

Abstract
The intrinsically disordered protein (IDP), α-synuclein (αS), is well-known for phospholipid membrane binding-coupled-folding into tunable helical conformers. Here, using single-molecule experiments in conjunction with ensemble assays and a theoretical model, we present a unique case demonstrating that the interaction-folding landscape of αS can be tuned by two-dimensional (2-D) crowding through simultaneous binding of a second protein on the bilayer surface. Unexpectedly, the experimental data show a clear deviation from a simple competitive inhibition model, but are consistent with a bimodal inhibition mechanism wherein membrane binding of a second protein (a membrane interacting chaperone, Hsp27, in this case) differentially inhibits two distinct modules of αS-membrane interaction. As a consequence, αS molecules are forced to access a hidden conformational state on the phospholipid bilayer in which only the higher-affinity module remains membrane-bound. Our results demonstrate that macromolecular crowding in two dimensions can play a significant role in shaping the conformational landscape of membrane binding IDPs with multiple binding modes.
Peripheral membrane binding of many intrinsically disordered proteins (IDPs) and/or disordered regions (IDRs) in multi-domain proteins is critical for proper subcellular localization and initiating downstream signaling cascades in biology[1]. Membrane binding of IDPs/IDRs often leads to folding, and is functionally tuned by post-translational modifications, interaction electrostatics and partner binding[1b, 2]. While it is known that multiple binding modes can coexist in protein–lipid association[3], little is known about how a common effector, such as a third component protein binding to the bilayer, modulates these individual modes.
In this study, we focus on the intrinsically disordered protein (IDP), α-synuclein (αS)[4] that undergoes binding-coupled-folding into a tunable ensemble of conformations upon interaction with anionic phospholipid membranes and membrane mimics[3b, 5]. Abundantly expressed in the central nervous system and linked to Parkinson's disease (PD)[6], αS is in dynamic equilibrium between disordered cytosolic and ordered membrane bound fractions[3b]. The primary structure of αS features 140 amino acid residues, where only the N-terminal ~ 100 residues are principally involved in membrane interactions[3b]. This segement of αS structure can adopt alternative partner-dependent helical conformations[7], while the C-terminal tail remains mostly unbound and disordered[5a, 5b, 8]. The N-terminal segment can be further subdivided into two motifs that interact with anionic phospholipid bilayers distinctively[3]. Here, we demonstrate that simultaneous membrane binding of a second protein (here a lipid interacting molecular chaperone) can differentially modulate the interaction-coupled-folding of these two αS motifs, giving rise to a bimodal inhibition in this ternary protein-lipid system.
Although molecular chaperones are well-known for their roles in folding and quality-control of native, functional states of proteins[9], their impact on IDP coupled interaction-folding landscapes remains elusive. Here, we tested the idea that a lipid-interacting chaperone from the small heat shock protein (sHSP) family may regulate αS-membrane binding and folding via interaction with the common partner, i.e., the phospholipid bilayer. To this end, we chose to investigate the effects of the membrane interacting Hsp27, one of the two ubiquitously expressed human sHSPs and a major stress inducible molecular chaperone in neurons[10] that has been biologically linked to αS by several reports[10a, 11]. To examine whether this chaperone has any effect on the membrane binding-induced-folding of αS, we performed steady-state ensemble fluorescence anisotropy experiments. We used Small Unilamellar phospholipid Vesicles (SUVs) that are composed of a well-characterized anionic phospholipid binding partner of αS, phosphatidylglycerol (PG) [12] with sizes comparable to synaptic vesicles [13] (Fig. S1). We found that αS binds to PG SUVs (signaled by an increase in anisotropy due to slower molecular tumbling; Fig. 1A) with a dissociation constant () of ~ 2.5 nM in SUVs (~ 50 μM in lipid; stoichiometry (Lipid/αS) ~ 120; SI Note-1). Interestingly, αS binding to the SUVs is substantially reduced in the presence of Hsp27 (Fig. 1A, B; Table S1). The apparent increased from ~ 2.5 nM (in SUVs) to ~ 15 nM (in SUVs) in response to an increase in [Hsp27] from 0 to 10 μM (Fig. 1B; Table S1), indicating a monotonic decrease in binding affinity as a function of increasing [Hsp27]. Next, we probed for the effect of Hsp27 on the coupled-folding of αS by single-molecule Förster Resonance Energy Transfer (smFRET) using PG SUVs as well as SUVs composed of other physiologically relevant phospholipids (Figs. S2 and S3). As anticipated from our ensemble fluorescence measurements, we observed that Hsp27 substantially increases the relative population of the unbound disordered state. Together, these results suggest that Hsp27 inhibits the coupled αS-membrane association and folding.
Figure 1. Characterization of the effect of Hsp27 on SUV binding-coupled-folding of αS.

(A) Steady state fluorescence anisotropy of dye-labeled αSG7C reporting on SUV-binding, with increasing [Hsp27]. Three representative [Hsp27] are shown for clarity. (B) Increasing [Hsp27] leads to a decreased αS binding to SUVs with a net increase in apparent (Supplementary Table S1). Error bars represent one sigma (σ) standard deviation (n = 3).
Next, we investigated the mechanistic basis of the inhibitory action of Hsp27. In agreement with previous literature reports on membrane-binding of other members of the sHSP family [14], Hsp27 binding to the phospholipid bilayer was confirmed by a bulk vesicle-to-protein FRET method (Fig. 2A), with an apparent dissociation constant () of ~ 12 nM in SUV (~ 250 μM in lipid; stoichiometry (Lipid/Hsp27) ~ 65; SI Note-1). Comparable binding affinities were also obtained with smFRET and fluorescence anisotropy (SI Note-2; Fig. S4). On the other hand, when we tested whether Hsp27 interacts with the monomeric disordered state of αS, both ensemble and single-molecule experiments failed to show evidence for a direct interaction (SI Note-3; Fig. S5). Thus, our data are consistent with a model where Hsp27 inhibits αS binding to membranes by sterically blocking the binding sites (SI Note-1). If that is the case, a competitive model should be sufficient to quantitatively describe the observed inhibitory effect of Hsp27. Numerical simulation (SI Note-4) using a mathematical model revealed a linear relationship between with [Hsp27] for simple competitive inhibition (Fig. S6), with the slope signifying the efficacy of the inhibition (Fig. S6b). In contrast, our experimental data (presented in Fig. 1B) revealed a clear nonlinearity, showing that has a steeper slope at lower Hsp27 concentrations (< 4 μM), compared to higher Hsp27 concentrations (> 5 μM). Given that previous NMR reports suggest αS binding to membranes is mediated by two distinct helical motifs [3, 8], we postulated that the nonlinearity observed in Fig. 1B may be due to differential inhibition of alternative αS binding motifs by Hsp27. To test this hypothesis, we next studied two αS fragments, referred to as the N-terminal motif (residues 1-25; αSN-ter) and the central motif (residues 26-97; αSNAC) [3]. In the absence of Hsp27, the αSNAC was observed to bind PG SUVs with ~ 15 fold higher affinity (KD = 0.61 nM in SUVs or 12 μM in lipid; Fig. 3A) than the αSN-ter (KD = 9.16 nM in SUVs or 178 μM in lipid; Fig. 3B), and when titrated with increasing [Hsp27], linear vs [Hsp27] relationships were observed for individual motifs (insets in Fig. 3A,B). These data suggest that a competitive inhibition mechanism is sufficient to explain the inhibitory action of Hsp27 for the individual motifs (Fig. S6b). Further analysis of the fragment data revealed a significantly different inhibition efficacies for αSN-ter vs αSNAC, as derived from the individual slopes (m) of the inhibition plot (mαS-N-ter /mαS-NAC ~ 100; Fig. 3A,B). This is consistent with the fact that αSN-ter has more than an order of magnitude lower binding affinity than the αSNAC for PG SUVs (SI Note-4). Therefore, consistent with this model (Figs. S6&7), we suggest that the observed nonlinearity for α vs [Hsp27] for the full-length αS (αSFL; Fig. 1B) is a manifestation of bimodal inhibition of αS-SUV interaction by Hsp27.
Figure 2. Characterization of Hsp27-membrane interaction by vesicle-to-protein FRET.
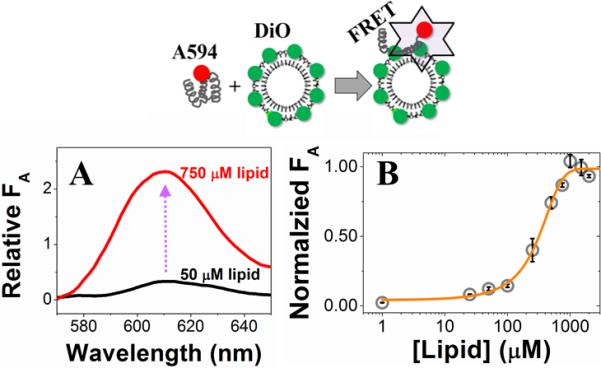
Schematic representation of the vesicle-to-protein FRET assay (top). (A) Representative emission spectra showing increased acceptor fluorescence (FA) as a result of FRET due to PG SUV binding of Hsp27 (bottom). (B) Binding isotherm of Hsp27-vesicle (POPG) interaction using vesicle-to-protein FRET assay with an apparent of 12.4±0.43 nM (242±84 αM in lipid). Error bars represent one sigma (σ) standard deviation (n = 3).
Figure 3. Hsp27 differentially modulates membrane interactions of the αSN-ter and αSNAC fragments, and drives αS to populate a “hidden” conformation on the lipid bilayer.

(A) Steady state fluorescence anisotropy of Alexa594-labeled αSN-ter-G7C reporting on PG SUV binding. The points are the experimental data, line is a fit to the Hill model, yielding KD = 9.16 ± 0.58 nM in SUV (178.78 ± 11.41 μM in lipid). The inset shows a linear relationship for KD vs [Hsp27]; slope (m) = 2178.6 (± 76.3). (B) Steady state fluorescence anisotropy of Alexa594-labeled αSNAC-G84C reporting on PG SUV binding. The points are the experimental data, line are fit to the Hill model, yielding KD = 0.61 ± 0.02 nM in SUV (11.98 ± 0.46 μM in lipid). The inset shows a linear relationship for KD vs [Hsp27]; slope (m) = 22.9 (± 0.9). Error bars represent one sigma (σ) standard deviation (n = 3). Adjusted R2 for the fits in the insets of A and B were ≈ 0.99. (C) smFRET histograms of the dual-labeled full-length αS3/51 showing transition of the U state (panel (i); EFRET 0.76) to F state (panel (ii); EFRET 0.45) in presence of 250 μM PG SUVs. In presence of Hsp27, the F state transitioned to the F* state (panel (v); EFRET 0.30)) with increasing [Hsp27]. Shown here are representative smFRET histograms at three different [Hsp27]: 0.5 μM (panel (iii)), 1 μM (panel (iv)) and 2.5 μM (panel (v)). Solid lines are Gaussian fits of the data. The smFRET histograms are compiled using triplicate data sets.
Our model indicates that the ratio between fragment KD values () will increase from ~15 to ~100 with increasing [Hsp27] from 0 to 10 μM (Fig. S6c), i.e., the two binding modes will be progressively decoupled by Hsp27. This implies that with increasing concentrations of Hsp27, the SUV-αSN-ter interaction will be selectively disrupted and αS molecules will be forced into a “hidden” (termed as F*) state where only the central helix is bound to the lipid bilayer (SI Note-4; Fig. S6c). To test this experimentally for full-length αS, next we performed two independent sets of ensemble vesicle-to-protein FRET experiments, where we placed the acceptor dye either at residue 7 for reporting on the N-terminal helix (αSFL-7), or at residue 84 for reporting on the central helix (αSFL-84; Fig. S8). Without Hsp27, both these labeling positions yielded similar binding affinities ( ~ 2.5 nM in SUVs or 50 μM in lipid). Increasing [Hsp27] resulted not only in an increase in , but also a concomitant decrease in the FRET signal for αSFL-7 at a pseudo-saturation level (~ 60%; Fig. S8a; Table S1). For αSFL-84, only an increase in the was observed with increasing [Hsp27] (Fig. S8b; Table S1). These observations are consistent with the bimodal competitive inhibition mechanism for Hsp27 discussed above. The inhibition mode primarily observed in the above titrations is one where αSN-ter is largely inhibited without disrupting the αSNAC, thereby populating the F* state (Fig. 4). This results in an increase in the average distance between the lipid-bilayer and the αSN-ter, signaled by a corresponding decrease in the FRET for αSFL-7 at pseudo-saturation, but not for αSFL-84.
Figure 4. A schematic representation of the modulation of membrane binding-coupled-folding of αS by Hsp27 via 2-D crowding.
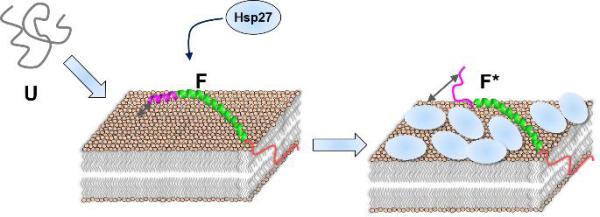
The disordered state of αS (U) transitions to an extended helical state when bound to the phospholipid bilayer. Hsp27 selectively inhibits the formation of the N-terminal helix without disrupting the central helix binding to the bilayer by bimodal inhibition.
To detect the F* state directly, next we employed smFRET experiments where we dual-labeled αSFL-3/51 with the Alexa488/Alexa594 dye pair. These labeling positions were designed to reduce the overlap of the bound population peaks with the zero peak in smFRET histograms (Fig. S2), and to provide better resolution of structural changes in bound αS. Similar to our previous smFRET results[7], the disordered (U) state of αS formed an extended helical (F) state upon SUV binding at high lipid concentrations (> apparent ; Fig. 3C). smFRET histograms for the dual-labelled αSFL-3/51 showed non-zero peaks at EFRET values of ~0.45 and ~0.80 for the U and F states, respectively. When we titrated with increasing [Hsp27], the population of αS gradually shifted to a new state at lower EFRET (~ 0.30, Fig. 3C) from the F state, which we assigned as the F* state. Finally, as predicted by the model, a peak corresponding to the F* state was also observed without Hsp27, but only at very high protein/lipid ratio (i.e., high packing density) (Fig. S9), here due to steric blocking by adjacent αS monomers. Therefore, our smFRET data directly validate the bimodal inhibition model where Hsp27 drives the membrane-bound αS molecules to populate a hidden conformation by differentially modulating two membrane binding motifs of αS.
In conclusion, our results reveal that two distinct regions of an IDP with substantially different affinities for the membrane provide an additional layer of complexity in the IDP interaction-folding landscape that could be tuned by a third component in protein-membrane interactions. Although our current study identified a lipid interacting molecular chaperone as a bimodal competitive inhibitor in IDP-lipid association, our mechanistic model is generally applicable for any peripheral membrane binding protein with comparable affinities. Furthermore, our results can be viewed as a unique example of population of an alternative conformational state due to partial escape of an IDP from enhanced crowding on a surface. While structural reorientation of proteins due to crowding on a bilayer surface was previously predicted by theory[15], our results provide a first direct demonstration of such an effect, here within a more complex context of multi-module IDP binding-coupled-folding. This interaction-folding landscape is likely to be modulated further via protein post-translational modifications[16] and membrane composition variation[3a, 17]. Given that several IDP systems bind and fold on biological membranes where many membrane proteins are typically present[18] occupying a significant fraction of the membrane surface area[19], the observed 2-D crowding effect is a key factor to consider for understanding the conformational ensembles of such dynamic protein systems.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grant (RO1 GM066833 to A.A.D.) from the NIGMS, NIH, and postdoctoral fellowship (15POST22520013 to P.R.B.) from AHA. We thank H. S. McHaourab for the Hsp27 plasmid, R. L. Nussbaum for the wild-type αS plasmid, S. Lindquist and J. Valastyan for the αS-9C/85C plasmid, and M. R. Wood for assistance with TEM.
REFERENCES
- 1.a Sigalov AB, Aivazian DA, Uversky VN, Stern LJ. Biochemistry. 2006;45:15731–15739. doi: 10.1021/bi061108f. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Perez Y, Maffei M, Amata I, Arbesú M, Pons M. Protocol Exchange. 2013 [Google Scholar]
- 2.a Strickfaden SC, Winters MJ, Ben-Ari G, Lamson RE, Tyers M, Pryciak PM. Cell. 2007;128:519–531. doi: 10.1016/j.cell.2006.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Sato I, Obata Y, Kasahara K, Nakayama Y, Fukumoto Y, Yamasaki T, Yokoyama KK, Saito T, Yamaguchi N. J Cell Sci. 2009;122:965–975. doi: 10.1242/jcs.034843. [DOI] [PubMed] [Google Scholar]; c Ikeda K, Nakayama Y, Ishii M, Obata Y, Kasahara K, Fukumoto Y, Yamaguchi N. Biochim Biophys Acta. 2009;1790:1345–1352. doi: 10.1016/j.bbagen.2009.07.009. [DOI] [PubMed] [Google Scholar]
- 3.a Bodner CR, Dobson CM, Bax A. J. Mol. Biol. 2009;390:775–790. doi: 10.1016/j.jmb.2009.05.066. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Fusco G, De Simone A, Gopinath T, Vostrikov V, Vendruscolo M, Dobson CM, Veglia G. Nat Commun. 2014;5:3827. doi: 10.1038/ncomms4827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Theillet FX, Binolfi A, Bekei B, Martorana A, Rose HM, Stuiver M, Verzini S, Lorenz D, van Rossum M, Goldfarb D, Selenko P. Nature. 2016;530:45–50. doi: 10.1038/nature16531. [DOI] [PubMed] [Google Scholar]
- 5.a Davidson WS, Jonas A, Clayton DF, George JM. J Biol Chem. 1998;273:9443–9449. doi: 10.1074/jbc.273.16.9443. [DOI] [PubMed] [Google Scholar]; b Jo E, McLaurin J, Yip CM, St George-Hyslop P, Fraser PE. J. Biol. Chem. 2000;275:34328–34334. doi: 10.1074/jbc.M004345200. [DOI] [PubMed] [Google Scholar]; c Trexler AJ, Rhoades E. Biochemistry. 2009;48:2304–2306. doi: 10.1021/bi900114z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Iwai A, Masliah E, Yoshimoto M, Ge N, Flanagan L, de Silva HA, Kittel A, Saitoh T. Neuron. 1995;14:467–475. doi: 10.1016/0896-6273(95)90302-x. [DOI] [PubMed] [Google Scholar]
- 7.Ferreon AC, Gambin Y, Lemke EA, Deniz AA. Proc. Natl. Acad. Sci. U. S. A. 2009;106:5645–5650. doi: 10.1073/pnas.0809232106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ulmer TS, Bax A, Cole NB, Nussbaum RL. J Biol Chem. 2005;280:9595–9603. doi: 10.1074/jbc.M411805200. [DOI] [PubMed] [Google Scholar]
- 9.Hartl FU, Bracher A, Hayer-Hartl M. Nature. 2011;475:324–332. doi: 10.1038/nature10317. [DOI] [PubMed] [Google Scholar]
- 10.a Zourlidou A, Payne Smith MD, Latchman DS. J Neurochem. 2004;88:1439–1448. doi: 10.1046/j.1471-4159.2003.02273.x. [DOI] [PubMed] [Google Scholar]; b Franklin TB, Krueger-Naug AM, Clarke DB, Arrigo AP, Currie RW. Int J Hyperthermia. 2005;21:379–392. doi: 10.1080/02656730500069955. [DOI] [PubMed] [Google Scholar]
- 11.Outeiro TF, Klucken J, Strathearn KE, Liu F, Nguyen P, Rochet JC, Hyman BT, McLean PJ. Biochem Biophys Res Commun. 2006;351:631–638. doi: 10.1016/j.bbrc.2006.10.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.a Rhoades E, Ramlall TF, Webb WW, Eliezer D. Biophys J. 2006;90:4692–4700. doi: 10.1529/biophysj.105.079251. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Middleton ER, Rhoades E. Biophys J. 2010;99:2279–2288. doi: 10.1016/j.bpj.2010.07.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hu Y, Qu L, Schikorski T. Synapse. 2008;62:953–957. doi: 10.1002/syn.20567. [DOI] [PubMed] [Google Scholar]
- 14.a Piotrowicz RS, Levin EG. J Biol Chem. 1997;272:25920–25927. doi: 10.1074/jbc.272.41.25920. [DOI] [PubMed] [Google Scholar]; b Tsvetkova NM, Horvath I, Torok Z, Wolkers WF, Balogi Z, Shigapova N, Crowe LM, Tablin F, Vierling E, Crowe JH, Vigh L. Proc Natl Acad Sci U S A. 2002;99:13504–13509. doi: 10.1073/pnas.192468399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Minton AP. Biophys J. 1999;76:176–187. doi: 10.1016/S0006-3495(99)77187-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.a Giasson BI, Duda JE, Murray IV, Chen Q, Souza JM, Hurtig HI, Ischiropoulos H, Trojanowski JQ, Lee VM. Science. 2000;290:985–989. doi: 10.1126/science.290.5493.985. [DOI] [PubMed] [Google Scholar]; b Dorval V, Fraser PE. J. Biol. Chem. 2006;281:9919–9924. doi: 10.1074/jbc.M510127200. [DOI] [PubMed] [Google Scholar]; c Oueslati A, Fournier M, Lashuel HA. Prog. Brain Res. 2010;183:115–145. doi: 10.1016/S0079-6123(10)83007-9. [DOI] [PubMed] [Google Scholar]; d Dikiy I, Eliezer D. J. Biol. Chem. 2014;289:3652–3665. doi: 10.1074/jbc.M113.512459. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Marotta NP, Lin YH, Lewis YE, Ambroso MR, Zaro BW, Roth MT, Arnold DB, Langen R, Pratt MR. Nat. Chem. 2015;7:913–920. doi: 10.1038/nchem.2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.a Sharon R, Goldberg MS, Bar-Josef I, Betensky RA, Shen J, Selkoe DJ. Proc. Natl. Acad. Sci. U. S. A. 2001;98:9110–9115. doi: 10.1073/pnas.171300598. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Fortin DL, Troyer MD, Nakamura K, Kubo S, Anthony MD, Edwards RH. J. Neurosci. 2004;24:6715–6723. doi: 10.1523/JNEUROSCI.1594-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Jo E, Darabie AA, Han K, Tandon A, Fraser PE, McLaurin J. Eur. J. Biochem. 2004;271:3180–3189. doi: 10.1111/j.1432-1033.2004.04250.x. [DOI] [PubMed] [Google Scholar]
- 18.Takamori S, Holt M, Stenius K, Lemke EA, Gronborg M, Riedel D, Urlaub H, Schenck S, Brugger B, Ringler P, Muller SA, Rammner B, Grater F, Hub JS, De Groot BL, Mieskes G, Moriyama Y, Klingauf J, Grubmuller H, Heuser J, Wieland F, Jahn R. Cell. 2006;127:831–846. doi: 10.1016/j.cell.2006.10.030. [DOI] [PubMed] [Google Scholar]
- 19.Lindén M, Sens P, Phillips R. PLoS Comput. Biol. 2012;8:e1002431. doi: 10.1371/journal.pcbi.1002431. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.