

SUMMARY
The recent Zika virus (ZIKV) outbreak in Brazil has been directly linked to increased cases of microcephaly in newborns. Current evidence indicates that ZIKV is transmitted vertically from mother to fetus. However, the mechanism of intrauterine transmission and the cell types involved remain unknown. We demonstrate that the contemporary ZIKV strain PRVABC59 (PR 2015) infects and replicates in primary human placental macrophages, called Hofbauer cells, and to a lesser extent in cytotrophoblasts, isolated from villous tissue of full-term placentae. Viral replication coincides with induction of type I interferon (IFN), pro-inflammatory cytokines, and antiviral gene expression, but with minimal cell death. Our results suggest a mechanism for intra-uterine transmission in which ZIKV gains access to the fetal compartment by directly infecting placental cells and disrupting the placental barrier.
In Brief
The currently circulating Zika virus strain is directly linked to fetal microcephaly. However, the mechanism of intrauterine ZIKV transmission is unknown. Quicke et al. demonstrate that a contemporary ZIKV strain infects and replicates in primary human placental macrophages and cytotrophoblasts, suggesting a route for ZIKV to cross the placental barrier.
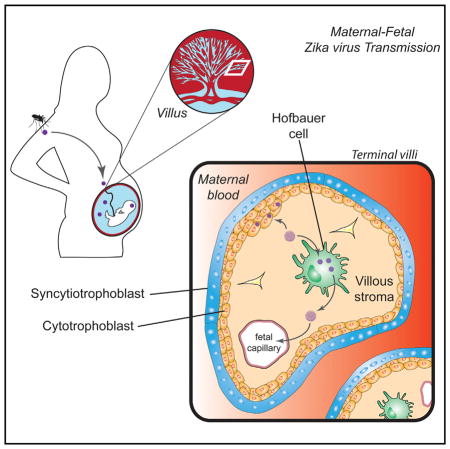
INTRODUCTION
Zika virus (ZIKV) is an emerging mosquito-borne flavivirus that has rapidly spread to over 30 countries in the Americas and causes illness with symptoms of fever, rash, joint pain, and conjunctivitis (Lazear and Diamond, 2016; Petersen et al., 2016). ZIKV is transmitted through several routes, including mosquito bites, sexual contact, and blood transfusion (Lazear and Diamond, 2016). Most notably, ZIKV can be vertically transmitted from an infected mother to the developing fetus in utero, resulting in adverse pregnancy outcomes that include fetal brain abnormalities and microcephaly, a condition characterized by a reduction in head circumference that is often associated with delayed or arrested brain development (Rasmussen et al., 2016). The mechanism by which ZIKV crosses the placenta to establish infection in the developing fetus is not well understood. Recent studies have identified ZIKV RNA in amniotic fluid and fetal and newborn brain tissue (Calvet et al., 2016; Driggers et al., 2016; Martines et al., 2016), and ZIKV-specific IgM antibodies have been detected in newborn cerebrospinal fluid (Cordeiro et al., 2016). Additionally, ZIKV antigen was found in the chronic villi of a human placenta from a mother who gave birth to an infant with microcephaly, and ZIKV RNA has been isolated from placental tissue of mice infected with ZIKV (Miner et al., 2016; Martines et al., 2016). Finally, a recent study detected ZIKV antigen in placental tissue from a mother diagnosed with ZIKV disease (Noronha et al., 2016). In particular, ZIKV antigen was detected in placental macrophages and histiocytes in the intervillous space.
Vertical transmission of ZIKV from an infected mother to the developing fetus in utero reflects tropism for placental cells. This organ is a target for a number of viruses by direct and contiguous infection of the cell layers, virion passage through a breach, or cell-associated transport. Examples include rubella, cytomegalovirus, herpes simplex, HIV-1, hepatitis B and C virus, and parvovirus B19 (Koi et al., 2001). The placenta is characterized by contact between the maternal blood and fetal chorionic villi. Each villus is lined by trophoblasts, which encase the fetal blood supply and placental macrophages (Hofbauer cells [HCs]). Several studies have confirmed HCs are targets of viral infection in vivo (Lewis et al., 1990) and in vitro (Johnson and Chakraborty, 2012). In contrast, syncytiotrophoblasts (differentiated cytotrophoblasts [CTBs]) have been shown to be resistant to infection by a wide range of viruses (Delorme-Axford et al., 2013). A recent study showed that syncytiotrophoblasts also appear to be resistant to infection by phylogenetically related, historic ZIKV strains at early times following infection (24 and 48 hr post-infection [hpi]) (Bayer et al., 2016).
Here we demonstrate that primary human HCs, and to a lesser extent CTBs, are permissive to productive infection by a contemporary strain of ZIKV, closely related to the strains currently circulating in Brazil. Upon infection, HCs are modestly activated and produce IFN-α and other pro-inflammatory cytokines. Analysis of antiviral gene expression shows upregulation of retinoic acid-inducible gene I (RIG-I)-like receptor (RLR) transcription as well as downstream antiviral effector genes, indicating that ZIKV induces an antiviral response in HCs and CTBs. Our results suggest that ZIKV gains access to the fetal compartment by infecting and proliferating in the cells of the placenta.
RESULTS
HCs and CTBs Are Permissive to Productive ZIKV Infection
To determine whether human placental cells are permissive to ZIKV infection, we isolated primary HCs and CTBs from villous tissue of full-term placentae and infected them with ZIKV (MOI 1). In this study, we used a low cell-culture-passaged and sequence-verified ZIKV strain, PRVABC59 (PR 2015), isolated from the sera of an infected patient in Puerto Rico in December 2015. This strain is closely related to the epidemic strains circulating in the Americas that have been linked to in utero ZIKV infection (Faria et al., 2016). Through multiple virologic assays, we demonstrate that HCs, and to a lesser extent CTBs, are permissive to productive ZIKV infection (Figure 1). Following infection of HCs, we performed a focus forming assay (FFA) on Vero cells and observed a steady decline in viral titers from 3 hpi through 24 hpi that was immediately followed by log phase virus growth through 72 hpi (Figure 1A). Notably, we observed donor-to-donor variation in viral kinetics and magnitude among HCs isolated from five donors. For donor 2, we detected an approximately 35-fold increase in virus in the supernatant between 3 and 48 hpi. In contrast, donor 5 showed about a 2.5-fold increase in virus in the supernatant between 48 and 96 hpi. We confirmed infection of HCs with viral qRT-PCR (Figure 1B) and immunofluorescence microscopy (Figures 1C–1E). In HCs, viral RNA was substantially increased in all donors by 48 or 72 hpi, reflecting an increase in virus release into the supernatant (Figure 1A). Furthermore, we detected viral envelope (E) protein, which localized to distinct, perinuclear regions within infected HCs (Figures 1C and 1D). This pattern may be indicative of viral localization to the endoplasmic reticulum (ER), or ER-associated vesicles, a staining pattern consistent with virus assembly (Welsch et al., 2009). Finally, we observed between 4.9% and 7.2% infected cells by immunofluorescence staining using a pan-flavivirus antibody (Figure 1E).
Figure 1. Hofbauer Cells Are Permissive to ZIKV Infection.

(A) HCs from five donors were infected with ZIKV (PR 2015) at an MOI of 1, and viral titers in supernatants determined by FFA. Viral inoculum for all donors was 1 × 106 ffu/mL. Data are represented as the mean of four technical replicates ± SD (top). Representative FFA staining (bottom). ffu, focus forming units.
(B) Viral RNA detected by qRT-PCR in HCs infected with ZIKV (PR 2015). Data are relative to GAPDH control and mock-infected cells (ΔΔCT).
(C) Confocal microscopy of mock- and ZIKV (PR 2015)-infected HCs at 72 hpi.
(D) 3D reconstruction of confocal images.
(E) Percent infected cells determined from five fields of view.
Data are represented as mean ± SD. See also Figure S2.
In contrast, we observed minimal viral replication in CTBs at early times post-infection (3–72 hpi; Figure S1A, available online). Of note, we found evidence of productive infection at 96 hpi, with all three donors exhibiting approximately 5-fold increase in viral load between 72 and 96 hpi, suggesting that CTBs may support productive virus infection, albeit at lower levels compared to HCs. We observed concurrent increases in viral RNA in all three donors between 72 and 96 hpi as well (Figure S1B). Most notably, we detected persistent viral RNA in CTBs at all time points through 72 hpi, further suggesting ZIKV infects and replicates in CTBs with delayed kinetics. Collectively, these findings demonstrate that HCs are permissive to ZIKV infection and represent a key target cell of ZIKV infection within the placenta.
To assess ZIKV replication in HCs at the single-cell level, flow cytometry was utilized to detect intracellular expression of viral E protein. Consistent with peak production of viral RNA and infectious virus (Figure 1), we detected 0.8%–6.8% and 0.4%–3.0% infected HCs at 48 and 72 hpi, respectively (Figure 2A). Minimal background staining was observed in donor- and time-matched uninfected cells and in ZIKV-infected cells stained with an IgG isotype control (Figure S2B). Consistent with our FFA findings, HCs isolated from donor 2 were the most permissive to infection, with an average of 5.6% and 2.3% infected cells at 48 and 72 hpi, respectively. This is consistent with infected cell counts observed by immunofluorescence microscopy (Figure 1E). In contrast to recent studies with neuronal progenitor cells (Garcez et al., 2016; Tang et al., 2016), we did not observe a significant loss of viability during ZIKV infection through 96 hpi (Figure S2C), suggesting that these cells may be more resistant to virus-induced cell death or that ZIKV (PR 2015) is a less cytopathic virus in HCs.
Figure 2. ZIKV Infection Induces Activation of HCs.

(A) HCs from three donors were infected with ZIKV (PR 2015) at an MOI of 1, or mock infected. Percentages of infected cells at 48 and 72 hpi were determined by intracellular viral E protein staining and flow cytometry (left panels). Horizontal bars indicate the mean of four technical replicates.
(B–D) Surface expression of (B) CD80, (C) CD86, and (D) MHC II was determined by flow cytometry. Data are represented as median fluorescence intensity (MFI). Horizontal bars indicate the mean of four technical replicates. Representative histograms are provided (right panels). hpi, hours post-infection. See also Figure S2.
Of note, percent infectivity and infectious virus production did not necessarily correspond to viral RNA levels (Figures 1 and 2A). Specifically, while donors 1 and 2 had a 6-fold difference in cellular infectivity at 48 hpi and a consistent 1-log-fold difference in infectious virus release between 24 and 96 hpi, both had similar viral RNA levels present at 48 and 72 hpi. Differences in infection between donor 1 and 2 may be explained by an enhanced rate of genome replication within HCs from donor 2, noted by an early increase in viral RNA at 24 hpi in donor 2, but not donor 1 (Figure 1). Overall, we observed variable levels of viral RNA at 24 and 48 hpi, despite similar levels of viral RNA at early (3 hpi) and late (48 and 72 hpi) time points, further supporting differential rates of genome replication between donors. Indeed, while donors 1, 3, and 4 had similar production of infectious virus at all time points assessed, notable differences in viral RNA levels were observed at 48 hpi between these donors (Figure 1). Furthermore, while donor 5 showed minimal production of infectious virus, we observed comparable RNA levels to the more permissive donors, further highlighting discordance between genome replication and release of infectious virus. Together, these results suggest that different donors may have the capacity to differentially regulate ZIKV replication and may be restricting replication at different stages of the viral life cycle.
ZIKV Infection Induces Modest Activation of HCs
Next, to determine if ZIKV-infected HCs are poised to interact with T cells, we measured cell surface expression of the co-stimulatory molecules CD80, CD86, and MHC II. In ZIKV-infected HCs from all three donors, we observed minimal upregulation of both CD80 and CD86 as compared to time-matched, mock-infected cells between 48 and 72 hpi (Figures 2B and 2C). Consistent with enhanced virus replication, ZIKV infection of HCs from donor 2 led to upregulation of both CD80 and CD86 by 72 hpi. Additionally, significant upregulation of MHC II was only observed with donor 2 between 48 and 72 hpi (Figure 2D). Overall, there appears to be donor-to-donor variability in terms of upregulation of co-stimulatory molecules; however, enhanced virus replication led to greater activation of HCs. These data suggest that ZIKV infection has the potential to program HCs for antigen presentation and T cell priming.
Type I IFN and Pro-inflammatory Cytokines Are Produced in Response to ZIKV Infection
When cells are infected with virus, pattern recognition receptors (PRRs) within the cell recognize the viral genetic material and trigger a potent innate immune response to control viral replication and spread. Upon binding viral RNA, PRRs initiate signaling cascades that result in the production of type I interferons (IFNs) and pro-inflammatory cytokines, and expression of antiviral effector genes that serve to limit virus replication. In order to further assess the immunostimulatory potential of HCs, we measured pro-inflammatory cytokines and chemokines in supernatants from infected cells by multiplex bead array. Following ZIKV infection, we observed increased IFNα secretion, but not IFNβ or IFNλ1 (IL-29; Figure 3; Table S1). We also found increased secretion of the pro-inflammatory cytokine IL-6 and chemokines MCP-1, involved in monocyte infiltration, and IP-10, involved in recruitment of activated effector T cells. Though these cytokines were induced in all five donors, there were individual differences in the magnitude of production. Donor 2, which had the highest viral load at 48 and 72 hpi (Figure 1A), tended to exhibit the highest overall levels of IFN-α, IL-6, MCP-1, and IP-10; however, donor 2 was not consistently the lead producer of cytokines over mock-infected controls. Of note, donor 5, which had the lowest viral load at 48 and 72 hpi, did not consistently show the lowest levels of cytokines, but did exhibit reduced induction over mock-infected controls at 72 hpi. No discernable patterns could be confidently drawn with CXCL-8, MIP-1α, MIP-1β, or IL-1RA. In contrast to HCs, we observed limited induction of type I IFN, IL-6, and IP-10, and no detectable type III IFN in CTBs at the time points assessed (Figure S3A; Table S2). Donor 1, while slightly less permissive to viral infection and replication (Figure S1), did not have correspondingly lower levels of cytokine production compared to donors 2 and 3. We did observe, however, that donor 1 tended to have reduced production of cytokines over mock-infected control cells at 72 hpi. These findings demonstrate that HCs are capable of initiating an inflammatory response to ZIKV infection.
Figure 3. ZIKV Infection of HCs Induces Type I IFN and Inflammatory Cytokines.

HCs from five donors were infected with ZIKV (PR 2015) at an MOI of 1, or mock infected. Cytokine levels in the supernatants were determined by multiplex bead array. All values are represented in pg/mL and shown with a connecting line between ZIKV-infected samples (48 and 72 hpi) and their respective donor- and time-matched, mock-infected samples. See also Table S1.
ZIKV Infection Provokes an Antiviral Immune Response in HCs and CTBs
To evaluate the antiviral potential of HCs and CTBs, we examined the expression of several antiviral effector genes. We observed increased expression of IFNA transcripts as early as 24 hpi in HCs (Figure 4A), concordant with increased IFNα secretion (Figure 3). While we did not observe IFNβ secretion, we detected an increase in IFNB1 transcripts over time-matched mock cells as early as 24 hpi (Figure 4A), suggesting possible discordance between transcript levels and translation/secretion of IFNβ (Schulz et al., 2010). In contrast, both IFNA and IFNB1 were induced at low levels in CTBs (Figure S3B). We next measured expression of the RLRs, a family of PRRs known to recognize flavivirus RNA and induce production of type I IFNs and pro-inflammatory cytokines (Daffis et al., 2009; Loo et al., 2008; Suthar et al., 2010, 2013). Expression of DDX58 (RIG-I), IFIH1 (MDA5), and DHX58 (LGP2) transcripts is induced above time-matched, mock-infected HCs across all donors by 72 hpi and remains highly expressed through 96 hpi (Figure 4B). RLR expression corresponds to kinetics of virus replication, suggesting that RLRs are induced in response to ZIKV infection of HCs. In CTBs, RLR transcription is modestly induced, and both IFIH1 and DHX58 return to near basal levels by 96 hpi, though DDX58 expression remains slightly elevated through 96 hpi (Figure S3B). We also evaluated expression of several antiviral genes produced downstream of the RLR and type I IFN signaling axes and found that RSAD2, IFIT1, IFIT2, IFIT3, and OAS1 were all induced by 72 hpi in HCs and remained elevated through 96 hpi (Figure 4C). In CTBs, these genes were modestly induced through 72 hpi (Figure S3B), likely corresponding to the low level of viral replication during this time period (Figure 1). By 96 hpi, a time point at which we observed productive virus replication, these cells also initiate an antiviral immune response. Importantly, we observed low levels of IFNA and ISG expression in mock-infected HCs and CTBs, likely induced by the cell isolation procedure, which may limit the percent of infected cells we see in our in vitro system. Taken together, these results show that both HCs and CTBs respond to ZIKV infection through initiation of antiviral signaling pathways.
Figure 4. ZIKV Infection Induces an Antiviral Response in HCs.

HCs from five donors were infected with ZIKV (PR 2015) at an MOI of 1, and antiviral gene expression determined by qRT-PCR. Gene expression data are represented as fold change relative to time-matched, mock-infected controls (gene expression normalized to GAPDH − ΔΔCT method). Individual donors are depicted as separate bars, organized from donor 1 to donor 5, within each time point block. Viral titers determined in Figure 1 are represented as a separate heat map below each group of genes. (A) shows type I IFNs, (B) shows RIG-I-like receptors, and (C) shows antiviral effector genes. hpi, hours post-infection.
The kinetics of the antiviral response are complex and variable, and we observed donor-to-donor variation in induction of antiviral gene expression. Of note, HCs from donor 2, which exhibited the highest viral loads, and donor 5, which exhibited the lowest viral loads, induced similar levels of antiviral effector genes by 96 hpi, although genes in donor 2 were induced at a faster rate (Figure 4). This may reflect the higher rate of replication and viral output by HCs from this donor (Figure 1). There is likely a multifactorial rationale for why viral load does not correlate with antiviral gene expression that likely encompasses differences in individual genetics and the antagonistic capabilities of the virus.
DISCUSSION
The present data demonstrate that primary HCs and CTBs isolated from full-term placentae are permissive to productive ZIKV infection by a contemporary strain currently circulating in the Americas. We also found that HCs respond to infection by triggering antiviral defense programs in the absence of overt cell death. In this limited study of five donors, we observed individual variability in kinetics and magnitude of virus replication, inflammation, and antiviral gene expression, likely reflecting differences in individual genetics (Querec et al., 2009; Thio, 2008). Though unlikely given the low number of cell passages PR 2015 has undergone, it is possible that minor cell culture adaptations or quasi-species may also be playing a role in donor-to-donor variability. These observations suggest that donors may have the capacity to restrict ZIKV at different stages of the viral replication cycle. This may also relate to observed differences in intrauterine transmission efficiency, where more susceptible HCs from a pregnant mother may support higher levels of virus replication and subsequent spread to the developing fetal nervous system. Additionally, it will be important in future studies to characterize when HCs and CTBs are most susceptible to ZIKV infection (i.e., first, second, or third trimester). Recent projections from the CDC based on data from Brazil indicate that virus infection during the first trimester or early in the second trimester of pregnancy is temporally associated with the observed increase in infants born with microcephaly (Reefhuis et al., 2016).
A recent study reported that primary syncytiotrophoblasts isolated from full-term placentae are resistant to ZIKV infection through a potential mechanism involving type III IFN-mediated antiviral immunity (Bayer et al., 2016). Similarly, in CTBs we observed a lack of productive virus replication through 48 hpi; however, we did observe persistent viral RNA through 72 hpi. By 96 hpi, we observed low-level virus replication as well as induction of antiviral effector genes, suggesting that ZIKV infects and persists in CTBs but is efficiently controlled at early times post-infection. Additionally, while Bayer et al. were able to identify IFN-λ (type III IFN) in the supernatant of uninfected syncytiotrophoblasts, we did not detect the presence of IFN-λ in the supernatants of ZIKV-infected HCs or CTBs. The discordance between these two studies may be attributed to differences in time points assessed and viral isolates used in each study (FSS13025 and MR766 as compared to PR 2015).
What are the possible mechanisms by which ZIKV crosses the placental barrier and infects HCs? One explanation is that ZIKV may initially infect trophoblasts and productively replicate and disseminate locally within the placenta to involve HCs, which then support more efficient ZIKV replication than CTBs. An alternative hypothesis is that non-neutralizing, cross-reactive antibodies bind ZIKV and traffic across the placenta, through a neonatal Fc-receptor-mediated mechanism, to infect placental macrophages. ZIKV crossing the placenta and replication in/release from HCs likely result in viral dissemination through the cord blood with subsequent infection of neural progenitor cells. At this time, it is uncertain whether maternal macrophages are infected or play a role in allowing ZIKV to cross the placental barrier. However, a recent report has directly identified the presence of viral antigen through immunohistochemistry in the placenta from a mother with an infant who developed ZIKV-related fetal anomalies (Martines et al., 2016). Of note, ZIKV antigen was detected within the chorionic villi and not in the maternal decidua. Based on these findings, it does not appear that decidual macrophages are key players in ZIKV transmission at the placenta.
HCs are likely programmed to limit inflammation following virus infection, a mechanism that is consistent with the immune-tolerant environment of the placenta and which would support higher infection of HCs compared to maternal macrophages. An alternative hypothesis is that the relative paucity of effector cells in the placenta that would otherwise readily kill infected macrophages (e.g., CD8+ T cells) contributes to a permissive environment for ZIKV infection and replication in HCs. Altogether, our data support the notion that HCs represent a key target cell within the placenta. These findings stress the importance of developing antiviral therapies directed against ZIKV replication within placental cells as a means to reduce vertical transmission in the mother-infant dyad and the incidence of adverse pregnancy outcomes and fetal abnormalities.
EXPERIMENTAL PROCEDURES
Ethics Statement
Human Placenta
Term (>37 weeks gestation) placentae from HIV-1 seronegative and hepatitis B-uninfected women (>18 years of age) were obtained immediately following elective cesarean section without labor from Grady Memorial and Emory Midtown Hospitals. Approval of the study was granted from the Emory University Institutional Review Board (IRB 00021715) and the Grady Research Oversight Committee. Written informed consent was obtained from donors before collection, and samples were de-identified prior to handling by laboratory personnel.
Isolation of Primary Placental Cells
HCs and CTBs were dissected from membrane-free villous placenta, as previously described (Johnson and Chakraborty, 2012). HCs were isolated and purified by positive selection with anti-CD14 magnetic beads per the manufacturer’s instructions. The purity of the HC population was assessed by CD14 staining and was, on average, greater than 97% (Figure S2A). CTBs were isolated and purified by negative selection with magnetic beads (Miltenyi Biotech). The purity of the CTB population was assessed by cytokeratin-7 staining and was, on average, greater than 97% (Chiuppesi et al., 2015). HCs were maintained in complete RPMI medium and CTBs were maintained in complete DMEM medium. A detailed protocol can be found in Supplemental Experimental Procedures.
Viruses and Infections
ZIKV (PR 2015) was isolated in 2015 from the serum of a patient who traveled to Puerto Rico, and it was passaged three times in Vero cells. PRVABC59 was obtained from the Centers for Disease Control and Prevention and passaged twice in Vero cells cultured in MEM (GIBCO) supplemented with 10% FBS (Optima, Atlanta Biologics) to generate working viral stocks. Viral stocks were titered by plaque assay on Vero cells and stored in MEM with 20% FBS. Vero cells (ATCC) were maintained in complete DMEM medium (Supplemental Experimental Procedures). HCs or CTBs were allowed to rest for ~24 hr before infecting with ZIKV (PR 2015) at an MOI of 1 for 1 hr at 37°C. Virus was washed off, and cells were resuspended in fresh complete media and incubated at 37°C for 3–96 hr. MOI of 1 was based on results of plaque assays as well as a recent paper where dendritic cells (DCs) (a similar cell type to macrophages) were infected with ZIKV at an MOI of 1 (Hamel et al., 2015). All work with infectious ZIKV was performed in an approved BSL-3 facility.
qRT-PCR
Total RNA was purified from mock- or ZIKV-infected HCs or CTBs (2 × 105 cells per condition) using the ZR-96 Quick-RNA Kit (Zymo Research) per the manufacturer’s instructions. Purified RNA was reverse transcribed using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems) using random hexamers. For quantitation of viral RNA and analysis of host gene expression, qRT-PCR was performed using TaqMan Gene Expression Master Mix (Applied Biosystems) per the manufacturer’s instructions. For quantitation of viral RNA, each 12.5 μL reaction contained 2.5 pmol of TaqMan probe directed against the amplified ZIKV E gene region. Host gene expression was performed using SYBR green with appropriate primer sets (Supplemental Experimental Procedures). All qRT-PCR results were normalized to GAPDH.
Flow Cytometry
The following mouse anti-human antibodies were purchased from BioLegend or Becton Dickinson: CD14 (M5E2), CD80 (2D10), CD86 (IT2.2), and HLA-DR (G46-6). Unconjugated 4G2 monoclonal antibody was kindly provided by Jens Wrammert and subsequently conjugated with APC (Novus Lightning-Link). In total, 2 × 105 HCs or CTBs were used per condition. Cells were stained for surface markers and permeabilized to stain for ZIKV E protein. A detailed protocol can be found in Supplemental Experimental Procedures.
Multiplex Bead Array
Cytokine analysis was performed on supernatants from mock- or ZIKV-infected HCs or CTBs (2 × 105 cells per condition) using a human cytokine 25-plex panel (ThermoScientific) and a custom two-plex panel with human IFNβ and IFNλ1 (eBioscience) per the manufacturer’s instructions, and read on a Luminex 100 Analyzer.
Statistical Analysis
Sample size was dependent on the number of donors. HCs were isolated from five donors and CTBs were isolated from three of these donors. Experiments with HCs were repeated twice (three donors in the first experiment and two donors in the second). Experiments with CTBs were repeated once (three donors in one experiment). All statistical analysis was performed in GraphPad Prism 6, with significance assessed by Mann-Whitney U test with p < 0.05. Infectivity as assessed by 4G2 staining utilized a one-tailed test. Cell activation as assessed by surface staining utilized a two-tailed test.
Supplementary Material
Highlights.
Zika virus (ZIKV) infects and replicates in primary human placental macrophages
ZIKV also infects human placental cytotrophoblasts, but with delayed replication kinetics
ZIKV replication coincides with IFN and antiviral gene induction, but minimal cell death
Acknowledgments
We thank A. Price for the OAS1 qRT-PCR reagents. This work was funded in part by NIH grants U19AI083019 (M.S.S), R56AI110516 (M.S.S), R21AI113485 (M.S.S.), 2U19AI090023 (B.P), 5R37DK057665 (B.P), 5R37AI048638 (B.P), and 2U19AI057266 (B.P); Children’s Healthcare of Atlanta (M.S.S); Emory Vaccine Center (M.S.S); The Georgia Research Alliance (M.S.S); Multi-Center NICHD International Maternal Pediatric Adolescent AIDS Clinical Trials Network (R.C); and P30AI050409 Center for AIDS Research at Emory University (R.F.S).
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, three figures, and two tables and can be found with this article online at http://dx.doi.org/10.1016/j.chom.2016.05.015.
AUTHOR CONTRIBUTIONS
K.M.Q. and J.R.B. contributed equally to this work. Both authors were involved in experimental design and execution, data analysis, figure generation, and manuscript preparation. B.H.R. collected and delivered full-term placenta from informed donors. E.L.J. and A.R. received placenta and isolated the HCs and CTBs used in these experiments. C.E.M. ran multiplex bead arrays for assessing IFN and cytokine secretion. H.M. performed confocal microscopy staining and imaging and helped generate the resulting figure panels. J.T.O. ran qRT-PCR for assessing viral RNA and antiviral gene expression. J.W. provided 4G2 monoclonal antibody used to detect ZIKV E protein. B.P., R.F.S., and R.C. provided valuable scientific discussion and mentorship. R.C. provided expertise in the field of placental immunology. M.S.S. contributed expertise in the fields of flavivirus virology and viral immunology, guidance for experimental design, and manuscript preparation and mentorship.
References
- Bayer A, Lennemann NJ, Ouyang Y, Bramley JC, Morosky S, Marques ET, Jr, Cherry S, Sadovsky Y, Coyne CB. Type III interferons produced by human placental trophoblasts confer protection against Zika virus infection. Cell Host Microbe. 2016;19:705–712. doi: 10.1016/j.chom.2016.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvet G, Aguiar RS, Melo AS, Sampaio SA, de Filippis I, Fabri A, Araujo ES, de Sequeira PC, de Mendonça MC, de Oliveira L, et al. Detection and sequencing of Zika virus from amniotic fluid of fetuses with microcephaly in Brazil: a case study. Lancet Infect Dis. 2016 doi: 10.1016/S1473-3099(16)00095-5. Published online February 17, 2016 http://dx.doi.org/10.1016/S1473-3099(16)00095-5. [DOI] [PubMed]
- Chiuppesi F, Wussow F, Johnson E, Bian C, Zhuo M, Rajakumar A, Barry PA, Britt WJ, Chakraborty R, Diamond DJ. Vaccine-derived neutralizing antibodies to the human cytomegalovirus gH/gL pentamer potently block primary cytotrophoblast infection. J Virol. 2015;89:11884–11898. doi: 10.1128/JVI.01701-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordeiro MT, Pena LJ, Brito CA, Gil LH, Marques ET. Positive IgM for Zika virus in the cerebrospinal fluid of 30 neonates with microcephaly in Brazil. Lancet. 2016 doi: 10.1016/S0140-6736(16)30253-7. Published online April 18, 2016. http://dx.doi.org/10.1016/S0140-6736(16)30253-7. [DOI] [PubMed]
- Daffis S, Suthar MS, Szretter KJ, Gale M, Jr, Diamond MS. Induction of IFN-beta and the innate antiviral response in myeloid cells occurs through an IPS-1-dependent signal that does not require IRF-3 and IRF-7. PLoS Pathog. 2009;5:e1000607. doi: 10.1371/journal.ppat.1000607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delorme-Axford E, Donker RB, Mouillet JF, Chu T, Bayer A, Ouyang Y, Wang T, Stolz DB, Sarkar SN, Morelli AE, et al. Human placental trophoblasts confer viral resistance to recipient cells. Proc Natl Acad Sci USA. 2013;110:12048–12053. doi: 10.1073/pnas.1304718110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driggers RW, Ho CY, Korhonen EM, Kuivanen S, Jääskeläinen AJ, Smura T, Rosenberg A, Hill DA, DeBiasi RL, Vezina G, et al. Zika virus infection with prolonged maternal viremia and fetal brain abnormalities. N Engl J Med. 2016 doi: 10.1056/NEJMoa1601824. Published online March 30, 2016 http://dx.doi.org/10.1056/NEJMoa1601824. [DOI] [PubMed]
- Faria NR, do Azevedo RS, Kraemer MU, Souza R, Cunha MS, Hill SC, Thézé J, Bonsall MB, Bowden TA, Rissanen I, et al. Zika virus in the Americas: early epidemiological and genetic findings. Science. 2016;352:345–349. doi: 10.1126/science.aaf5036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcez PP, Loiola EC, Madeiro da Costa R, Higa LM, Trindade P, Delvecchio R, Nascimento JM, Brindeiro R, Tanuri A, Rehen SK. Zika virus impairs growth in human neurospheres and brain organoids. Science. 2016;352:816–818. doi: 10.1126/science.aaf6116. [DOI] [PubMed] [Google Scholar]
- Hamel R, Dejarnac O, Wichit S, Ekchariyawat P, Neyret A, Luplertlop N, Perera-Lecoin M, Surasombatpattana P, Talignani L, Thomas F, et al. Biology of Zika virus infection in human skin cells. J Virol. 2015;89:8880–8896. doi: 10.1128/JVI.00354-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson EL, Chakraborty R. Placental Hofbauer cells limit HIV-1 replication and potentially offset mother to child transmission (MTCT) by induction of immunoregulatory cytokines. Retrovirology. 2012;9:101. doi: 10.1186/1742-4690-9-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koi H, Zhang J, Parry S. The mechanisms of placental viral infection. Ann N Y Acad Sci. 2001;943:148–156. doi: 10.1111/j.1749-6632.2001.tb03798.x. [DOI] [PubMed] [Google Scholar]
- Lazear HM, Diamond MS. Zika virus: new clinical syndromes and its emergence in the Western Hemisphere. J Virol. 2016;90:4864–4875. doi: 10.1128/JVI.00252-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis SH, Reynolds-Kohler C, Fox HE, Nelson JA. HIV-1 in trophoblastic and villous Hofbauer cells, and haematological precursors in eight-week fetuses. Lancet. 1990;335:565–568. doi: 10.1016/0140-6736(90)90349-a. [DOI] [PubMed] [Google Scholar]
- Loo YM, Fornek J, Crochet N, Bajwa G, Perwitasari O, Martinez-Sobrido L, Akira S, Gill MA, García-Sastre A, Katze MG, Gale M., Jr Distinct RIG-I and MDA5 signaling by RNA viruses in innate immunity. J Virol. 2008;82:335–345. doi: 10.1128/JVI.01080-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martines RB, Bhatnagar J, Keating MK, Silva-Flannery L, Muehlenbachs A, Gary J, Goldsmith C, Hale G, Ritter J, Rollin D, et al. Notes from the field: evidence of Zika virus infection in brain and placental tissues from two congenitally infected newborns and two fetal losses—Brazil, 2015. MMWR Morb Mortal Wkly Rep. 2016;65:159–160. doi: 10.15585/mmwr.mm6506e1. [DOI] [PubMed] [Google Scholar]
- Miner JJ, Cao B, Govero J, Smith AM, Fernandez E, Cabrera OH, Garber C, Noll M, Klein RS, Noguchi KK, et al. Zika virus infection during pregnancy in mice causes placental damage and fetal demise. Cell. 2016;165:1081–1091. doi: 10.1016/j.cell.2016.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noronha L, Zanluca C, Azevedo ML, Luz KG, Santos CN. Zika virus damages the human placental barrier and presents marked fetal neurotropism. Mem Inst Oswaldo Cruz. 2016 doi: 10.1590/0074-02760160085. Published online April 29, 2016 http://dx.doi.org/10.1590/0074-02760160085. [DOI] [PMC free article] [PubMed]
- Petersen LR, Jamieson DJ, Powers AM, Honein MA. Zika virus. N Engl J Med. 2016;374:1552–1563. doi: 10.1056/NEJMra1602113. [DOI] [PubMed] [Google Scholar]
- Querec TD, Akondy RS, Lee EK, Cao W, Nakaya HI, Teuwen D, Pirani A, Gernert K, Deng J, Marzolf B, et al. Systems biology approach predicts immunogenicity of the yellow fever vaccine in humans. Nat Immunol. 2009;10:116–125. doi: 10.1038/ni.1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen SA, Jamieson DJ, Honein MA, Petersen LR. Zika virus and birth defects—reviewing the evidence for causality. N Engl J Med. 2016;374:1981–1987. doi: 10.1056/NEJMsr1604338. [DOI] [PubMed] [Google Scholar]
- Reefhuis J, Gilboa SM, Johansson MA, Valencia D, Simeone RM, Hills SL, Polen K, Jamieson DJ, Petersen LR, Honein MA. Projecting month of birth for at-risk infants after Zika virus disease outbreaks. Emerg Infect Dis. 2016;22:828–832. doi: 10.3201/eid2205.160290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz O, Pichlmair A, Rehwinkel J, Rogers NC, Scheuner D, Kato H, Takeuchi O, Akira S, Kaufman RJ, Reis e Sousa C. Protein kinase R contributes to immunity against specific viruses by regulating inter-feron mRNA integrity. Cell Host Microbe. 2010;7:354–361. doi: 10.1016/j.chom.2010.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suthar MS, Ma DY, Thomas S, Lund JM, Zhang N, Daffis S, Rudensky AY, Bevan MJ, Clark EA, Kaja MK, et al. IPS-1 is essential for the control of West Nile virus infection and immunity. PLoS Pathog. 2010;6:e1000757. doi: 10.1371/journal.ppat.1000757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suthar MS, Diamond MS, Gale M., Jr West Nile virus infection and immunity. Nat Rev Microbiol. 2013;11:115–128. doi: 10.1038/nrmicro2950. [DOI] [PubMed] [Google Scholar]
- Tang H, Hammack C, Ogden SC, Wen Z, Qian X, Li Y, Yao B, Shin J, Zhang F, Lee EM, et al. Zika virus infects human cortical neural progenitors and attenuates their growth. Cell Stem Cell. 2016;18:587–590. doi: 10.1016/j.stem.2016.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thio CL. Host genetic factors and antiviral immune responses to hepatitis C virus. Clin Liver Dis. 2008;12:713–726. xi. doi: 10.1016/j.cld.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsch S, Miller S, Romero-Brey I, Merz A, Bleck CK, Walther P, Fuller SD, Antony C, Krijnse-Locker J, Bartenschlager R. Composition and three-dimensional architecture of the dengue virus replication and assembly sites. Cell Host Microbe. 2009;5:365–375. doi: 10.1016/j.chom.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.