

Abstract
To investigate how the complex organization of heterochromatin is reproduced at each replication cycle, we examined the fate of HP1-rich pericentric domains in mouse cells. We find that replication occurs mainly at the surface of these domains where both PCNA and chromatin assembly factor 1 (CAF-1) are located. Pulse–chase experiments combined with high-resolution analysis and 3D modeling show that within 90 min newly replicated DNA become internalized inside the domain. Remarkably, during this time period, a specific subset of HP1 molecules (α and γ) coinciding with CAF-1 and replicative sites is resistant to RNase treatment. Furthermore, these replication-associated HP1 molecules are detected in Suv39 knockout cells, which otherwise lack stable HP1 staining at pericentric heterochromatin. This replicative pool of HP1 molecules disappears completely following p150CAF-1 siRNA treatment. We conclude that during replication, the interaction of HP1 with p150CAF-1 is essential to promote delivery of HP1 molecules to heterochromatic sites, where they are subsequently retained by further interactions with methylated H3-K9 and RNA.
Keywords: chromatin assembly, heterochromatin, nuclear organization, replication in vivo
Introduction
Many aspects of genome function are controlled by epigenetic parameters not encoded within the DNA sequence, yet stably transmitted through multiple cell divisions. Recent advances have identified several important marks involved in gene regulation, including DNA methylation, post-translational histone modifications, and chromatin-associated factors (Strahl and Allis, 2000; Grewal and Moazed, 2003; Vermaak et al, 2003). Whereas transmission of DNA methylation patterns can be explained by coupling enzymatic modification to semiconservative DNA replication, it is not clear how the epigenetic marks and higher order structure of heterochromatin domains are maintained and faithfully transmitted.
Constitutive heterochromatin domains near centromeres exemplify the complexity of the problem. In mammalian cells and fission yeast, these domains are critical for proper chromosome segregation. They contain generally hypoacetylated histones (Jeppesen et al, 1992; Ekwall et al, 1997; Taddei et al, 2001) in combination with histone H3 methylated on lysine 9 (Rea et al, 2000; Nakayama et al, 2001), a modification that provides binding sites for the HP1 proteins (Bannister et al, 2001; Lachner et al, 2001). In addition to H3-K9 methylation, an unidentified RNA is required for accumulation of HP1 proteins at these domains (Maison et al, 2002; Muchardt et al, 2002). Although these observations have better characterized a specific pericentric heterochromatin architecture, it remains to be defined how its higher order structure can be inherited.
During DNA replication, the parental nucleosomes ahead of the replication fork are transiently disrupted and distributed behind it onto daughter duplex DNA (Randall and Kelly, 1992; Gruss et al, 1993). Newly synthesized histones are additionally incorporated to obtain the full nucleosome complement on the nascent DNA (Verreault, 2000). In the case of pericentric heterochromatin domains, not only the nucleosomal organization but also its post-translational modifications, associated proteins, such as HP1, and the RNA component will suffer the disruptive effect of the replication fork passage. These different components that participate in this higher order structure must be re-established after replication in order to reproduce the complete subnuclear domains of heterochromatin. Given such complexity, it appears necessary to examine the mechanism that allows both the local disruption and the maintenance of this higher order structure during DNA replication. Replication sites corresponding to HP1-rich heterochromatin domains can be visualized as foci in S-phase mouse nuclei (Fox et al, 1991; O'Keefe et al, 1992; Taddei et al, 1999; Dimitrova and Berezney, 2002). We set out to define the specific organization of HP1-rich domains during replication, in order to characterize the architecture of these regions, and to determine which parameters are essential to their establishment and maintenance. Although general models for replication ‘factories' have been proposed (Cook, 1999), their exact nature (Dimitrova and Gilbert, 2000) and a detailed organization of the replication machinery, chromatin assembly factors, and heterochromatin proteins at foci corresponding to specific heterochromatin regions are still poorly understood.
Candidate factor(s) involved in the inheritance of heterochromatin domains must fulfill at least two criteria: (i) they must be present during replication and (ii) they must permit a link between nucleosome formation and proteins, such as HP1, involved in defining the higher order structure of pericentric heterochromatin. In this context, the conserved chromatin assembly factor 1 (CAF-1) seems a prime candidate. This complex, containing three subunits (p150, p60, and p48), has been identified based on its ability to specifically assemble nucleosomes in vitro onto newly synthesized DNA during replication (Smith and Stillman, 1989) or nucleotide excision repair (Gaillard et al, 1996), and it is found in vivo at replication foci in human cells (Krude, 1995; Taddei et al, 1999). The interaction of its large subunit (p150) with PCNA provides a molecular link with DNA synthesis (Shibahara and Stillman, 1999; Moggs et al, 2000). Furthermore, a role in silencing, often considered as a hallmark of heterochromatin, was reported in Saccharomyces cerevisiae strains lacking the orthologs of CAF-1 (Kaufman et al, 1995; Enomoto et al, 1997). These findings prompted further investigations in mammals concerning a link between CAF-1 and heterochromatin. In this respect, the fact that the p150 subunit of CAF-1 (p150CAF-1) could interact with HP1 proteins from mammals (Murzina et al, 1999) was particularly interesting; however, the functional relevance of this interaction in vivo has remained a puzzle.
Our goal was to gain insight into the role of CAF-1 in the faithful inheritance of the heterochromatin state. We show that in mouse cells, replication of pericentric heterochromatin domains involves a dedicated infrastructure, in which we have localized PCNA, sites of DNA synthesis, and p150CAF-1. Within this specific architecture, we found that most of the DNA is replicated at the periphery of the HP1-rich domain and then relocated inside the domain. Importantly, we find evidence for the existence of distinct pools of HP1 molecules during replication. In particular, we have identified a replication-specific pool that requires p150CAF-1 but not H3-K9 methylation nor an RNA component. These data highlight the critical importance of p150CAF-1 for the heritability of HP1 in the heterochromatin domains.
Results
Dynamics of DNA at HP1-rich heterochromatin domains in mouse cells
In mouse cells, HP1-rich domains found in pericentric heterochromatin can be easily identified by dense DAPI staining (Minc et al, 2000; Nielsen et al, 2001). To examine how these domains duplicate during S-phase, we labeled cells with bromodeoxyuridine (BrdU) for 10 min. Within the BrdU-positive cells (30% of the total cell population), we revealed replication profiles typical of early (dense BrdU labeling throughout the nucleus, 60%), mid-late (ring-shaped labeling, 30%), and late S-phase (a few large dots mainly at the nuclear periphery, 10%) (Figure 1A) as reported in rat (Nakamura et al, 1986) and mouse cells (Fox et al, 1991; Dimitrova and Berezney, 2002). Importantly, only the mid-late S-phase profiles often described as ‘horse-shoe' replication foci (Nakamura et al, 1986; Dimitrova and Berezney, 2002) coincided with the DAPI dense regions that correspond to pericentric heterochromatin (Figure 1A). The exact nature of these types of foci has remained elusive (Dimitrova and Gilbert, 2000). We thus examined such foci more closely. Remarkably, analysis of several hundred such profiles, obtained with an asynchronous population of cells for a 10 min BrdU pulse, reproducibly displayed a ring-shaped appearance around the DAPI domains, and never marked the most internal part (Figure 1A, 10 min). To determine whether this ring-shaped staining pattern reflects sites of DNA synthesis, rather than a restricted accessibility of the BrdU antibody to the interior of the domain, we performed a 90 min BrdU pulse. In this case, the entire pericentric heterochromatin domain could be stained (Figure 1A, 90 min) eliminating the possibility of limited antibody accessibility. These observations suggest that replicated DNA moves from the periphery into the interior of the domain. To better investigate these DNA dynamics, we followed the relocation of newly synthesized DNA with respect to sites of ongoing DNA synthesis using pulse–chase–pulse labeling with the two halogenated deoxyuridines Chloro-dU (CldU) and Iodo-dU (IdU) (Aten et al, 1992). When both labels are added at the same time (no chase, 0 min), they yield a similar staining pattern forming a ring at the periphery of the heterochromatin domain (Figure 1B, left). After 45 min of chase, the second pulse (IdU) visualized in green still displays a ring-shaped pattern, whereas the first pulse (CldU) visualized in red shows a diffuse pattern in the interior part of the ring pattern (Figure 1B, left). As a further confirmation, we used BrdU to follow synthesized DNA and PCNA to locate the replication machinery at sites of ongoing synthesis. Cells were pulsed for 10 min with BrdU and chased for 45, 60, and 90 min to allow relocation of synthesized DNA in the interior of the domain. Figure 1B (right) shows that after a 45 min chase, the BrdU-labeled DNA is detected within the interior of the DAPI dense domain (60 and 90 min), whereas sites of ongoing DNA synthesis, visualized by PCNA staining, display a clear ring-shaped pattern. The time required for this relocation process to be completed is within the time range of the mid-late S-phase (O'Keefe et al, 1992; Dimitrova and Berezney, 2002). For a more accurate localization of the replicated DNA, we used high-resolution imaging and 3D reconstruction for a 10 min BrdU pulse either without or after 90 min chase (Figure 1C and D). A representative image of a single domain reveals that a 10 min pulse of BrdU labels DNA at the periphery of the DAPI dense domain (Figures 1C and 2B, BrdU+DAPI). In contrast, after 90 min chase, most of the BrdU label is detected inside the DAPI dense domain (Figure 1D). These results argue for the existence of a majority of active sites for DNA synthesis operating as stable entities at the periphery of the domain combined with specific DNA dynamics: DNA moves to the periphery to get replicated and then back, inside the domain.
Figure 1.
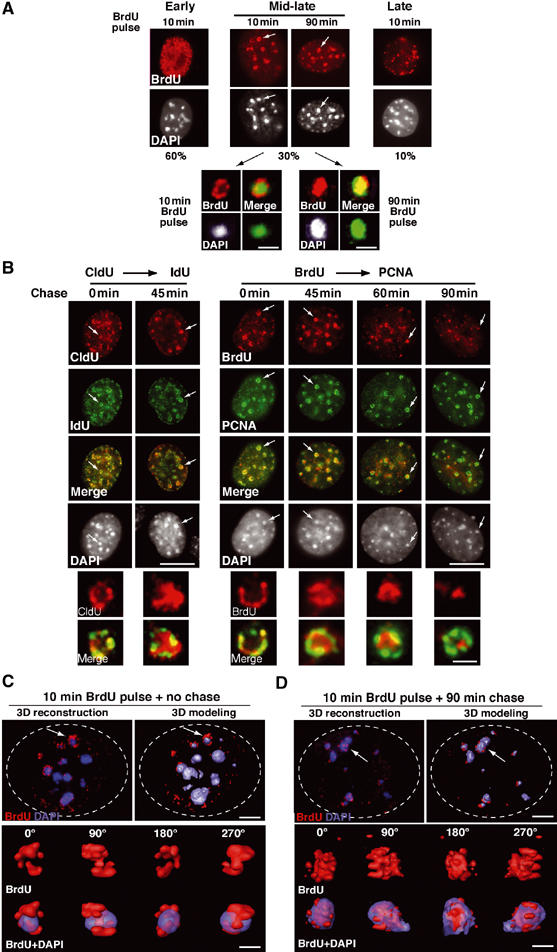
Specific dynamics of DNA synthesis in replicating pericentric heterochromatin. (A) Typical BrdU-labeled S-phase patterns. Top: 3T3 cells were pulsed for 10 or 90 min with BrdU (red, top) and stained with DAPI (bottom). Scale bar, 10 μm. Bottom: Magnification (six-fold) of individual replicating pericentric heterochromatin domain indicated by arrows on top. Corresponding BrdU (red), DAPI, and merge images are presented. In merge, DAPI was pseudocolored in green. Scale bar, 0.5 μm. (B) Newly replicated DNA is located at the periphery of the DAPI dense region and then becomes internalized. Left: Pulse–chase–pulse on 3T3 cells with first a CldU pulse followed by 45 min chase before a second IdU pulse was given, or both CldU and IdU were added together (0 min). Representative images (CldU, red; IdU, green) are shown with merge and DAPI staining. Right: 3T3 cells pulsed for 10 min with BrdU were chased for various times as indicated. Typical BrdU (red) and PCNA (green) patterns with merge and their corresponding DAPI staining are presented. Scale bar, 10 μm. The arrows point to the pericentric heterochromatin domain magnified six-fold below. Scale bar, 0.5 μm. (C) 3D reconstruction and modeling of DNA synthesis at pericentric heterochromatin domains. 3T3 cell were pulse labeled with BrdU for 10 min, and after immuno- and DAPI staining, z stack images of a single 3T3 cell nucleus in mid-late S-phase were used for 3D reconstruction and modeling. Localization of BrdU incorporation (red) and DNA (blue) in the whole nucleus. The dashed line represents the periphery of the nucleus. The arrow indicates the heterochromatin domain selected for magnification below. Scale bar, 10 μm. Several rotation angles are presented and ongoing DNA synthesis (red) is shown with DAPI staining (transparent blue). Scale bar, 0.5 μm. (D) As above, for 90 min chase.
Figure 2.
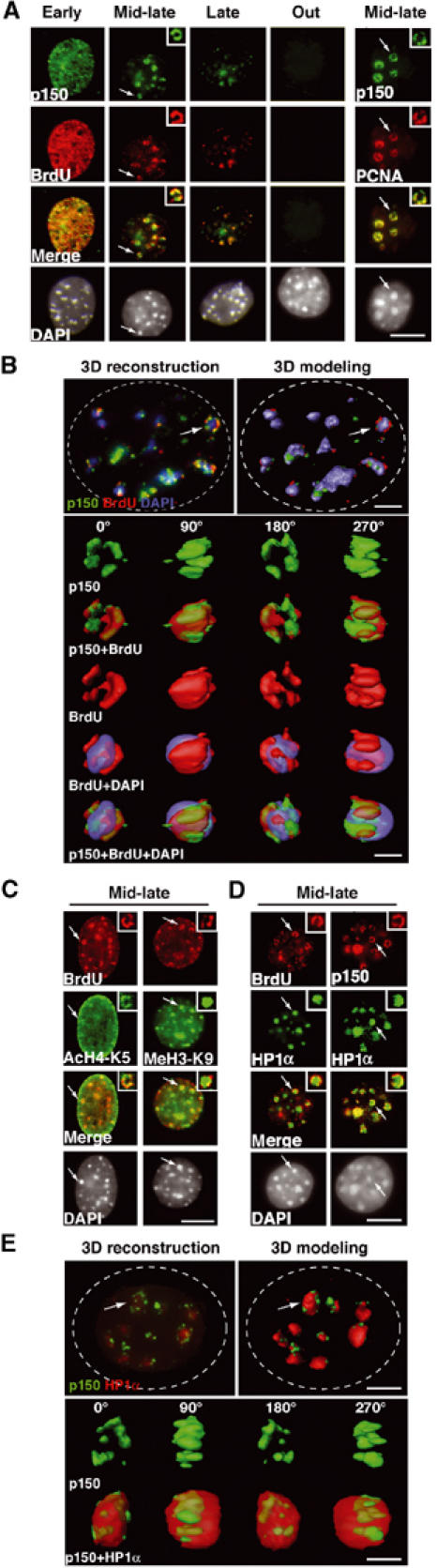
3D architecture of p150CAF-1, HP1, PCNA, and histones in replicating pericentric heterochromatin domains. (A) Mouse p150CAF-1 and DNA synthesis. p150CAF-1 (green) in combination with a 10 min pulse BrdU labeling (red, left), both during and outside S-phase, is shown in parallel with a double staining with PCNA (red, right) for mid-late S-phase only. Merge and DAPI images are presented. See typical foci in mid-late S-phase identified by an arrow at a three-fold magnification (insets). Scale bar, 10 μm. (B) 3D analysis of p150CAF-1 colocalizing with active DNA synthesis at the periphery of the pericentric heterochromatin domains. As in Figure 1C with p150CAF-1 (green), BrdU incorporation after a 10 min pulse (red), and DNA (blue). Scale bar, 1 μm. Bottom: The heterochromatin domain (see arrow above) is magnified four-fold and p150CAF-1 (green), BrdU (red), and DNA (blue) are shown either separately or merged for several rotation angles. The BrdU and DAPI stainings are shown transparently in the merge images. Scale bar, 0.5 μm. (C) In mid-late S-phase, AcH4-K5 and MeH3-K9 (green) were visualized in combination with BrdU incorporation (red) in mid-late S-phase. The arrow indicates typical foci magnified four-fold in the inset. Scale bar is as in (A). (D) Mouse p150CAF-1 and BrdU incorporation (red) were visualized in combination with HP1α (green) in mid-late S-phase. The arrow indicates typical foci magnified four-fold in the inset. Scale bar is as in (A). (E) 3D analysis as in (B) with p150CAF-1 (green) and HP1α (red and transparent).
p150CAF-1, PCNA, and HP1α define a specific architecture for replication foci at pericentric heterochromatin
We then located sites of DNA replication either by BrdU incorporation or by PCNA staining, and detected CAF-1 using a novel antibody raised against the mouse p150CAF-1 protein (Supplementary Figure S1). This antibody revealed that p150CAF-1 colocalized both with BrdU and PCNA in all S-phase profiles in mouse cells as shown in human cell lines (Krude, 1995; Taddei et al, 1999) (Figure 2A and Supplementary Figure S2A). Importantly, closer examination of the mid-late S-phase profiles revealed that BrdU incorporation, PCNA, and p150CAF-1 were associated at the periphery of DAPI dense regions (Figure 2A, insets). A similar pattern was obtained with a GFP-p150CAF-1 fusion (Supplementary Figure S2B), ruling out the possibility that the observed p150CAF-1 staining could result from a restricted accessibility to antibodies within heterochromatin. High-resolution images were collected to perform 3D reconstruction in mid-late S-phase (Figure 2B). Computer modeling of the reconstructed nuclei revealed that p150CAF-1 colocalized with sites of ongoing DNA synthesis (10 min BrdU) at the periphery of DAPI dense regions (Figure 2B) within the resolution limits of light microscope. Rotation of single domains showed that p150CAF-1 staining and sites of DNA synthesis (BrdU) occupy the same volume and form an irregular ribbon in contact with the volume of the DAPI dense domains (Figure 2B). To examine the fate of ubiquituous chromatin proteins in these domains during replication, we chose modified histones such as H4 acetylated on lysine 5 (AcH4-K5) typically found in newly synthesized histones (Taddei et al, 1999) and methylated H3-K9 histones (MeH3-K9), a hallmark of pericentric heterochromatin domain (Peters et al, 2001). Figure 2C shows that AcH4-K5 is detected at sites of active DNA synthesis within the periphery of the domain. In contrast, MeH3-K9 does not colocalize as a ring-like pattern and rather stains the entire core domain (Figure 2C). We then examined the fate of HP1α, one of the three isoforms of HP1 proteins in mammals associated with pericentric regions in various species (Gilbert et al, 2003). In mid-late S-phase, we found a unique organization in which newly synthesized DNA (BrdU), together with PCNA (not shown) and CAF-1, is localized at the periphery of a central domain enriched in HP1α and densely stained with DAPI (Figure 2D). The part of the heterochromatin domain that is subject to disruption during replication seems to be restricted to the periphery, thus explaining how the overall stability could be preserved. High-resolution imaging and analysis (Figure 2E) confirmed that the p150CAF-1 staining tightly fits with the HP1 staining at the periphery of the HP1-enriched domain, within the resolution limit of our set-up. The above observations reveal a sophisticated 3D structure dedicated to the duplication of HP1-rich pericentric heterochromatin.
A distinct pool of HP1α proteins resistant to RNase treatment during replication of heterochromatin
In an asynchronous population, HP1 staining at pericentric heterochromatin domains is lost in the vast majority of cells after RNase treatment (Maison et al, 2002; Muchardt et al, 2002). This change in staining does not result from a loss of HP1 from the nuclei, since nuclear amounts were found to be unmodified (Maison et al, 2002). Rather it reflects a structural reorganization that does not allow HP1 proteins to be maintained at a high local concentration at pericentric heterochromatin, possibly by redistribution to other sites within the nucleus (Maison et al, 2002).
Interestingly, we observed systematically that a few cells (9% with n>500) always retained significant HP1α signal (Figure 3A), even with the highest doses of RNase or the longest incubation times. In these cells, the HP1α staining pattern is significantly altered, indicating that the enzymatic treatment had worked effectively. Instead of a homogenous spherical shaped staining, RNase treatment left a rim of HP1α staining around the DAPI dense heterochromatin domain, reminiscent of the pattern found with p150CAF-1, PCNA, and BrdU in mid-late S-phase (Figure 3A, higher magnification insets). Figure 3B shows that HP1α staining at pericentric heterochromatin domains (in early or late S, and out of S-phase) was lost upon RNase treatment in most cells except for the mid-late S-phase cells positively marked with BrdU (9% of the total cells). Remarkably, the HP1α staining pattern resistant to RNase treatment in mid-late S-phase colocalized both with BrdU and p150CAF-1 in a ring-like structure (Figure 3B and C).
Figure 3.
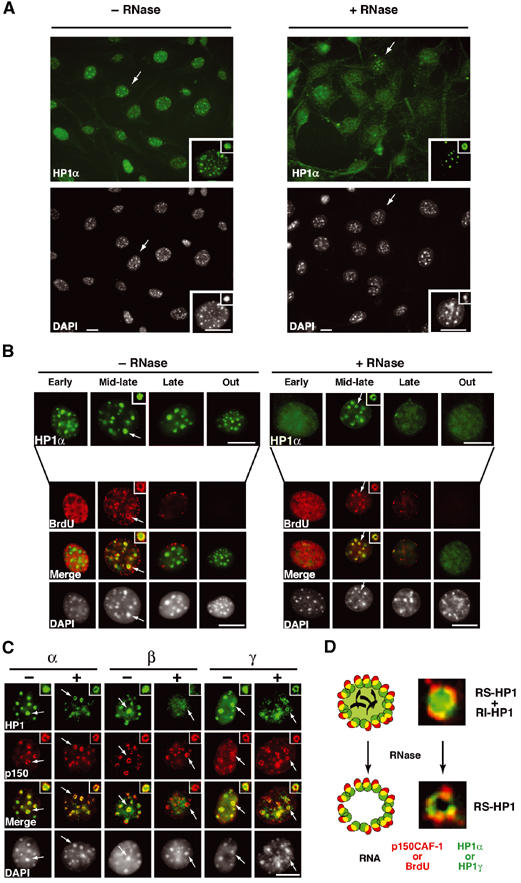
Replication-specific RNase-resistant HP1α and HP1γ but not HP1β pools around pericentric loci colocalizes with p150CAF-1 and BrdU. (A) A subset of cells retain some HP1α staining at pericentric heterochromatin domains following RNase treatment. A wide field showing HP1α labeling (green, top) and DAPI staining (bottom) in 3T3 cells without or with RNase treatment before fixation. The insets show higher magnifications focused on the cell indicated by the arrow as well as on individual foci magnified four-fold. Scale bar, 10 μm. (B) Only cells in mid-late S-phase retain significant HP1α staining as a rim around pericentromeric heterochromatin and sites of DNA synthesis. Double labeling in 3T3 cells of HP1α (green, top isolated) and BrdU (red) in 3T3 cells treated or not with RNase is shown for the early, mid-late, late, and out of S-phase. The arrow indicates typical foci magnified four-fold in the inset. The merge and corresponding DAPI staining are presented. Scale bar as in (A). (C) In mid-late S-phase cells, a fraction of HP1α and HP1γ but not HP1β is RNase-resistant and forms a rim around pericentromeric heterochromatin colocalizing with p150CAF-1. Double labeling of mid-late S-phase 3T3 cells with the three HP1 isoforms (green) and p150CAF-1 (red) without (−) or with (+) RNase treatment. Scale bar and arrows as in (B). (D) Model for HP1α and HP1γ distribution during replication of pericentromeric heterochromatin in mid-late S-phase. Top: Replicating pericentric heterochromatin is represented with a replication-independent pool of HP1 (RI-HP1, large light green circle) in the inner region interacting with RNA (black), and a replication-specific pool of HP1 at the periphery (RS-HP1, small dashed green circles). The location of p150CAF-1 or BrdU incorporation is shown (red circle). Colocalization of the DNA synthesis/p150CAF-1 site and RS-HP1 is represented in yellow. Bottom: Upon RNase treatment, the replication-independent pool of HP1 (RI-HP1) in the inner part is removed following RNA degradation without affecting the replication-specific pool of HP1 (RS-HP1) at the periphery. Corresponding stainings for p150CAF-1 (red) and HP1α (green) are shown as magnified images.
Given that HP1β and HP1γ colocalize with DAPI dense domains, we also examined whether these isoforms of HP1 (HP1β and HP1γ) behaved similarly following RNase treatment. During replication, in control cells, we found that p150CAF-1 localized around both the HP1β and HP1γ isoforms. However, upon RNase treatment, while HP1γ staining, similar to HP1α, was retained and colocalized with p150CAF-1, the HP1β staining at pericentric heterochromatin domains was completely erased (Figure 3C).
A simple interpretation for these observations (Figure 3D) is that, at the time of replication, HP1α and HP1γ molecules behave in a similar manner and are found in two pools: (i) one pool that is RNase-sensitive and replication-independent (RI-HP1), constituting the core domain, and (ii) another pool that is RNase-insensitive, replication-specific (RS-HP1) located at the periphery of the domain. Given the previously reported in vitro interaction between the p150CAF-1 subunit and HP1 proteins (Murzina et al, 1999), we hypothesized that this latter pool of HP1α and HP1γ molecules, but not HP1β, may be maintained through interactions with p150CAF-1. We thus decided to isolate native cellular complexes containing HP1α to verify that p150CAF-1 and specific HP1 molecules physically interact in vivo.
An HP1α/γ–CAF-1 complex that does not contain histones
Although mouse cells represent an ideal model to follow microscopically pericentric heterochromatin domains, human cell systems are better developed for biochemical studies. Given the conservation of the HP1 interaction domain (MIR, MOD1 Interacting Region) in the mouse and human p150CAF-1 (Murzina et al, 1999), we decided to produce a stable HeLa cell line (Nakatani and Ogryzko, 2003) with adequate epitope-tagged proteins to isolate HP1 complexes. An HP1α fusion protein was stably expressed with FLAG and HA epitope tags (e-HP1α) in HeLa cells by retroviral transduction. We could then prepare extracts to purify by affinity the complexes of interest. Although HP1 proteins can be found in several fractions, given our data obtained by immunofluorescence, we chose to focus our attention on nuclear extracts as the most relevant to search for CAF-1 interaction (Figure 4A). Mock purification from untransduced HeLa cells did not yield any polypeptides detectable by silver staining or Western blotting (Figure 4B and not shown). By mass spectrometry and immunoblotting analyses, we identified HP1γ and the three subunits of CAF-1(p150, p60, and p48) associated with HP1α (Figure 4A). Further fractionation of the FLAG-purified complex by glycerol gradient confirmed the presence of CAF-1 in the complex (Figure 4A). Interestingly, the HP1β isoform was not found in this complex, correlating with the fact that only HP1α and HP1γ isoforms showed a replication-specific pool resistant to RNase, RS-HP1 pool (Figure 3C). Similarly, following immunoprecipitation of CAF-1 from a mouse cell extract, we found that HP1α but not HP1β was co-immunoprecipitated with the p150 and p60 subunits (Supplementary Figure S1B). The existence in vivo of such a complex strongly argues that during replication of HP1-rich domains, p150CAF-1–HP1 complexes could ensure the maintenance of a specific replicative pool of HP1 molecules at the sites of pericentric DNA replication.
Figure 4.
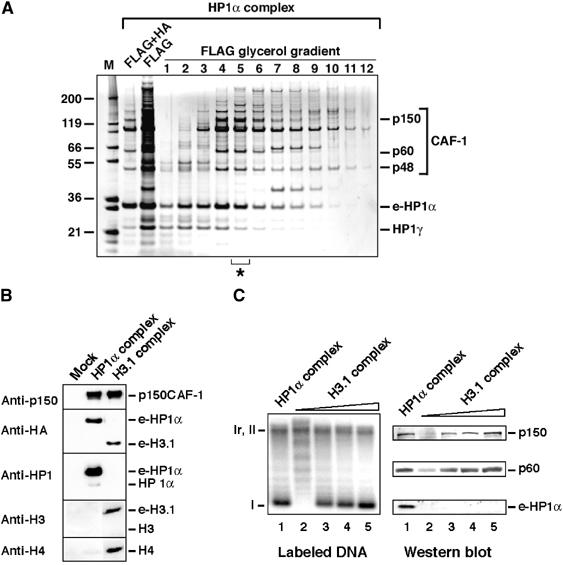
CAF-1 is found in HP1α/γ- and H3.1-containing complexes. (A) Analysis of e-HP1α-containing complexes. SDS–PAGE with fractions purified by FLAG and HA affinity (FLAG+HA), FLAG affinity (FLAG), and from glycerol gradient with the FLAG affinity-purified complex (FLAG glycerol gradient) stained with silver. The asterisk identifies the fraction used in (C). Positions corresponding to CAF-1 p150, p60, p48, HP1γ, and e-HP1α identified by mass spectrometry are indicated. Molecular weight markers (M) are shown. (B) Histones are not detected in the HP1α–CAF-1 complex. Western blot of complexes purified from mock, or e-HP1α- or e-H3.1-transduced HeLa cells. Proteins identified (right) and antibodies used (left) are indicated. (C) Comparison of the HP1α and H3.1 complexes in promoting nucleosome assembly coupled to DNA synthesis. Left: Supercoiling analysis. Cytosolic extract deficient in CAF-1 activity (p150) is used in combination with the HP1α–CAF-1 complex (fraction 5 of the glycerol gradient from (A), lane 1) and increasing amounts of the H3.1–CAF-1 complex (lanes 2–5). The migration of relaxed/nicked (Ir/II) and supercoiled circular DNA (I) is indicated. Right: Western blot analysis with samples corresponding to reactions on the left. Revelation with antibodies is as indicated.
In an independent set of experiments, we observed that CAF-1, which interacts with newly synthesized histones H3 and H4 (Kaufman et al, 1995; Verreault et al, 1996), is also part of a complex that contains the epitope-tagged major histone H3, H3-1 (Tagami et al, 2004) (Figure 4B). We therefore tested whether the HP1α–CAF-1 complex also contains histones. For comparable amounts of p150CAF-1, histones H3 and H4 were not detected in the HP1α–CAF-1 complex, whereas histone H4 (and e-H3.1) was clearly present in the H3.1–CAF-1 complex (Figure 4B). Conversely, HP1α was not detected in the H3.1–CAF-1 complex (Figure 4B). Thus CAF-1 is present in at least two distinct complexes. We then compared the ability of HP1α–CAF-1 and H3.1–CAF-1 complexes to promote nucleosome formation coupled to DNA synthesis. Cytosolic extracts competent for UV-induced DNA repair synthesis necessitate the addition of CAF-1 to ensure nucleosome assembly (Gaillard et al, 1996). Remarkably, when we used the HP1α–CAF-1 complex as the source of CAF-1, chromatin assembly coupled to DNA synthesis was efficiently achieved, as revealed by the accumulation of supercoiled DNA (Figure 4C). By careful titration of the amount of CAF-1, we compared the efficiency of nucleosome formation with both complexes (Figure 4C, Western blot) and found that it correlated directly with the amount of CAF-1 in the complex whatever may be its origin. Thus, the CAF-1 subunits within the HP1α–CAF-1 complex are able to accept and deliver histones. The existence of an HP1α–CAF-1 complex that does not contain histones but yet can accept and deliver them establishes CAF-1 as a good candidate to coordinate nucleosome formation to the maintenance of HP1-rich regions.
A replication-specific pool of HP1α is present at the periphery of pericentric heterochromatin domains in Suv39h double-knockout cells
Since HP1 domains are compromised in cells deficient for the Suv39 histone methyltransferase in a manner that resembles the situation following RNase treatment (Maison et al, 2002), we wondered whether we could find in such cells a replicative pool of HP1. We thus made use of mouse embryonic fibroblast (MEFs) that carried a double-null mutation for the two H3-K9 histone methyltransferases Suv39h1 and Suv39h2 (Suv39h dn) (Peters et al, 2001). Strikingly, we observed a significant HP1α staining at pericentric heterochromatin specifically at mid-late S-phase in Suv39h dn cells (Figure 5, top, −RNase, cells marked by an arrow). Closer examination of these mid-late S-phase Suv39h dn cells revealed that the HP1α staining formed a ring-like structure colocalizing with p150CAF-1 (Figure 5, bottom, −RNase). We thus had a situation that mirrored exactly the one obtained after RNase treatment in control cells. Indeed, in wild-type (WT) control cells, the HP1α staining pattern occupies the entire pericentric heterochromatin domain as found before and harbored a ring-like structure only after RNase treatment (Figure 5, compare bottom, −RNase, +RNase). Taken together, these results show that, although H3-K9 methylation and RNA are required for the stable maintenance of HP1α at sites of pericentric heterochromatin (RI-HP1 pool), they are dispensable for the replicative RS-HP1 pool, which rather depends on p150CAF-1.
Figure 5.
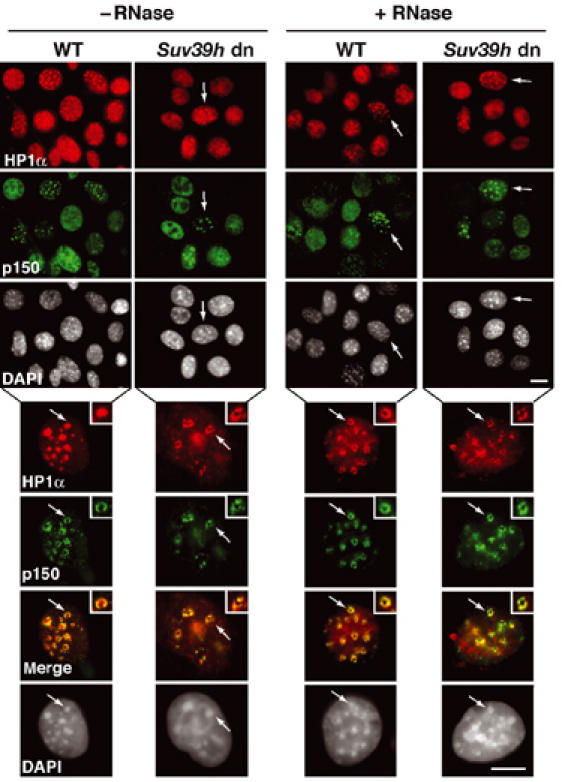
In Suv39h double mutant cells, a replication-specific pool of HP1α is colocalizing with p150CAF-1 at pericentric heterochromatin domains. Top: A wide field showing HP1α labeling (green), and p150CAF-1 (red) and DAPI staining in WT or Suv39h double-null (Suv39h dn) MEFs treated or not by RNase before fixation. The arrows indicate cells in mid-late S-phase based on p150CAF-1 staining. Scale bar, 10 μm. Bottom: As above with a mid-late S-phase nucleus. Merge and DAPI staining are presented. The arrow indicates typical foci magnified three-fold in the inset. Scale bar, 10 μm.
p150CAF-1 knock-down induces the loss of the replication-specific pool of HP1α from mid-late S-phase foci
To determine whether p150CAF-1 is critical for the RS-HP1 pool, we used an siRNA strategy to deplete p150CAF-1 in vivo. We verified knock-down efficiency in our assays (Supplementary Figure S3A). Consistent with previous reports in which p150CAF-1 was altered (Hoek and Stillman, 2003; Ye et al, 2003), we found that cells accumulated in S-phase (Supplementary Figure S3B and we detected only a few mid-late or late S-phase cells as identified by BrdU staining.
We then went on to detect HP1α, DNA synthesis (10 min BrdU pulse), and p150CAF-1 simultaneously in cells following p150CAF-1 knock-down. Cells that were in mid-late S-phase (based on BrdU staining) but p150CAF-1 negative were examined for their HP1α localization. We found HP1α within the pericentric heterochromatin domains in these cells (Figure 6A), but strikingly, upon RNase treatment, all HP1α staining was erased, and no resistant residual HP1α located in a ring-like structure (the RS-HP1 pool) could be seen (Figure 6A). Thus, the RS-HP1α pool, in mid-late S-phase, defined on the basis of RNase-insensitive cells depends on the presence of p150CAF-1. A magnification of the data with an interpretative scheme is shown in Figure 6B. These data argue for a dedicated role of p150CAF-1 in the establishment of a replication-specific pool of HP1 molecules at sites of pericentric heterochromatin replication.
Figure 6.
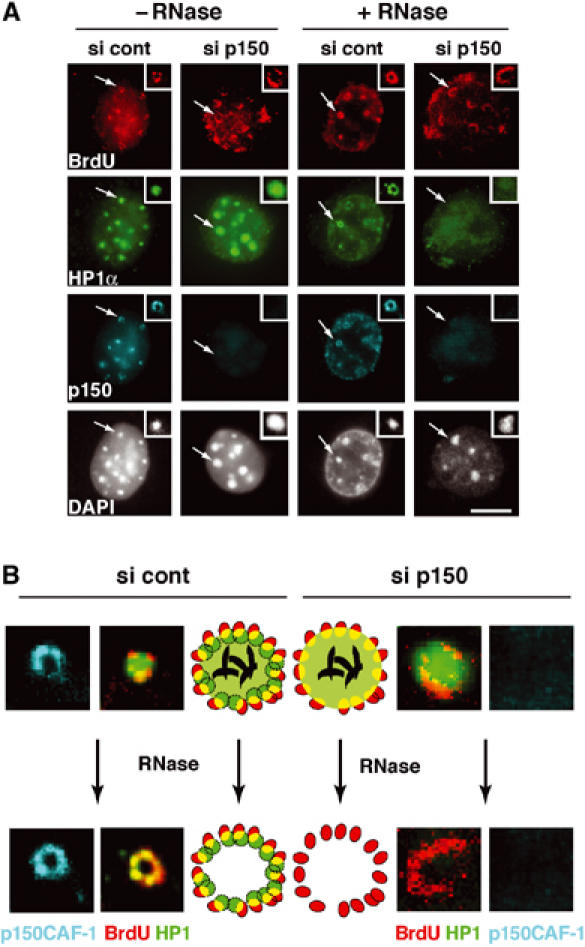
p150CAF-1 knock-down leads to a loss of HP1α at mid-late replication foci. (A) A concordant loss of p150CAF-1 at mid-late S-phase and the HP1 RNase-resistant fraction. Triple immunofluorescence labelings with BrdU (red, 10 min pulse), p150CAF-1 (green), and HP1α (blue) are shown for control or p150CAF-1 siRNA-treated 3T3 cells. Cells with mid-late S-phase profiles for various conditions are shown: nontreated (−RNase, left) or treated by RNase (+RNase, right). The arrow indicates a spot shown at a three-fold magnification in insets. Corresponding DAPI stainings are shown. Scale bar, 10 μm. (B) Model for the role of p150CAF-1 and HP1 localization at sites of DNA synthesis during replication of pericentromeric heterochromatin in mid-late S-phase. Left: In the presence of p150CAF-1 (si cont), replicating pericentric heterochromatin is represented with a replication-independent pool of HP1 (RI-HP1, large light green circle) in the inner region interacting with RNA (black), and a replication-specific pool of HP1 at the periphery (RS-HP1, small dashed green circles) colocalizing with p150CAF-1 (red circles). Colocalization of DNA synthesis and RS-HP1 is represented in yellow. Upon RNase treatment, the RI-HP1 in the inner part is removed following RNA degradation without affecting the RS-HP1 at the periphery. Right: In the absence of p150CAF-1 (si p150), the RS-HP1 (small dashed green circles) at the periphery is lost whereas the RI-HP1 (large light green circle) maintained by RNA (black) remains. Upon RNase treatment, all HP1 is thus lost. Corresponding magnified images from insets of (A) are shown. Stainings are p150CAF-1 (blue), and a merged image of HP1α (green) and BrdU (red).
Discussion
We provide evidence for a unique 3D organization at pericentric heterochromatin domains in mouse cells during replication. Although we cannot formally exclude that alternative models may be envisaged, a simple functional organization can be described in a model where both DNA replication and heterochromatin assembly occur in a single operative unit specific to pericentric heterochromatin that we term the ‘pericentric heterochromatin duplication body' (pHDB) (Figure 7). From our data, we interpret that within a pHDB, parental chromatin is pulled at the periphery, becomes transiently disrupted during replication, and then the coordinated action of the assembly machinery allows both nucleosome formation and HP1 retention/deposition. These data thus support the vision of factories not only for DNA replication but also for higher order duplication. PCNA as a stable component in replication foci (Sporbert et al, 2002) provides the ‘landing pad' for p150CAF-1 (Shibahara and Stillman, 1999; Moggs et al, 2000). In turn, p150CAF-1 would then be used as a critical platform for HP1 (Figure 7). p150CAF-1, as a component of the replicative factories, would be key to maintaining the replication-specific pool of HP1 molecules. Such a pool would ensure the self-perpetuation of the pre-existing structure. This pool is strictly dependent on CAF-1 (Figure 6) and does not require RNA (Figure 3) or Suv39h mediated H3-K9 methylation (Figure 5). Indeed, loss of CAF-1 leads to a loss of this replication-specific stable HP1 subset. In addition, CAF-1 could also participate in the transfer of HP1 onto newly replicated material (Murzina et al, 1999). Importantly, we found that HP1α–CAF-1 and H3.1–CAF-1 complexes are distinct entities, yet both of them efficiently complement cytosolic extracts to promote nucleosome assembly (Figure 4). This clearly indicates the ability of CAF-1 to dynamically exchange/accept partners to form two types of complexes. Such dynamic properties of CAF-1 would be adapted for a dual function of CAF-1 to accept and deliver either histones or HP1 as proposed in our model. Furthermore, a continuous supply of new HP1 molecules required to maintain a constant HP1/chromatin ratio could be provided by exchange with soluble CAF-1–HP1 complex as those found in our nuclear extracts.
Figure 7.
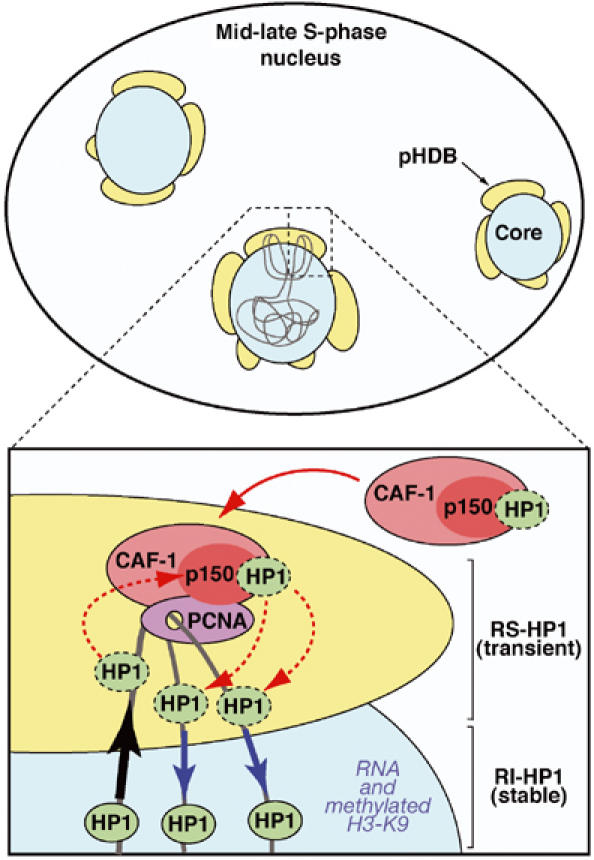
Hypothetical model for duplication of pericentric heterochromatin domains. Top: Scheme of a mouse cell nucleus in mid-late S-phase during replication of pericentric heterochromatin. A central core domain (blue) is surrounded by pHDB (yellow). Chromatin drawn in gray contains two forks emanating from one replication origin. Active synthesis takes place within this pHDB. Bottom: A model for a functional pHDB. For simplification, chromatin (gray) from only one fork is represented with one PCNA complex and no distinction is made between leading or lagging strand. The parental chromatin is pulled at the periphery (black arrow) to get replicated in pHDB (yellow). At the replication fork, PCNA (purple) targets p150CAF-1 (red) within the CAF-1 complex (pink), which could accept/transfer HP1 at the site of DNA synthesis and chromatin assembly (red dashed line). Constant HP1/chromatin ratio would be ensured by exchange with soluble CAF-1–HP1 complex (red solid arrow). Thereby a transient pool of replication-specific HP1 (RS-HP1, green, dashed line) localizes within pHDB. Synthesized and assembled daughter DNA is then pushed back inside the domain (blue arrows). The HP1 molecules from the parental chromatin can redistribute onto the newly replicated/assembled DNA (red dashed arrows) and would be stabilized in the inner domain as a stable pool of replication-independent HP1 (RI-HP1, green, solid line) by RNA and methylated H3-K9 as indicated.
Although some assembly may occur within the interior of the domain, an attractive hypothesis based on our results would be that coordinated with DNA synthesis newly synthesized histones together with parental histones should be incorporated in a peripheral compartment. Then, HP1 association would follow. It is noteworthy that HP1 can bind to the Suv39h histone H3 methyltransferase (Aagaard et al, 1999); thus, a controlled transfer of HP1 may also help to attract the necessary enzyme to modify newly incorporated histones. The daughter molecules are then pushed back inside the domain where both RNA and pre-existing H3-K9 methylation could participate in stabilizing a dynamic HP1 domain to form the replication-independent pool (Cheutin et al, 2003; Festenstein et al, 2003). This overall mechanism would permit to reproduce faithfully the epigenetic state of the domain.
Interestingly, we did not identify HP1β in the replication-specific subset of HP1 by immunofluorescence nor in the HP1α–CAF-1 complex, although it is detected within the pericentric heterochromatin domain in the replication-independent pool. This may indicate that among the three isoforms only HP1α and HP1γ are involved in replication-dependent dynamics through p150CAF-1 in vivo. Thus the HP1β isoform is likely to integrate the domain by other means, independently of the replication event per se. This is entirely consistent with the fact that targeting HP1β (MOD1) to heterochromatin does not require heterochromatin replication (Murzina et al, 1999). In addition, the binding of Orc2 to HP1α and HP1β, recently uncovered, may also contribute to their location (Prasanth et al, 2004).
Although for simplification, we have not represented the histones in our scheme (Figure 7), their importance should not be overlooked, especially since their specific modification is important for HP1 binding (Bannister et al, 2001; Lachner et al, 2001). It will thus be important to understand how methylation on H3-K9 can be imposed, whether it is on a hypothetic dimeric intermediate as recently proposed (Tagami et al, 2004) or on a nucleosomal template, and how this is coordinated with acetylation/deacetylation cycle on histone H4-K5/12.
Taken together, our data support an integrated model in which CAF-1, as a key component, uses its dual binding properties to HP1 and histones as a specific requirement for heterochromatin duplication. Most importantly, the specific organization established at the mid-late S-phase transition has further implications for genome stability. It facilitates the confinement of heterochromatic domains to avoid aberrant spreading of HP1-associated silencing. In addition, this architecture may also help to minimize recombinational events between repetitive sequences by restricting the extent to which these regions might interact. The concept of specific entities adapted for the replication of complex architectures could also apply to the duplication of other specialized chromatin domains (Palstra et al, 2003). It would be particularly interesting to find out whether chromocenters in Drosophila cells make use of similar mechanisms for their duplication. Whether this is specific to constitutive heterochromatin or facultative as exemplified by the inactive X chromosome in female mammals and developmentally regulated heterochromatin domains will be a future challenge.
Materials and methods
Purification of FLAG–HA-tagged complexes
Stable HeLa cell lines expressing FLAG–HA-tagged HP1α (e-HP1α) or FLAG–HA tagged H3.1 (e-H3.1) (Tagami et al, 2004) were established (Nakatani and Ogryzko, 2003). e-HP1α-containing complexes were used after immunopurification or an additional purification on a 10–40% glycerol gradient again followed by a second immunopurification. Proteins were identified by mass spectrometry and Western blotting.
In vitro nucleosome assembly
Nucleosome assembly coupled to DNA synthesis during nucleotide excision repair (NER) was performed with UV-irradiated plasmid (Gaillard et al, 1996) incubated in HeLa cytosolic extracts (Martini et al, 1998) and [α-32P]dCTP at 37°C for 3 h in the presence of various sources of CAF-1. DNA synthesis and supercoiling were analyzed after DNA purification by agarose gel electrophoresis and labeled DNA was visualized by autoradiography.
Cell synchronization, siRNA treatment, and in situ immuno-biochemical assays
Cells were grown in DMEM (Gibco BRL) with 10% fetal calf serum (FCS) and antibiotics in 5% CO2. They were transfected with 20 nM siRNA duplex (Harborth et al, 2001) and harvested 72 h post-transfection. For immunofluorescence with the p150CAF-1 antibody, cells were extracted with the nonionic detergent Triton X-100 to remove soluble proteins (Martini et al, 1998). Immunodetection and RNase treatment were performed as described (Maison et al, 2002). Methanol fixation was used for PCNA staining. IdU and CldU (10 μM) were added to the cell medium for 5 min.
Image acquisition, 3D reconstruction and modeling, and colocalization
We used a DMR-HC (Leica) epifluorescence microscope with a × 63 objective and a CCD camera (CoolSnap Fx, Photometrics) the resolution limit of which is about 200 nm. For 3D reconstruction, using a DMIRE 2 (Leica) epifluorescence photo-microscope with a DG4 illumination device (Sutter Instruments), a × 100 objective and a piezoelectric objective positioning device (PIFOC, Physics instruments), stacks of images (200 nm step) were recorded using a chilled CCD camera (CoolSnap HQ, Photometrics) and deconvolved with Metamorph software (Universal Imaging). 3D modeling of nuclei was achieved by building a 3D texture from an iso-surface reconstruction of the deconvolved stack of images with the Amira software (TGS, www.tgs.com). At least 1000 nuclei were examined and representative images are shown.
Antibodies
For immunofluorescence, we used the following antibodies: anti-HP1α 2HP1H5 (Euromedex), anti-HP1β 1MOD1A9 (Euromedex), and anti-HP1γ 2MOD1G6 (Euromedex), anti-PCNA PC10 (DAKO), anti-mouse p150CAF-1 from rabbit serum immunized with recombinant mouse p150 (AgroBio), anti-tri-Me H3-K9 (Abcam), anti-AcH4-K5 (Turner et al, 1989), and secondary antibodies coupled to Texas red, FITC, Cy-3, and Cy-5 (Jackson Immuno Research Laboratories Inc.). After 10 min denaturation in 4 M HCl, the following antibodies were used: anti-BrdU OBT0030 (AbCys) to detect BrdU, anti-BrdU supernatant OBT0030S (AbCys) for CldU, anti-BrdU (clone B44, Becton Dickinson) for IdU. For Western blots, we used anti-human p150CAF-1 H300 (Santa Cruz) or anti-human p150CAF-1 Ab765 (Abcam), anti-human p60CAF-1 (Agrobio), anti-HA tag 12CA5 (Roche), anti-H3 06-0755, anti-H4 07-108, and anti-HP1α 07-333 (Upstate).
siRNA synthesis
siRNA duplexes were constructed with the Silencer siRNA construction kit (Ambion) using phenol/chloroform and a G-25 spin column RNA grade (Roche) for purification. The sequence of p150CAF-1 duplex was 5′AAGGAGAAGGCGGAGAAGCAG3′ and 5′AAGCTGGAGTACAACTACAACCCTGTCTC3′ for GFP control duplex.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Acknowledgments
We thank members of UMR218 for discussions, P Le Baccon and S Huart for 3D reconstruction, S Ross for the mouse p150 antibody, D Rouillard for FACS analysis, F Sangrado for technical help, A Verreault for recombinant CAF-1 and mouse p150 cDNA, T Jenuwein for the Suv39h double null MEFs, B Turner for AcH4-K5 antibodies, M Guenatri for early observations, and E Heard, P Hanawalt, and PA Defossez for critical reading. DK was supported by a European network grant to GA. (Disclaimer: the European Commission is not responsible for any views or results expressed.) GA is supported by la Ligue Nationale contre le Cancer (Equipe labellisée la Ligue), Euratom (FIGH-CT-1999-00010, FIGH-CT-2002-00207), Commissariat à l'Energie Atomique (LRC #. 26), and Curie Program on epigenetic parameters and YN by grants from NIH (GM065939-02).
References
- Aagaard L, Laible G, Selenko P, Schmid M, Dorn R, Schotta G, Kuhfittig S, Wolf A, Lebersorger A, Singh PB, Reuter G, Jenuwein T (1999) Functional mammalian homologues of the Drosophila PEV-modifier Su(var)3-9 encode centromere-associated proteins which complex with the heterochromatin component M31. EMBO J 18: 1923–1938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aten JA, Bakker PJ, Stap J, Boschman GA, Veenhof CH (1992) DNA double labelling with IdUrd and CldUrd for spatial and temporal analysis of cell proliferation and DNA replication. Histochem J 24: 251–259 [DOI] [PubMed] [Google Scholar]
- Bannister AJ, Zegerman P, Partridge JF, Miska EA, Thomas JO, Allshire RC, Kouzarides T (2001) Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature 410: 120–124 [DOI] [PubMed] [Google Scholar]
- Cheutin T, McNairn AJ, Jenuwein T, Gilbert DM, Singh PB, Misteli T (2003) Maintenance of stable heterochromatin domains by dynamic HP1 binding. Science 299: 721–725 [DOI] [PubMed] [Google Scholar]
- Cook PR (1999) The organization of replication and transcription. Science 284: 1790–1795 [DOI] [PubMed] [Google Scholar]
- Dimitrova DS, Berezney R (2002) The spatio-temporal organization of DNA replication sites is identical in primary, immortalized and transformed mammalian cells. J Cell Sci 115: 4037–4051 [DOI] [PubMed] [Google Scholar]
- Dimitrova DS, Gilbert DM (2000) Temporally coordinated assembly and disassembly of replication factories in the absence of DNA synthesis. Nat Cell Biol 2: 686–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekwall K, Olsson T, Turner BM, Cranston G, Allshire RC (1997) Transient inhibition of histone deacetylation alters the structural and functional imprint at fission yeast centromeres. Cell 91: 1021–1032 [DOI] [PubMed] [Google Scholar]
- Enomoto S, McCune-Zierath PD, Gerami-Nejad M, Sanders MA, Berman J (1997) RLF2, a subunit of yeast chromatin assembly factor-I, is required for telomeric chromatin function in vivo. Genes Dev 11: 358–370 [DOI] [PubMed] [Google Scholar]
- Festenstein R, Pagakis SN, Hiragami K, Lyon D, Verreault A, Sekkali B, Kioussis D (2003) Modulation of heterochromatin protein 1 dynamics in primary mammalian cells. Science 299: 719–721 [DOI] [PubMed] [Google Scholar]
- Fox MH, Arndt-Jovin DJ, Jovin TM, Baumann PH, Robert-Nicoud M (1991) Spatial and temporal distribution of DNA replication sites localized by immunofluorescence and confocal microscopy in mouse fibroblasts. J Cell Sci 99: 247–253 [DOI] [PubMed] [Google Scholar]
- Gaillard PH, Martini EM, Kaufman PD, Stillman B, Moustacchi E, Almouzni G (1996) Chromatin assembly coupled to DNA repair: a new role for chromatin assembly factor I. Cell 86: 887–896 [DOI] [PubMed] [Google Scholar]
- Gilbert N, Boyle S, Sutherland H, de Las Heras J, Allan J, Jenuwein T, Bickmore WA (2003) Formation of facultative heterochromatin in the absence of HP1. EMBO J 22: 5540–5550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grewal SI, Moazed D (2003) Heterochromatin and epigenetic control of gene expression. Science 301: 798–802 [DOI] [PubMed] [Google Scholar]
- Gruss C, Wu J, Koller T, Sogo JM (1993) Disruption of the nucleosomes at the replication fork. EMBO J 12: 4533–4545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harborth J, Elbashir SM, Bechert K, Tuschl T, Weber K (2001) Identification of essential genes in cultured mammalian cells using small interfering RNAs. J Cell Sci 114: 4557–4565 [DOI] [PubMed] [Google Scholar]
- Hoek M, Stillman B (2003) Chromatin assembly factor 1 is essential and couples chromatin assembly to DNA replication in vivo. Proc Natl Acad Sci USA 100: 12183–12188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeppesen P, Mitchell A, Turner B, Perry P (1992) Antibodies to defined histone epitopes reveal variations in chromatin conformation and underacetylation of centric heterochromatin in human metaphase chromosomes. Chromosoma 101: 322–332 [DOI] [PubMed] [Google Scholar]
- Kaufman PD, Kobayashi R, Kessler N, Stillman B (1995) The p150 and p60 subunits of chromatin assembly factor I: a molecular link between newly synthesized histones and DNA replication. Cell 81: 1105–1114 [DOI] [PubMed] [Google Scholar]
- Krude T (1995) Chromatin assembly factor 1 (CAF-1) colocalizes with replication foci in HeLa cell nuclei. Exp Cell Res 220: 304–311 [DOI] [PubMed] [Google Scholar]
- Lachner M, O'Carroll D, Rea S, Mechtler K, Jenuwein T (2001) Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature 410: 116–120 [DOI] [PubMed] [Google Scholar]
- Maison C, Bailly D, Peters AH, Quivy JP, Roche D, Taddei A, Lachner M, Jenuwein T, Almouzni G (2002) Higher-order structure in pericentric heterochromatin involves a distinct pattern of histone modification and an RNA component. Nat Genet 30: 329–334 [DOI] [PubMed] [Google Scholar]
- Martini E, Roche DM, Marheineke K, Verreault A, Almouzni G (1998) Recruitment of phosphorylated chromatin assembly factor 1 to chromatin after UV irradiation of human cells. J Cell Biol 143: 563–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minc E, Courvalin JC, Buendia B (2000) HP1gamma associates with euchromatin and heterochromatin in mammalian nuclei and chromosomes. Cytogenet Cell Genet 90: 279–284 [DOI] [PubMed] [Google Scholar]
- Moggs JG, Grandi P, Quivy JP, Jonsson ZO, Hubscher U, Becker PB, Almouzni G (2000) A CAF-1–PCNA-mediated chromatin assembly pathway triggered by sensing DNA damage. Mol Cell Biol 20: 1206–1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muchardt C, Guilleme M, Seeler JS, Trouche D, Dejean A, Yaniv M (2002) Coordinated methyl and RNA binding is required for heterochromatin localization of mammalian HP1alpha. EMBO Rep 3: 975–981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murzina N, Verreault A, Laue E, Stillman B (1999) Heterochromatin dynamics in mouse cells: interaction between chromatin assembly factor 1 and HP1 proteins. Mol Cell 4: 529–540 [DOI] [PubMed] [Google Scholar]
- Nakamura H, Morita T, Sato C (1986) Structural organizations of replicon domains during DNA synthetic phase in the mammalian nucleus. Exp Cell Res 165: 291–297 [DOI] [PubMed] [Google Scholar]
- Nakatani Y, Ogryzko V (2003) Immunoaffinity purification of mammalian protein complexes. Methods Enzymol 370: 430–444 [DOI] [PubMed] [Google Scholar]
- Nakayama J, Rice JC, Strahl BD, Allis CD, Grewal SI (2001) Role of histone H3 lysine 9 methylation in epigenetic control of heterochromatin assembly. Science 292: 110–113 [DOI] [PubMed] [Google Scholar]
- Nielsen AL, Oulad-Abdelghani M, Ortiz JA, Remboutsika E, Chambon P, Losson R (2001) Heterochromatin formation in mammalian cells: interaction between histones and HP1 proteins. Mol Cell 7: 729–739 [DOI] [PubMed] [Google Scholar]
- O'Keefe RT, Henderson SC, Spector DL (1992) Dynamic organization of DNA replication in mammalian cell nuclei: spatially and temporally defined replication of chromosome-specific alpha-satellite DNA sequences. J Cell Biol 116: 1095–1110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palstra RJ, Tolhuis B, Splinter E, Nijmeijer R, Grosveld F, de Laat W (2003) The beta-globin nuclear compartment in development and erythroid differentiation. Nat Genet 35: 190–194, Epub 2003 Sep 2021 [DOI] [PubMed] [Google Scholar]
- Peters AH, O'Carroll D, Scherthan H, Mechtler K, Sauer S, Schofer C, Weipoltshammer K, Pagani M, Lachner M, Kohlmaier A, Opravil S, Doyle M, Sibilia M, Jenuwein T (2001) Loss of the Suv39h histone methyltransferases impairs mammalian heterochromatin and genome stability. Cell 107: 323–337 [DOI] [PubMed] [Google Scholar]
- Prasanth SG, Prasanth KV, Siddiqui K, Spector DL, Stillman B (2004) Human Orc2 localizes to centrosomes, centromeres and heterochromatin during chromosome inheritance. EMBO J 23: 2651–2663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randall SK, Kelly TJ (1992) The fate of parental nucleosomes during SV40 DNA replication. J Biol Chem 267: 14259–14265 [PubMed] [Google Scholar]
- Rea S, Eisenhaber F, O'Carroll D, Strahl BD, Sun ZW, Schmid M, Opravil S, Mechtler K, Ponting CP, Allis CD, Jenuwein T (2000) Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature 406: 593–599 [DOI] [PubMed] [Google Scholar]
- Shibahara K, Stillman B (1999) Replication-dependent marking of DNA by PCNA facilitates CAF-1-coupled inheritance of chromatin. Cell 96: 575–585 [DOI] [PubMed] [Google Scholar]
- Smith S, Stillman B (1989) Purification and characterization of CAF-I, a human cell factor required for chromatin assembly during DNA replication in vitro. Cell 58: 15–25 [DOI] [PubMed] [Google Scholar]
- Sporbert A, Gahl A, Ankerhold R, Leonhardt H, Cardoso MC (2002) DNA polymerase clamp shows little turnover at established replication sites but sequential de novo assembly at adjacent origin clusters. Mol Cell 10: 1355–1365 [DOI] [PubMed] [Google Scholar]
- Strahl BD, Allis CD (2000) The language of covalent histone modifications. Nature 403: 41–45 [DOI] [PubMed] [Google Scholar]
- Taddei A, Maison C, Roche D, Almouzni G (2001) Reversible disruption of pericentric heterochromatin and centromere function by inhibiting deacetylases. Nat Cell Biol 3: 114–120 [DOI] [PubMed] [Google Scholar]
- Taddei A, Roche D, Sibarita JB, Turner BM, Almouzni G (1999) Duplication and maintenance of heterochromatin domains. J Cell Biol 147: 1153–1166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagami H, Ray-Gallet D, Almouzni G, Nakatani Y (2004) Histone H3.1 and H3.3 complexes mediate nucleosome assembly pathways dependent or independent of DNA synthesis. Cell 116: 51–61 [DOI] [PubMed] [Google Scholar]
- Turner BM, O'Neill LP, Allan IM (1989) Histone H4 acetylation in human cells. Frequency of acetylation at different sites defined by immunolabeling with site-specific antibodies. FEBS Lett 253: 141–145 [DOI] [PubMed] [Google Scholar]
- Vermaak D, Ahmad K, Henikoff S (2003) Maintenance of chromatin states: an open-and-shut case. Curr Opin Cell Biol 15: 266–274 [DOI] [PubMed] [Google Scholar]
- Verreault A (2000) De novo nucleosome assembly: new pieces in an old puzzle. Genes Dev 14: 1430–1438 [PubMed] [Google Scholar]
- Verreault A, Kaufman PD, Kobayashi R, Stillman B (1996) Nucleosome assembly by a complex of CAF-1 and acetylated histones H3/H4. Cell 87: 95–104 [DOI] [PubMed] [Google Scholar]
- Ye X, Franco AA, Santos H, Nelson DM, Kaufman PD, Adams PD (2003) Defective S phase chromatin assembly causes DNA damage, activation of the S phase checkpoint, and S phase arrest. Mol Cell 11: 341–351 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3