa and Cos-7 cells, and also with the introduction of cloned espG or orf3 (pEGFP-EspG, pEGFP-Orf3) into Swiss 3T3 cells (data not shown). In addition, the time course of microtubule destabilization after EPEC WT or espG/orf3 mutant infection was measured, and the percentage of destabilization beneath EPEC microcolonies was calculated (Figure 3B). Destabilization of the local microtubule structures was greatly increased as a result of WT infection. In contrast, no such effect was detected in infection with the espG/orf3 mutant. Like EPEC WT, the espG or orf3 single mutant still possessed the ability to destabilize the microtubule networks, and complementation of the espG/orf3 double-knockout mutant with a cloned espG or orf3 restored the ability to destabilize microtubule networks due to functional redundancy (Figure 3C and D). Collectively, both EPEC effectors, EspG and Orf3, are involved in the induction of actin stress fiber formation and the destabilization of microtubule networks in vivo.
Figure 3.
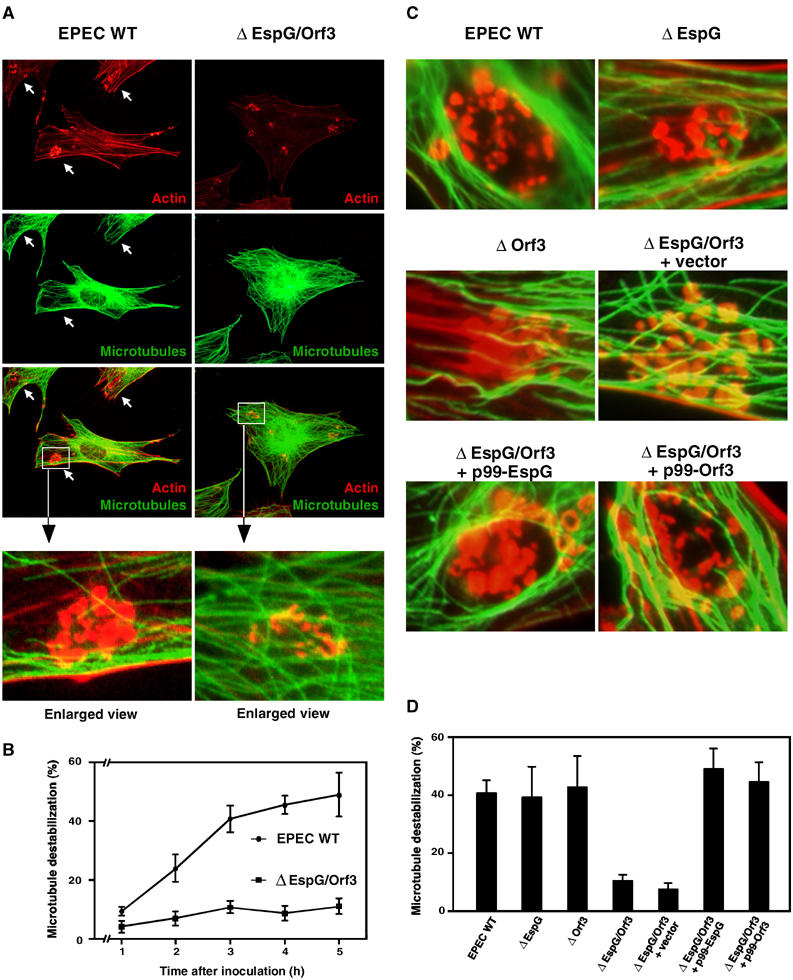
Effect of EPEC infection on microtubule dynamics. (A) Swiss 3T3 cells were infected with EPEC WT and the espG/orf3 mutant for 3 h, and the cells were fixed and stained with rhodamine-labeled phalloidin and fluorescently labeled anti-tubulin antibodies in order to detect the actin cytoskeleton and microtubule networks, respectively. The arrows indicate disruptions of the microtubule network beneath the adherent EPEC microcolonies. (B) Time course of the local destabilization of microtubule networks after EPEC infection. The percentages of microtubule destabilization beneath the EPEC microcolonies were based on a count of at least 150 microcolonies, and the values given are the means±standard deviation from three independent experiments. (C) Swiss 3T3 cells were infected with EPEC WT and its mutants for 3 h and the cells were fixed and visualized as in (A). (D) The percentage of local destabilization of microtubule networks beneath the EPEC microcolonies using various EPEC derivatives. After 3 h infection, percentages were calculated as in (B).
Next, using a GST pull-down assay, we investigated GST-EspG and GST-Orf3 for their ability to interact with tubulins contained in Swiss 3T3 or HeLa cell lysates. Analysis of the precipitated proteins with GST-EspG or GST-Orf3 in an immunoblot analysis with anti-tubulin antibodies revealed that the tubulins were co-precipitated with GST-EspG or GST-Orf3, but not with GST alone (Figure 4A). The direct binding of EspG/Orf3 to tubulins was further confirmed by a ligand overlay assay (see Materials and methods; data not shown) and by a pull-down assay with purified twice-cycled microtubule proteins (Yoshida et al, 2002) (Figure 4B). Moreover, we considered whether or not EspG/Orf3 would be able to affect either microtubule stabilization or destabilization using twice-cycled microtubule proteins (Figure 4C and D). Twice-cycled microtubule proteins were polymerized at 37°C for 1 h, and the resulting polymerized microtubules (15 μM) were incubated with an equal volume of GST alone, GST-EspG, or GST-Orf3 (15 μM each), and the microtubule dynamics were monitored by spectrophotometry (absorbance at 340 nm) (Figure 4C). In the absence of GST-EspG or GST-Orf3, the destabilization of the microtubules was slightly induced after 30 min (Figure 4C, circles), and similar results were observed in an incubation with GST alone (Figure 4C, triangles). In contrast, depolymerization of the microtubules was triggered by the addition of GST-EspG (Figure 4C, squares) or GST-Orf3 (Figure 4C, crosses). Furthermore, microtubule assembly was inhibited by the addition of GST-EspG and GST-Orf3, but not GST alone (Figure 4D). These results clearly demonstrate that EspG/Orf3 has the ability to bind directly to tubulins, and that it promotes the destabilization of microtubules in vitro.
Figure 4.
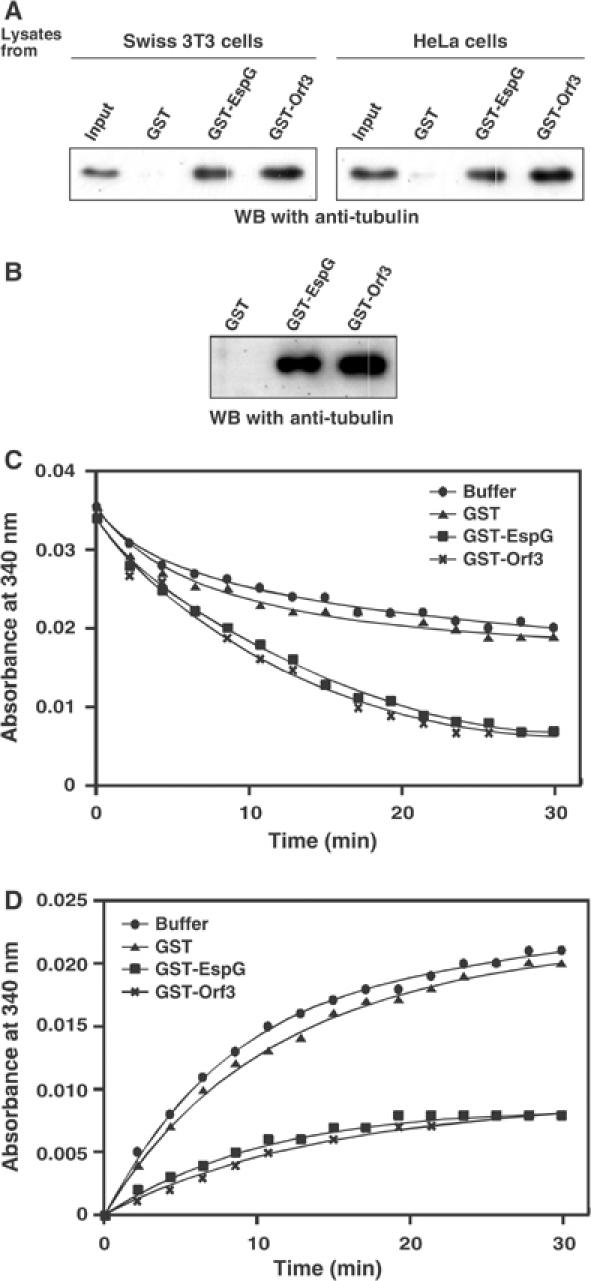
EspG/Orf3 bound to tubulins and induced the destabilization of microtubules in vitro. (A) GST fusion proteins bound to glutathione–Sepharose 4B beads were incubated with Swiss 3T3 and HeLa cell lysates for 2 h at 4°C. The beads were washed, and then bound proteins were analyzed by immunoblotting using anti-tubulin antibodies. (B) GST fusion proteins bound to glutathione–Sepharose 4B beads were incubated with purified twice-cycled microtubule proteins for 2 h at 4°C. The beads were washed, and then each sample was subjected to immunoblotting as in (A). (C) EspG/Orf3 induces microtubule destabilization. Purified twice-cycled microtubule proteins (15 μM) were polymerized at 37°C for 1 h, and then equal volumes of PM-1.3 M buffer alone (circles) or PM-1.3 M buffer each containing 15 μM of GST alone (triangles), GST-EspG (squares), or GST-Orf3 (crosses) were added. Destabilization of microtubules was monitored spectrophotometrically (absorbance at 340 nm) at 37°C for 30 min. (D) EspG/Orf3 inhibited microtubule polymerization. Purified twice-cycled microtubule proteins (7.5 μM) were polymerized with PM-1.3 M buffer alone (circles) or PM-1.3 M buffer each containing 7.5 μM of GST alone (triangles), GST-EspG (squares), and GST-Orf3 (crosses).
EspG/Orf3-induced rearrangement of the actin cytoskeleton is dependent on GEF-H1
Regulation of the actin cytoskeleton by microtubules relies on the activity of the Rho family GTPases (Wittmann and Waterman-Storer, 2001). Microtubule disassembly results in the activation of RhoA, which enhances myosin contractility and actin stress fiber formation (Enomoto, 1996; Liu et al, 1998; Ren et al, 1999). The newly identified GEF family members, including murine Lfc (Glaven et al, 1999), murine p190RhoGEF (van Horck et al, 2001), and human GEF-H1 (Lfc homolog) (Krendel et al, 2002), are associated with microtubules and function as RhoA/Rac activators. Since the destruction of local microtubule networks and the formation of actin stress fibers were the most pronounced host responses to EPEC infection, we investigated the involvement of GEF-H1 (human) in EspG- or Orf3-induced stress fiber formation in human epithelial HeLa cells, instead of in mouse Swiss 3T3 cells. When HeLa cells were infected with EPEC WT, extensive actin accumulation was detected beneath a substantial number of adherent bacteria. However, actin stress fiber formation was very weak in HeLa cells (data not shown). (In fact, this has been one of the major reasons as to why EPEC-induced stress fiber formation has not been characterized to date.) It is possible that actin may be preferentially supplied for the formation of a pedestal structure, rather than for stress fiber formation in EPEC-infected HeLa cells. To facilitate the detection of pathogen-induced stress fiber formation, including GEF-H1 activation, we created tir/espG/orf3 triple mutants carrying either p99-EspG or p99-Orf3. Our preliminary data indicated that mutation of the tir gene turned off the formation of F-actin aggregation beneath the EPEC adherent; this finding allowed us to detect the F-actin architecture in epithelial cells such as HeLa cells. As shown in Figure 5A, upon infection of HeLa cells with the tir/espG/orf3 triple mutant overexpressing EspG, actin stress fibers appeared, a result that was not observed in HeLa cells infected with the triple mutant carrying the vector alone. Importantly, EspG-induced actin cytoskeletal rearrangement was almost completely abolished by the expression of a dominant-negative form of GEF-H1 (GEF-H1 DHmut) in HeLa cells. Indeed, the percentage of cells showing a marked formation of actin stress fibers increased greatly among HeLa cells infected with the tir/espG/orf3 triple mutant carrying p99-EspG or p99-Orf3 (Figure 5B). To further analyze the direct involvement of GEF-H1 in the EspG/Orf3-mediated actin stress fiber formation, a dominant-negative form of p115-RhoGEF (p115-RGS) (Johnson et al, 2003) was also used in this study, since p115-RhoGEF is well characterized as a RhoA-specific activator. RhoA is activated by the treatment of lysophosphatidic acid (LPA) via p115-RhoGEF activity (Hart et al, 1998; Kozasa et al, 1998). Indeed, LPA-induced stress fiber formation was inhibited by the expression of p115-RGS (Figure 5B). As expected, the formation of EspG-induced stress fibers was not affected by the expression of p115-RGS (Figure 5A and B). In contrast, GEF-H1 was found to be modulated by the addition of nocodazole, which induces the depolymerization of microtubules. Moreover, the nocodazole-, but not LPA-, induced stress fiber formation was inhibited by the expression of GEF-H1 DHmut. The impact of actin stress fiber formation elicited by EspG or Orf3 in HeLa cells was similar to that elicited via the destruction of microtubule structures by nocodazole treatment, but not by LPA treatment (Figure 5B). The EspG/Orf3–GEF-H1 signaling pathway was further confirmed by a small interfering RNA (siRNA) assay (Figure 6). Two siRNAs homologous to genes coding for GEF-H1 in different regions (GEF-H1-siRNA-I and GEF-H1-siRNA-II; Benais-Pont et al, 2003) were used; nonsilencing siRNA was used as a negative control. These siRNAs were introduced into HeLa cells, and the reduction in the transcription of GEF-H1 mRNA was confirmed by RT–PCR (Figure 6A). EspG- and Orf3-induced actin cytoskeletal rearrangement was almost completely abolished by the introduction of GEF-H1-siRNA-I or GEF-H1-siRNA-II into HeLa cells (Figure 6B and C). Collectively, these data clearly demonstrate that GEF-H1 is absolutely required for EspG- and Orf3-induced actin cytoskeletal rearrangement.
Figure 5.
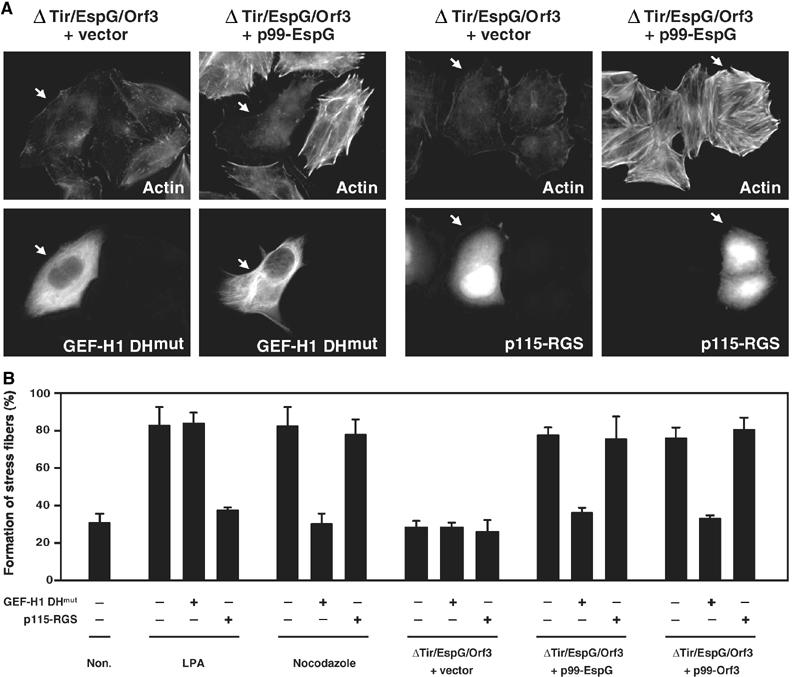
Effect of GEF-H1 on EPEC-induced actin stress fiber formation. (A) HeLa cells were transfected with a plasmid encoding the dominant-negative form of GEF-H1 (GEF-H1 DHmut), containing a Tyr-to-Ala substitution at residue 393 in the DH domain of GEF-H1, or RGS domain of p115-RhoGEF (p115-RGS). Transfected cells were infected with a tir/espG/orf3 mutant complemented with a cloned espG (p99-EspG) or with vector alone for 3 h, at a final concentration of 1 mM of IPTG in order to induce EspG expression. The cells were fixed and stained with rhodamine-labeled phalloidin in order to detect F-actin, and the transfected cells were visualized by EGFP fluorescence. The arrows indicate GEF-H1 DHmut or p115-RGS. (B) HeLa cells were transfected with GEF-H1 DHmut or p115-RGS, and then transfected cells were infected with the EPEC mutant complemented with a cloned espG (p99-EspG), orf3 (p99-Orf3), or vector alone (vector). Otherwise, transfected cells were treated with 1 μg/ml of LPA for 3 min or 10 μM nocodazole for 1 h. The percentage of HeLa cells showing marked formation of actin stress fibers is indicated. Percentages were based on counts of at least 75 cells, and the values given are the means±standard deviation from three independent experiments.
Figure 6.
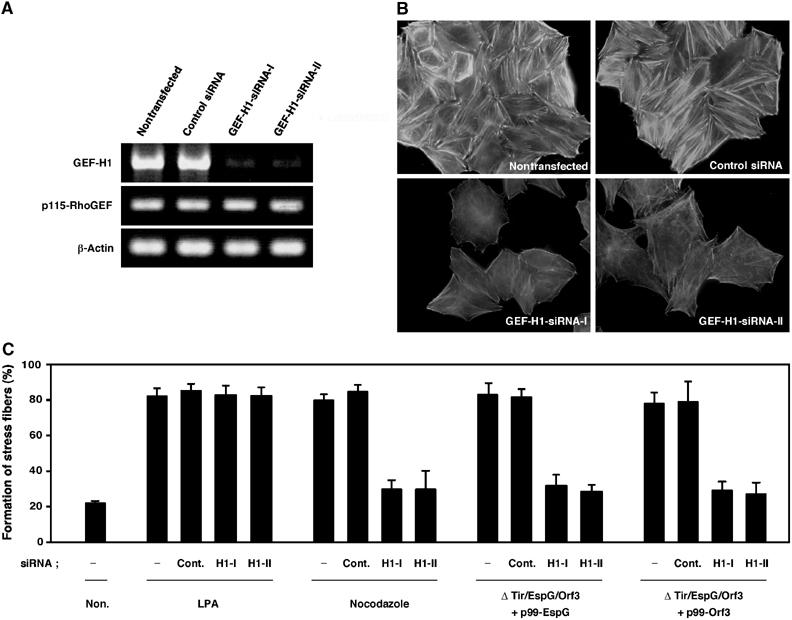
The EspG/Orf3-induced stress fiber formation was inhibited by reduced expression of GEF-H1. (A) Total RNA was purified from HeLa cells transfected with nonsilencing control siRNA, GEF-H1-siRNA-I, or GEF-H1-siRNA-II, and mRNAs for GEF-H1, p115-RhoGEF, and β-actin were analyzed by quantitative RT–PCR. (B) HeLa cells transfected with GEF-H1-siRNA-I, GEF-H1-siRNA-II, or nonsilencing control siRNA were infected with a tir/espG/orf3 mutant complemented with a cloned espG (p99-EspG) for 3 h, at a final concentration of 1 mM of IPTG in order to induce EspG expression. The cells were fixed and stained with rhodamine-labeled phalloidin in order to detect F-actin. (C) The percentage of HeLa cells showing marked formation of actin stress fibers is indicated. HeLa cells were transfected with or without GEF-H1-siRNA-I (H1-I), GEF-H1-siRNA-II (H1-II), or nonsilencing control siRNA (Cont.). Otherwise, the transfected cells were treated with 1 μg/ml of LPA for 3 min or 10 μM nocodazole for 1 h. Percentages were based on counts of at least 150 cells, and the values are the means±standard deviation from three independent experiments.
EspG/Orf3 induce GEF-H1 release from the cytoskeleton to the cytosol
In a previous study, Krendel et al reported that GEF-H1 is localized in the microtubules in its inactive form, and it switches to an active form when it is released from microtubule networks. Hence, to investigate whether GEF-H1 is released from the cytoskeleton to the cytosol by EPEC infection, both cytoskeletal and cytosolic fractions were separated from the lysates of HeLa cells stably expressing GFP-tagged GEF-H1, and then GFP-GEF-H1 was detected by using anti-GFP antibodies (Figure 7). As was the case with the treatment with nocodazole, the relative amount of GEF-H1 associated with the cytoskeletal components was greatly reduced by infection with EPEC WT. The same results were observed in cells infected with the espG or orf3 mutant (data not shown), but not with the espG/orf3 mutant, or with the escF mutant (defective in the type III secretion system); these findings indicate that both EspG and Orf3 effectors possess the ability to release GEF-H1 from the cytoskeleton to the cytosol, and this effect presumably contributes to switching GEF-H1 to its active form.
Figure 7.
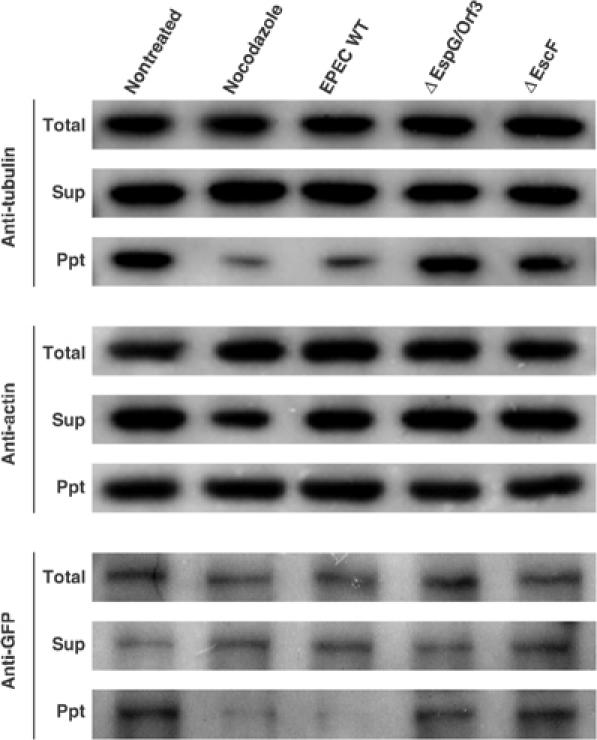
EspG/Orf3 induces GEF-H1 release from the cytoskeleton to the cytosol. HeLa cells stably expressing GFP-GEF-H1 were infected with EPEC WT or its mutants for 3 h. Otherwise, HeLa cells were treated with 10 μM nocodazole for 1 h as a positive control. After infection, the cells were washed and lysed for the preparation of total protein (Total). The cytosolic fraction (supernatant; Sup) and the cytoskeletal fraction (precipitate; Ppt) were separated by centrifugation. In all, 5% of the total recovered whole-cell lysates or the cytosolic fraction and 20% of the cytoskeletal fraction were subjected to SDS–PAGE. The amount of tubulin, actin, and GEF-H1 was analyzed by Western blotting using anti-actin antibodies, anti-tubulin antibodies, or anti-GFP antibodies, respectively.
EspG and Orf3 stimulate the RhoA–ROCK signaling pathway
Since GEF-H1 specifically promotes nucleotide exchange on RhoA, but not on Rac1 or Cdc42 (Krendel et al, 2002), cells transfected with pFLAG-N19 (a RhoA dominant-negative mutant), pFLAG-N17-1 (a Rac1 dominant-negative mutant), and pFLAG-N17-42 (a Cdc42 dominant-negative mutant) were infected with EPEC WT, and the effects of the respective dominant-negative forms on bacterial-induced actin stress fiber formation were investigated by F-actin staining. Figure 8A shows the representative data, which reveal that the formation of EPEC-induced actin stress fibers was blocked when the RhoA dominant-negative form (N19) was expressed but not when the Rac1 (N17-1) or Cdc42 (N17-42) dominant-negative form was expressed. To further confirm these observations, at least 150 FLAG-positive cells with F-actin accumulation underneath the adherent bacteria were randomly chosen from each series, and the percentage of cells showing extensive stress fiber formation was calculated (Figure 8B). The number of cells with extensive actin stress fibers decreased greatly in the EPEC-infected Swiss 3T3 cells overexpressing FLAG-N19 (Figure 8B). This finding was reaffirmed by a pull-down assay using the Rho-binding domain (RBD) of rhotekin, which can bind to active RhoA (GTP-bound form). The level of the active form of RhoA in EPEC WT-infected cells was significantly higher than that in cells infected with the espG/orf3 double mutant or the escF mutant (Figure 8C). Moreover, the espG as well as the orf3 single mutants still possessed the ability to activate RhoA, and complementation of the espG/orf3 double-knockout mutant with p99-EspG or p99-Orf3 restored this ability. These findings indicate that RhoA was activated by EPEC WT infection, and that this event was dependent on the delivery of EspG/Orf3 into host cells by the type III secretion system.
Figure 8.

Activation of the RhoA signal pathway by EPEC infection. (A) Swiss 3T3 cells were transfected with plasmids encoding WT or dominant-negative forms (N19 for RhoA, N17-1 for Rac1, and N17-42 for Cdc42) of the Rho family GTPases, and the cells were infected with the EPEC WT for 3 h. The cells were then fixed and stained with rhodamine-labeled phalloidin and fluorescently labeled anti-FLAG antibodies in order to detect F-actin and transfected cells, respectively. FLAG-positive cells are shown in (A). (B) FLAG-positive cells with F-actin accumulation beneath the adherent bacteria were obtained, and the percentage of cells showing stress fiber formation is indicated. Percentages were based on counts of at least 150 cells, and the values given are the means±standard deviation from three independent experiments. (C) Cos-7 cells were transfected with FLAG-tagged WT RhoA, and then the cells were infected with the EPEC WT or mutants for 3 h, or they were treated with 1 μg/ml of LPA for RhoA activation as a positive control for 3 min. Cells were lysed and the GTP-bound RhoA, the active form of RhoA, was co-precipitated with the GST-Rho binding domain (RBD) of rhotekin. The amount of RhoA bound to RBD and RhoA in the whole-cell lysates was analyzed by Western blotting with anti-FLAG antibodies.
Since these results strongly suggest that EspG- and Orf3-induced stress fiber formation is stimulated by activating the RhoA signaling pathway, we treated Swiss 3T3 cells with Y27632, a ROCK inhibitor, and we then analyzed the resulting effects on stress fiber formation. ROCK, a downstream effector of RhoA, is an excellent candidate for mediating RhoA-induced changes in the actin cytoskeleton (Kimura et al, 1996; Leung et al, 1996; Hall, 1998). As expected, Tir-induced F-actin accumulation beneath the adherent bacteria, caused by EPEC WT infection, was not affected by the addition of Y27632 (Figure 9A and B). However, the formation of actin stress fibers in parallel bundles, induced by EPEC WT and the tir mutant, was completely inhibited in the presence of Y27632. Collectively, these results demonstrate that stress fiber formation, when induced by the RhoA–ROCK signaling pathway, proceeds via a mechanism that is independent of the Tir–intimin interaction. This process is instead triggered via the stimulation of GEF-H1 activity, which is elicited by the destruction of host microtubule networks by EspG and Orf3 effectors.
Figure 9.
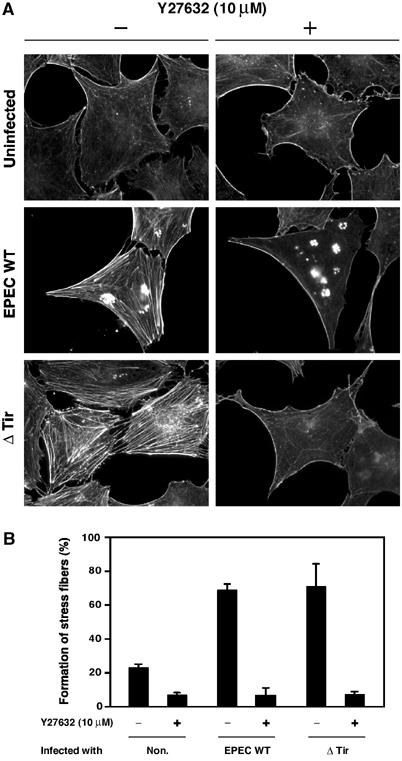
Effect of Y27632 on EPEC-induced actin stress fiber formation. (A) Swiss 3T3 cells were maintained in the presence or absence of 10 μM Y27632 for 1 h, and were infected with EPEC WT and the tir mutant for 3 h in the presence or absence of 10 μM Y27632. The cells were fixed and stained with rhodamine-labeled phalloidin in order to detect F-actin. All fluorescence images were taken with the same exposure time. (B) The percentage of Swiss 3T3 cells showing marked formation of actin stress fibers is indicated. Swiss 3T3 cells were infected with EPEC WT and the tir mutant for 3 h in the presence or absence of 10 μM Y27632 as in (A). Percentages are based on counts of at least 150 cells, and the values are the means±standard deviation from three independent experiments.
Discussion
We demonstrated in this series of experiments that both EspG and Orf3 can modulate GEF-H1-mediated signaling by the disruption of host microtubules. EspG- or Orf3-induced GEF-H1 modulation might thus contribute to an increase in paracellular permeability, rather than to the disruption of tight junctions. A recent study has shown that GEF-H1 is associated with tight junctions and that it regulates size-selective paracellular permeability by the activation of the RhoA–ROCK signaling pathway (Benais-Pont et al, 2003). Furthermore, GEF-H1 was not found to have an effect on overall cell morphology, including tight junction assembly, as was determined by measuring the transepithelial electrical resistance (TER). The disruption of tight junctions has been observed in studies of EPEC WT infection, but not in those of espF mutant infection, indicating that the EspF effector is involved in tight junction disruption (McNamara et al, 2001). These findings are supported by data from a previous report (Elliott et al, 2001). As was the case with the infection of EPEC WT, infection of an espG/orf3 double mutant equally induced a reduction in TER, indicating that neither EspG nor Orf3 is involved in tight junction disruption (Elliott et al, 2001). Currently, the effects of EPEC infection on paracellular permeability are under investigation in a study introducing purified EspG and Orf3 into MDCK cells; EPEC effectors that regulate epithelial barrier function may be involved in this type of virulence. Indeed, a recent study demonstrated that both EspG and EspF are required for C. rodentium to achieve full virulence (Deng et al, 2004).
When EspG and Orf3 were translocated into host cells, both proteins appeared to interact with tubulins; this effect was found to promote the disruption of microtubule networks. A GST pull-down assay showed that EspG and Orf3 had the ability to bind to the soluble form of tubulins (Figure 4). In order to investigate in more detail whether or not EspG and Orf3 could bind to microtubules, taxol-stabilized tubulins, prepared as previously described (Yoshida et al, 2002), were used for the pull-down assay. However, we were unable to detect the binding of EspG or Orf3 to taxol-stabilized tubulins (data not shown). It should be noted that similar results have been reported in the case of the Shigella VirA effector (Yoshida et al, 2002). The oncoprotein Op18/stathmin is known to sequester tubulins, and the presence of this protein resulted in a reduction in the tubulin concentration required for microtubule assembly, followed by a promotion of microtubule destabilization (Cassimeris, 2002). EspG and Orf3 may exert an effect on microtubules much like that of Op18/stathmin (Cassimeris, 2002). Although EspG and Orf3 in EPEC, as well as VirA in Shigella, can exert effects on microtubule networks, EspG and Orf3 induce the formation of actin stress fibers and VirA induces membrane ruffling (Yoshida et al, 2002). The different mechanisms underlying the cytoskeletal rearrangement of host cells, induced by these factors, remain to be elucidated.
Figure 10 depicts the action of EPEC type III effectors (e.g., Tir, EspF, and EspG/Orf3) in host cells. In EPEC-infected cells, the rearrangement of actin organization is promoted by two different pathways, that is, by the formation of a pedestal structure induced by Tir, and by actin stress fiber assembly induced by EspG and Orf3. The tir and eae mutants still possessed the ability to induce stress fiber formation, but they could not form the pedestal structure (Figure 1B). In contrast, the formation of a pedestal structure was still induced by infection with the espG/orf3 double mutant (Figure 2A). Although actin stress fiber formation was inhibited by the introduction of dominant-negative RhoA, and also by treatment with Y27632, Tir-induced pedestal formation was not affected (Figures 8 and 9). These results indicate that Tir and EspG/Orf3 independently contribute to the induction of host cytoskeletal rearrangement using different host signal pathways. Like Tir, EspG and its homolog Orf3 are secreted and injected into host cells via type III machinery. EspG/Orf3 can in turn bind directly to tubulins, and thereby induce the destabilization of microtubules (Figures 3 and 4). These effects of EspG/Orf3 on tubulins cause the release of GEF-H1 from the cytoskeleton to the cytosol, and may activate a host cell signaling pathway involving GEF-H1, RhoA, and ROCK (Figures 5, 6, 7, 8 and 9).
Figure 10.

A presumed model for the action of EPEC type III effectors in host cells. EPEC injects the effectors (e.g., Tir, EspF, EspG, and the EspG homolog Orf3) via a type III secretion machinery into the host cells. The interaction of a bacterial membrane protein, intimin, and translocated Tir triggers the host signal transduction pathway, which includes the recruitment of Nck, N-WASP, and the Arp2/3 complex, resulting in the formation of a pedestal structure beneath the adherent bacteria. On the other hand, it is possible that EspF and EspG/Orf3 influence the function of cell junctions (CJs). Although the precise function of EspF is not clear at present, EspF is known to exert an effect on the morphology of tight junction strands. EspG and Orf3 associate with tubulins, resulting in the disruption of microtubule networks. GEF-H1 switches to the active form as a result of its dissociation from microtubule networks. Activated GEF-H1 promotes the binding of GTP to RhoA, resulting in the activation of RhoA. Finally, the activation of ROCK, which is located downstream of the RhoA signal, induces actin stress fiber assembly and probably exerts influence on paracellular permeability in EPEC-infected cells.
We demonstrated here that EspG and Orf3 effectors regulate the crosstalk between microtubules and actin stress fibers by exploiting GEF-H1. These events are likely to be involved in an increase in paracellular permeability. A/E lesion formation due to Tir–intimin interactions, and the disruption of tight junctions by EspF have an effect on the overall morphology and physiology of host cells. The synergistic effects caused by multiple effectors are expected to contribute to triggering EPEC-mediated diarrhea.
Materials and methods
Bacterial strains, plasmids, transfection, infection assay, in vitro tubulin binding assay, and ligand overlay assay
See Supplementary Materials and methods available at The EMBO Journal Online.
In vitro microtubule-destabilization assay
Polymerization of twice-cycled microtubule proteins was monitored spectrophotometrically (absorbance at 340 nm) at 37°C as described previously (Gaskin, 1982; Horwitz et al, 1997; Yoshida et al, 2002). To assess the effect of EspG or Orf3 on microtubule stabilization, equal volumes of PM-1.3 M buffer (PM buffer containing 1.3 M glycerol) alone or PM-1.3 M buffer containing GST alone, GST-EspG, or GST-Orf3 were added to the cuvettes either before or after inducing the polymerization of the microtubules.
Small-interference RNA assay
Duplex siRNAs were constructed against sequences coding for different GEF-H1 regions (GEF-H1-siRNA-I, 5′-AACAAGAGCATCACAGCCAAGdTdT-3′; GEF-H1-siRNA-II, 5′-AATGTGACTATCCACAACCGCdTdT-3′), as described elsewhere (Benais-Pont et al, 2003), and nonsilencing control siRNA with a random sequence was purchased from QIAGEN. These siRNA oligonucleotides were introduced into cultured cells with RNAiFect transfection reagent (QIAGEN) and the transfection efficiency (approximately 90%) was confirmed by using a fluorescein-labeled siRNA. The total RNA was purified from siRNA-transfected cells using Concert Micro-to-Midi Total RNA Purification System (Invitrogen, Carlsbad, CA), and then reverse transcription was performed using a Superscript III First Strand Synthesis system (Invitrogen). Transcription of GEF-H1, p115-RhoGEF, and β-actin mRNA was analyzed by agarose gel electrophoresis after RT–PCR using the respective primers.
Differential cytoskeletal extraction
Differential cytoskeletal extraction was performed as described previously (van Bergen en Henegouwen et al, 1992; Ding et al, 1996). HeLa cells stably expressing GFP-tagged GEF-H1 were infected with EPEC WT or its mutants for 3 h, or the cells were treated with 10 μM nocodazole for 1 h as a positive control. After infection, the cells were washed four times in PBS and lysed in cytoskeleton-stabilizing buffer (10 mM PIPES–NaOH pH 6.8, 250 mM sucrose, 3 mM MgCl2, 150 mM KCl, 1 mM EGTA, and 1 mM PMSF) containing 0.15% Triton X-100 for 5 min at 37°C. Cell lysates were then centrifuged at 14 000 g at room temperature for 10 min, and the supernatant and the pellet were considered as the cytosolic and cytoskeletal fractions, respectively. The relative amounts of actin, tubulin, and GEF-H1 were analyzed by Western blotting using anti-actin antibodies (CHEMICON, Temecula, CA), anti-tubulin antibodies (Sigma, St Louis, MO), or anti-GFP antibodies (Molecular Probes, Eugene, OR).
GST-RBD pull-down assay
See Supplementary Materials and methods available at The EMBO Journal Online.
Supplementary Material
Supplementary Materials and methods
Acknowledgments
We thank GM Bokoch for the generous gift of plasmids encoding GEF-H1 and GEF-H1 DHmut, Y Horiguchi for the plasmids encoding the Rho family GTPases, S Narumiya for a plasmid encoding GST-rhotekin (RBD), and BB Finlay for the EPEC tir mutant strain. This research was partially supported by operating grants from the All Kitasato Project Study, the Waksman Foundation of Japan Inc., and Ministry of Education, Science, Sports and Culture, Grant-in-Aid for Scientific Research on Priority Areas, 14021109, 2002; Young Scientists (B), 14770123, 2002; Scientific Research (C), 16590370, 2004; COE Research.
References
- Abe A, Heczko U, Hegele RG, Finlay BB (1998) Two enteropathogenic Escherichia coli type III secreted proteins, EspA and EspB, are virulence factors. J Exp Med 188: 1907–1916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Ami G, Ozeri V, Hanski E, Hofmann F, Aktories K, Hahn KM, Bokoch GM, Rosenshine I (1998) Agents that inhibit Rho, Rac, and Cdc42 do not block formation of actin pedestals in HeLa cells infected with enteropathogenic Escherichia coli. Infect Immun 66: 1755–1758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benais-Pont G, Punn A, Flores-Maldonado C, Eckert J, Raposo G, Fleming TP, Cereijido M, Balda MS, Matter K (2003) Identification of a tight junction-associated guanine nucleotide exchange factor that activates Rho and regulates paracellular permeability. J Cell Biol 160: 729–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassimeris L (2002) The oncoprotein 18/stathmin family of microtubule destabilizers. Curr Opin Cell Biol 14: 18–24 [DOI] [PubMed] [Google Scholar]
- Deng W, Puente JL, Gruenheid S, Li Y, Vallance BA, Vazquez A, Barba J, Ibarra JA, O'Donnell P, Metalnikov P, Ashman K, Lee S, Goode D, Pawson T, Finlay BB (2004) Dissecting virulence: systematic and functional analyses of a pathogenicity island. Proc Natl Acad Sci USA 101: 3597–3602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVinney R, Gauthier A, Abe A, Finlay BB (1999) Enteropathogenic Escherichia coli: a pathogen that inserts its own receptor into host cells. Cell Mol Life Sci 55: 961–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding A, Chen B, Fuortes M, Blum E (1996) Association of mitogen-activated protein kinases with microtubules in mouse macrophages. J Exp Med 183: 1899–1904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnenberg MS, Kaper JB (1992) Enteropathogenic Escherichia coli. Infect Immun 60: 3953–3961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnenberg MS, Tacket CO, James SP, Losonsky G, Nataro JP, Wasserman SS, Kaper JB, Levine MM (1993a) Role of the eaeA gene in experimental enteropathogenic Escherichia coli infection. J Clin Invest 92: 1412–1417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnenberg MS, Tzipori S, McKee ML, O'Brien AD, Alroy J, Kaper JB (1993b) The role of the eae gene of enterohemorrhagic Escherichia coli in intimate attachment in vitro and in a porcine model. J Clin Invest 92: 1418–1424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott SJ, Krejany EO, Mellies JL, Robins-Browne RM, Sasakawa C, Kaper JB (2001) EspG, a novel type III system-secreted protein from enteropathogenic Escherichia coli with similarities to VirA of Shigella flexneri. Infect Immun 69: 4027–4033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enomoto T (1996) Microtubule disruption induces the formation of actin stress fibers and focal adhesions in cultured cells: possible involvement of the rho signal cascade. Cell Struct Funct 21: 317–326 [DOI] [PubMed] [Google Scholar]
- Gaskin F (1982) Techniques for the study of microtubule assembly in vitro. Methods Enzymol 85 (Part B): 433–439 [DOI] [PubMed] [Google Scholar]
- Giron JA, Ho AS, Schoolnik GK (1991) An inducible bundle-forming pilus of enteropathogenic Escherichia coli. Science 254: 710–713 [DOI] [PubMed] [Google Scholar]
- Glaven JA, Whitehead I, Bagrodia S, Kay R, Cerione RA (1999) The Dbl-related protein, Lfc, localizes to microtubules and mediates the activation of Rac signaling pathways in cells. J Biol Chem 274: 2279–2285 [DOI] [PubMed] [Google Scholar]
- Gruenheid S, DeVinney R, Bladt F, Goosney D, Gelkop S, Gish GD, Pawson T, Finlay BB (2001) Enteropathogenic E. coli Tir binds Nck to initiate actin pedestal formation in host cells. Nat Cell Biol 3: 856–859 [DOI] [PubMed] [Google Scholar]
- Hall A (1998) Rho GTPases and the actin cytoskeleton. Science 279: 509–514 [DOI] [PubMed] [Google Scholar]
- Hart MJ, Jiang X, Kozasa T, Roscoe W, Singer WD, Gilman AG, Sternweis PC, Bollag G (1998) Direct stimulation of the guanine nucleotide exchange activity of p115 RhoGEF by Galpha13. Science 280: 2112–2114 [DOI] [PubMed] [Google Scholar]
- Hecht G (2001) Microbes and microbial toxins: paradigms for microbial-mucosal interactions. VII. Enteropathogenic Escherichia coli: physiological alterations from an extracellular position. Am J Physiol Gastrointest Liver Physiol 281: G1–G7 [DOI] [PubMed] [Google Scholar]
- Horwitz SB, Shen HJ, He L, Dittmar P, Neef R, Chen J, Schubart UK (1997) The microtubule-destabilizing activity of metablastin (p19) is controlled by phosphorylation. J Biol Chem 272: 8129–8132 [DOI] [PubMed] [Google Scholar]
- Ide T, Laarmann S, Greune L, Schillers H, Oberleithner H, Schmidt MA (2001) Characterization of translocation pores inserted into plasma membranes by type III-secreted Esp proteins of enteropathogenic Escherichia coli. Cell Microbiol 3: 669–679 [DOI] [PubMed] [Google Scholar]
- Jarvis KG, Giron JA, Jerse AE, McDaniel TK, Donnenberg MS, Kaper JB (1995) Enteropathogenic Escherichia coli contains a putative type III secretion system necessary for the export of proteins involved in attaching and effacing lesion formation. Proc Natl Acad Sci USA 92: 7996–8000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson EN, Seasholtz TM, Waheed AA, Kreutz B, Suzuki N, Kozasa T, Jones TL, Brown JH, Druey KM (2003) RGS16 inhibits signalling through the G alpha 13-Rho axis. Nat Cell Biol 5: 1095–1103 [DOI] [PubMed] [Google Scholar]
- Kalman D, Weiner OD, Goosney DL, Sedat JW, Finlay BB, Abo A, Bishop JM (1999) Enteropathogenic E. coli acts through WASP and Arp2/3 complex to form actin pedestals. Nat Cell Biol 1: 389–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenny B, DeVinney R, Stein M, Reinscheid DJ, Frey EA, Finlay BB (1997) Enteropathogenic E. coli (EPEC) transfers its receptor for intimate adherence into mammalian cells. Cell 91: 511–520 [DOI] [PubMed] [Google Scholar]
- Kenny B, Ellis S, Leard AD, Warawa J, Mellor H, Jepson MA (2002) Co-ordinate regulation of distinct host cell signalling pathways by multifunctional enteropathogenic Escherichia coli effector molecules. Mol Microbiol 44: 1095–1107 [DOI] [PubMed] [Google Scholar]
- Kimura K, Ito M, Amano M, Chihara K, Fukata Y, Nakafuku M, Yamamori B, Feng J, Nakano T, Okawa K, Iwamatsu A, Kaibuchi K (1996) Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase). Science 273: 245–248 [DOI] [PubMed] [Google Scholar]
- Kozasa T, Jiang X, Hart MJ, Sternweis PM, Singer WD, Gilman AG, Bollag G, Sternweis PC (1998) p115 RhoGEF, a GTPase activating protein for Galpha12 and Galpha13. Science 280: 2109–2111 [DOI] [PubMed] [Google Scholar]
- Krendel M, Zenke FT, Bokoch GM (2002) Nucleotide exchange factor GEF-H1 mediates cross-talk between microtubules and the actin cytoskeleton. Nat Cell Biol 4: 294–301 [DOI] [PubMed] [Google Scholar]
- Leung T, Chen XQ, Manser E, Lim L (1996) The p160 RhoA-binding kinase ROK alpha is a member of a kinase family and is involved in the reorganization of the cytoskeleton. Mol Cell Biol 16: 5313–5327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu BP, Chrzanowska-Wodnicka M, Burridge K (1998) Microtubule depolymerization induces stress fibers, focal adhesions, and DNA synthesis via the GTP-binding protein Rho. Cell Adhes Commun 5: 249–255 [DOI] [PubMed] [Google Scholar]
- McDaniel TK, Jarvis KG, Donnenberg MS, Kaper JB (1995) A genetic locus of enterocyte effacement conserved among diverse enterobacterial pathogens. Proc Natl Acad Sci USA 92: 1664–1668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamara BP, Koutsouris A, O'Connell CB, Nougayrede JP, Donnenberg MS, Hecht G (2001) Translocated EspF protein from enteropathogenic Escherichia coli disrupts host intestinal barrier function. J Clin Invest 107: 621–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon HW, Whipp SC, Argenzio RA, Levine MM, Giannella RA (1983) Attaching and effacing activities of rabbit and human enteropathogenic Escherichia coli in pig and rabbit intestines. Infect Immun 41: 1340–1351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren XD, Kiosses WB, Schwartz MA (1999) Regulation of the small GTP-binding protein Rho by cell adhesion and the cytoskeleton. EMBO J 18: 578–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Y, Li R, Zheng Y, Busch H (1998) Cloning and characterization of GEF-H1, a microtubule-associated guanine nucleotide exchange factor for Rac and Rho GTPases. J Biol Chem 273: 34954–34960 [DOI] [PubMed] [Google Scholar]
- Rosenshine I, Donnenberg MS, Kaper JB, Finlay BB (1992) Signal transduction between enteropathogenic Escherichia coli (EPEC) and epithelial cells: EPEC induces tyrosine phosphorylation of host cell proteins to initiate cytoskeletal rearrangement and bacterial uptake. EMBO J 11: 3551–3560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekiya K, Ohishi M, Ogino T, Tamano K, Sasakawa C, Abe A (2001) Supermolecular structure of the enteropathogenic Escherichia coli type III secretion system and its direct interaction with the EspA-sheath-like structure. Proc Natl Acad Sci USA 98: 11638–11643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonovic I, Rosenberg J, Koutsouris A, Hecht G (2000) Enteropathogenic Escherichia coli dephosphorylates and dissociates occludin from intestinal epithelial tight junctions. Cell Microbiol 2: 305–315 [DOI] [PubMed] [Google Scholar]
- Tu X, Nisan I, Yona C, Hanski E, Rosenshine I (2003) EspH, a new cytoskeleton-modulating effector of enterohaemorrhagic and enteropathogenic Escherichia coli. Mol Microbiol 47: 595–606 [DOI] [PubMed] [Google Scholar]
- van Bergen en Henegouwen PM, den Hartigh JC, Romeyn P, Verkleij AJ, Boonstra J (1992) The epidermal growth factor receptor is associated with actin filaments. Exp Cell Res 199: 90–97 [DOI] [PubMed] [Google Scholar]
- van Horck FP, Ahmadian MR, Haeusler LC, Moolenaar WH, Kranenburg O (2001) Characterization of p190RhoGEF, a RhoA-specific guanine nucleotide exchange factor that interacts with microtubules. J Biol Chem 276: 4948–4956 [DOI] [PubMed] [Google Scholar]
- Wittmann T, Waterman-Storer CM (2001) Cell motility: can Rho GTPases and microtubules point the way? J Cell Sci 114: 3795–3803 [DOI] [PubMed] [Google Scholar]
- Yoshida S, Katayama E, Kuwae A, Mimuro H, Suzuki T, Sasakawa C (2002) Shigella deliver an effector protein to trigger host microtubule destabilization, which promotes Rac1 activity and efficient bacterial internalization. EMBO J 21: 2923–2935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuhan R, Koutsouris A, Savkovic SD, Hecht G (1997) Enteropathogenic Escherichia coli-induced myosin light chain phosphorylation alters intestinal epithelial permeability. Gastroenterology 113: 1873–1882 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Materials and methods