

Abstract
AIM: To investigate whether chromosomal instability (CIN) is associated with tumor phenotypes and/or with global genomic status based on MSI (microsatellite instability) and CIMP (CpG island methylator phenotype) in early-onset colorectal cancer (EOCRC). METHODS: Taking as a starting point our previous work in which tumors from 60 EOCRC cases (≤45 years at the time of diagnosis) were analyzed by array comparative genomic hybridization (aCGH), in the present study we performed an unsupervised hierarchical clustering analysis of those aCGH data in order to unveil possible associations between the CIN profile and the clinical features of the tumors. In addition, we evaluated the MSI and the CIMP statuses of the samples with the aim of investigating a possible relationship between copy number alterations (CNAs) and the MSI/CIMP condition in EOCRC. RESULTS: Based on the similarity of the CNAs detected, the unsupervised analysis stratified samples into two main clusters (A, B) and four secondary clusters (A1, A2, B3, B4). The different subgroups showed a certain correspondence with the molecular classification of colorectal cancer (CRC), which enabled us to outline an algorithm to categorize tumors according to their CIMP status. Interestingly, each subcluster showed some distinctive clinicopathological features. But more interestingly, the CIN of each subcluster mainly affected particular chromosomes, allowing us to define chromosomal regions more specifically affected depending on the CIMP/MSI status of the samples. CONCLUSIONS: Our findings may provide a basis for a new form of classifying EOCRC according to the genomic status of the tumors.
Abbreviations: aCGH, array comparative genomic hybridization; CIMP, CpG island methylator phenotype; CIN, chromosomal instability; CNA, copy number alteration; CRC, colorectal cancer; DFS, disease-free survival; EOCRC, early-onset colorectal cancer; GII, genomic Instability Index; LS, Lynch syndrome; MACS, microsatellite and chromosome stable tumors; MMR, mismatch repair; MSI, microsatellite instability; MSS, microsatellite stability; OS, overall survival
Introduction
Colorectal cancer (CRC) has a great impact on the world population, since it represents the third most common malignancy and the second leading cause of death in developed countries [1], [2]. Its pathogenesis is a multistep process in which the accumulation of different genetic and epigenetic alterations leads to the transformation of healthy colonic epithelial cells into malignant cells [3]. The loss of genomic stability is a key molecular pathogenic step that occurs early in tumorigenesis, and it can be caused by at least three major molecular pathways: chromosomal instability (CIN), microsatellite instability (MSI) and CpG island methylator phenotype (CIMP).
Early-onset CRC (EOCRC) represents a relatively unusual entity commonly related with hereditary forms of the disease. Thus, it is estimated that about 11% of colon cancers and 18% of rectal cancers arise in individuals younger than 50 years [4], [5], [6], [7]. In comparison with late-onset CRC, EOCRC is more frequently associated with poor clinical features and it is considered as a high-risk group within CRC [8], [9].
The clinicopathological features of tumors can differ significantly depending on the type of genomic alterations, which makes CRC a heterogeneous disease in which it is difficult to determine the clinical consequences of individual alterations. Although some studies have attempted to correlate the clinicopathological features and the molecular profile in late-onset tumors [10], [11], [12], this relationship has not been fully investigated in EOCRC, possibly because of the low frequency of CRC in young people [4], [5]. In our previous work, we performed a comprehensive analysis of the DNA copy number alterations (CNAs) that occur in two groups of patients differing in age at onset, and observed substantial dissimilarities regarding the CIN pattern as well as the most frequent CNAs arising in each group [13]. Taking this as a starting point, the purpose of the present study was to investigate whether the CIN profile is also associated with the biological characteristics and/or with the global genomic status (based on MSI and CIMP) in EOCRC, when an unsupervised hierarchical clustering analysis is performed according to the similarity of the CNAs detected.
Materials and Methods
Patients, Samples and Data Collection
A total of 88 individuals diagnosed with CRC at an age of 45 years or younger (range: 16–45 years) were collected at the 12 de Octubre University Hospital in Madrid. Family history of cancer (including at least three generations) and clinicopathological information was obtained for each patient, with a follow-up of at least 60 months from surgery. All patients (or a first degree relative in case of death of the index case) provided written consent, and the study was approved by the Ethics Committee of our Institution.
Six patients were excluded because familial adenomatous polyposis was diagnosed. Material for array comparative genomic hybridization (aCGH) analysis could be obtained from 60 of the remaining 82 patients. In our series, the left location was considerably more common than the right one (53.3% vs. 20%) (Supplementary Table S1). Moreover, and as expected given the early-onset of the disease, the percentage of sporadic cases was lower than the percentage of patients who had a familial component or fulfilled the clinical criteria for Lynch syndrome (LS). Additional clinical, pathological and familial features are shown in Supplementary Table S1.
Assessment of Genomic Instability: Molecular Classification
A tissue specimen was obtained from each index case. Microscopic inspection of paraffin-embedded samples was performed by a pathologist, and samples with more than 70% of tumor cells in the neoplastic material were considered adequate for further analysis. The protocol for DNA isolation was as previously reported [13].
We used the Bethesda panel to assess the MSI status, and considered a result positive when two or more markers were altered. Blood samples were taken from the MSI index cases to assess germline mutations in MLH1, MSH2 and MSH6. Moreover, MSI tumors were analyzed for the BRAF V600E mutation in order to identify possible sporadic cases. For the assessment of CIMP, we investigated the methylation status of the promoter regions of CACNA1G, CDKN2A, CRABP1, IGF2, MLH1, NEUROG1, RUNX3 and SOCS1. CIMP-High was defined as the presence of ≥6/8 methylated promoters, CIMP-Low as 1/8 to 5/8 methylated promoters and CIMP-0 as the absence of methylated promoters [14]. We classified tumors into four categories according to the MSI and CIMP status as described by Ogino and Goel: (1) MSI/CIMP-High; (2) MSI/CIMP-Low/0; (3) MSS/CIMP-High; (4) MSS/CIMP-Low/0 [15]. Finally, the degree of CIN was evaluated by aCGH, considering tumors with more than 3 whole chromosomes affected as CIN+, tumors with 1–3 whole chromosomes affected as MACS (microsatellite and chromosome stable), and tumors with no whole chromosome affected as CIN-.
The procedures for the evaluation of CIN, MSI, and CIMP were as previously reported [9], [13].
Unsupervised Analysis of aCGH Data
Tumors were clustered based on the copy number states of their windowed probes [13]. Unsupervised analysis was performed using hierarchical clustering algorithms (squared Euclidean distances) implemented in Multi Experiment Viewer 4.8.1 (www.tm4.org/mev.html).
Statistical Analysis
Comparison of continuous variables was done using Student's two-tailed t test (for normal distributions) or the Mann–Whitney U test (for nonparametric distributions), whereas comparison of categorical variables was done using Pearson's chi square (χ2) test. For comparisons between more than two groups, analysis of variance (ANOVA) (for normal distributions) or the Kruskal-Wallis test (for nonparametric distributions) were used. Statistical analysis was performed using SPSS 17.0 (SPSS Inc., Chicago, IL, USA), and differences were considered statistically significant when the P < .05.
The study of association and correlation was performed employing R Statistical Software [16] using Fisher's exact test, analysis of variance and the independent sample t test. P < .05 was considered statistically significant.
Results
Unsupervised Analysis According to the CIN Profile
The unsupervised hierarchical clustering stratified samples into two main clusters. The first of them (cluster A) was composed of 38 cases while the second one (cluster B) was composed of 22 cases (Fig. 1; Supplementary Fig. 1). Both clusters, in turn, were divided into two secondary clusters: subcluster A1 (n = 25) and A2 (n = 13) derived from A, and subcluster B3 (n = 10) and B4 (n = 12) derived from B (Fig. 1; Supplementary Fig. 1).
Fig. 1.

Unsupervised hierarchical clustering based on the genomic instability profiles of the studied tumors. The studied samples were stratified into two main clusters (A, B) and four secondary clusters (A1, A2, B3, B4) according to the similarity of the CNAs detected.
Clinicopathological Features in the Different Clusters
Cluster A included a larger percentage of very young patients (<36 years at diagnosis) and all tumor recurrences (P = .035), and was associated with a worse prognosis (Fig. 2; Table 1). In the comparison of subgroups A1, A2, B3 and B4, statistical significance was only reached when tumor histology was considered (Table 1). However, it should be emphasized that there were some distinctive clinicopathological features between the different subgroups. On the one hand, subgroup A1 was associated with the highest percentage of tumor recurrences. On the other hand, the subgroup including the highest proportion of stage IV tumors (B4) showed the highest death rates as well as the highest prevalence of rectal location (Table 1). As expected, the right-sided location (typically related with LS) was predominant within the subcluster with the highest percentage of individuals with familial antecedents of the disease (A2) (Table 1).
Fig. 2.

Kaplan–Meier survival plots: (A) Comparison of the DFS between the two main clusters; (B) comparison of the DFS between the four subclusters; (C) comparison of the OS between the two main clusters; (D) comparison of the OS between the four subclusters. Note the significantly higher proportion of tumor recurrence within the cluster A.
Table 1.
Clinicopathological correlation of the different subgroups obtained by unsupervised hierarchical clustering analysis
| Cluster A1 | Cluster A2 | Cluster B3 | Cluster B4 | P-value [1] | |
|---|---|---|---|---|---|
| No. of tumors | 25 | 13 | 10 | 12 | |
| 36–45 years old | 19 | 10 | 9 | 11 | NS |
| <36 years old | 6 (54.5%) | 3 (27.3%) | 1 (9.1%) | 1 (9.1%) | |
| Location | |||||
| Right colon | 5 (20%) | 5 (38.4%) | 1 (10%) | 1 (8.3%) | |
| Left colon | 15 (60%) | 4 (30.8%) | 7 (70%) | 6 (50%) | NS |
| Rectum | 5 (20%) | 4 (30.8%) | 2 (20%) | 5 (41.7%) | |
| Histology | |||||
| Adenocarcinoma | 22 (88%) | 7 (53.8%) | 9 (90%) | 11 (91.7%) | 0.034 |
| Malignant adenoma | 3 (12%) | 6 (46.2%) | 1 (10%) | 1 (8.3%) | |
| T. differentiation: | |||||
| High | 10 (45.5%) | 3 (42.9%) | 4 (44.4%) | 2 (18.2%) | |
| Moderate | 8 (36.4%) | 4 (57.1%) | 5 (55.6%) | 9 (81.8%) | NS |
| Low | 4 (18.1%) | - | - | - | |
| Mucin production | 7 (31.8%) | 1 (14.3%) | 3 (33.3%) | 2 (18.2%) | NS |
| “Signet ring” cells | 1 (4.5%) | - | 1 (11.1%) | - | NS |
| TNM stage | |||||
| I | 6 (24%) | 6 (46.2%) | 2 (20%) | 3 (25%) | |
| II | 9 (36%) | 4 (30.8%) | 7 (70%) | 1 (8.3%) | NS |
| III | 5 (20%) | 1 (7.6%) | 1 (10%) | 3 (25%) | |
| IV | 5 (20%) | 2 (15.4%) | - | 5 (41.7%) | |
| Average No. of polyps | 3.76 [1.00] | 6.00 [4.00] | 2.40 [2.00] | 1.33 [0.00] | NS[2] |
| Synchronous T. | 2 (8%) | 1 (7.7%) | - | - | NS |
| Metachronous T. | - | 2 (15.4%) | - | - | NS |
| OS ± SD | 87.12 ± 50.85 | 99.08 ± 63.18 | 86.10 ± 10.03 | 59.50 ± 36.07 | NS[3] |
| DFS ± SD | 72.60 ± 53.90 | 78.38 ± 65.68 | 84.50 ± 9.48 | 51.42 ± 44.26 | NS[3] |
| T. recurrence | 5 (20%) | 2 (15.4%) | - | - | NS |
| Mortality | 7 (28%) | 3 (23.1%) | - | 4 (33.3%) | NS |
| Sporadic | 13 (52%) | 3 (23.1%) | 5 (50%) | 6 (50%) | |
| Familial aggregation | 9 (36%) | 6 (46.2%) | 1 (10%) | 4 (33.3%) | NS |
| Amsterdam II positive | 3 (12%) | 4 (30.7%) | 4 (40%) | 2 (16.7%) | |
| Lynch syndrome | 2/7 (28.6%) | 3/7 (42.8%) | 2/7 (28.6%) | - | NS |
Data shown in brackets represent median values. [1]Statistical comparison was performed using Pearson's Chi Square (χ2) test.[2] Statistical comparison was performed using the Kruskal-Wallis test. [3]Statistical comparison was performed using analysis of variance (ANOVA). ⁎DFS: Disease-free survival. †No.: Number. ‡NS: Not significant. §OS: Overall survival. ᵟSD: Standard deviation. ᵠT.: Tumor.
Distribution of MSI and CIMP
Thirteen patients (16%) of a total of 81 showed MSI and the concomitant loss of expression of one or more of the DNA mismatch repair (MMR) proteins. Of the 13 patients with MSI, 7 had a pathogenic germline mutation in one of the MMR genes, 4 had variants of unknown significance in at least one of these genes, and 1 showed lack of expression of MLH1 and hypermethylation of its gene promoter (data not shown).
Of the 60 tumors that could be evaluated by aCGH, MSI was present in 10 cases, 7 of which were confirmed as LS patients (1 had a mutation in MLH1, 4 in MSH2 and 2 in MSH6) (Table 2). There was only 1 patient with hypermethylation of the MLH1 promoter; however, the BRAF V600E mutation was absent in this patient. MSI was homogeneously distributed among clusters A and B, with a complete correlation with the immunohistochemical results (Table 2). Regarding the methylator phenotype, we observed a clear prevalence of CIMP-High tumors within cluster A, with all but one of the tumors contained therein showing this genotype (Table 2). When we compared the subclusters derived from the main clusters, we observed that only B4 showed absence of MSI tumors (being also the cluster with the lowest proportion of right-sided tumors), and that B3 was the only subgroup entirely composed of CIMP-Low/0 tumors (Table 2).
Table 2.
Global distribution of MSI and CIMP among the different subgroups obtained by unsupervised hierarchical clustering analysis
| Cluster A1 | Cluster A2 | Cluster B3 | Cluster B4 | P-value[1] | |
|---|---|---|---|---|---|
| No. of tumors | 25 | 13 | 10 | 12 | - |
| MSI | 2 (8%) | 3 (23.1%) | 5 (50%) | - | 0.007 |
| Expression of MMR: | |||||
| Normal | 23 | 10 | 5 | - | 0.007 |
| Absence | 2 | 3 | 5 | - | |
| Lynch Syndrome | 2 | 3 | 2 | - | NS |
| MMR genes affected: | |||||
| MLH1 | 1 | - | - | - | - |
| MSH2 | - | 3 | 1 | - | |
| MSH6 | 1 | - | 1 | - | |
| BRAF mutation | - | - | - | 1 (100%) | - |
| CIMP-High | 7 (28%) | 4 (30.8%) | - | 1 (8.3%) | NS |
| CIMP-Low/0 | 18 (72%) | 9 (69.2%) | 10 (100%) | 11 (91.7%) | |
| Mol. classification: | |||||
| MSI/CIMP-High | 1 (4%) | 2 (15.4%) | - | - | 0.004 |
| MSI/CIMP-Low/0 | 1 (4%) | 1 (7.7%) | 5 (50%) | - | |
| MSS/CIMP-High | 6 (24%) | 2 (15.4%) | - | 1 (8.3%) | |
| MSS/CIMP-Low/0 | 17 (68%) | 8 (61.5%) | 5 (50%) | 11 (91.7%) |
Statistical comparison was performed using Pearson's Chi Square (χ2) test. ⁎CIMP: CpG island methylator phenotype. †MMR: Mismatch repair. ‡Mol.: Molecular. §MSI: Microsatellite instability. ᵟMSS: Microsatellite instability. ᵠNo.: Number. ᵜNS: Not significant.
Distribution of CIN
Clusters A and B did not differ in terms of CNAs nor with respect to the total genomic instability index (GII), even though cluster B showed a slightly higher value of GII total and a lower number of CNAs per sample. However, the average aneuploidy frequency was significantly higher in cluster B (P = .026; Supplementary Table S2). Interestingly, subgroup A1 showed significantly higher values for GII total (P = .001), GII gains (P = .0001), GII losses (P = .030) and average number of CNAs per sample (P = .002), and contained almost the entire instability associated with cluster A (Supplementary Table S3). By contrast, we did not find significant differences between B3 and B4, despite the higher degree of instability in the first subgroup (Supplementary Table S4). In the comparison of the four secondary subgroups, A2 demonstrated significantly lower values for almost all parameters suggestive of CIN. As a result, it was the subgroup with the highest percentage of CIN- tumors (P = .003; Table 3).
Table 3.
Global description and comparison of genomic instabilities observed for the different subgroups obtained by unsupervised hierarchical clustering analysis
| Cluster A1 | Cluster A2 | Cluster B3 | Cluster B4 | P-value[1] | |
|---|---|---|---|---|---|
| No. of tumors | 25 | 13 | 10 | 12 | - |
| GII total Mean [SD] | 0.40010 [0.26115] | 0.11184 [0.21733] | 0.39149 [0.27826] | 0.30166 [0.31020] | 0.017 |
| Average CNAs/tumor | 135.16 (118.00) | 45.69 (42.00) | 95.30 (104.00) | 68.50 (32.50) | 0.048 |
| GII gains Mean [SD] | 0.18963 [0.10511] | 0.03881 [0.09584] | 0.19251 [0.22203] | 0.15778 [0.15866] | 0.016 |
| Average CNAs gained/tumor: | 72.32 (58.00) | 11.38 (7.00) | 39.10 (29.50) | 32.42 (14.50) | 0.0001[2] |
| > 1 Mb | 58.28 (48.00) | 6.08 (4.00) | 26.50 (18.00) | 24.42 (7.50) | 0.0001[2] |
| < 1 Mb | 14.04 (11.00) | 5.31 (5.00) | 11.60 (11.50) | 8.00 (4.50) | 0.047[2] |
| GII losses Mean [SD] | 0.21047 [0.21216] | 0.07303 [0.14926] | 0.19898 [0.11768] | 0.14388 [0.16880] | NS[2] |
| Average CNAs lost/tumor: | 62.84 (58.00) | 34.31 (42.00) | 56.20 (72.00) | 36.08 (16.50) | NS |
| > 1 Mb | 42.12 (20.00) | 11.46 (14.00) | 40.80 (51.00) | 23.75 (12.00) | 0.024[2] |
| < 1 Mb | 20.72 (15.00) | 22.85 (24.00) | 15.40 (15.00) | 12.33 (3.00) | NS |
| Average of aneuploidy: | 2.83 (3.00) | 1.46 (0.00) | 4.44 (4.00) | 4.08 (3.00) | NS |
| Whole chromosomes gained | 1.76 (2.00) | 0.76 (0.00) | 1.80 (0.50) | 2.58 (1.50) | 0.010[2] |
| Whole chromosomes lost | 0.96 (0.00) | 0.69 (0.00) | 2.20 (3.00) | 1.50 (1.00) | NS[2] |
| CIN- tumors | 4 (20%) | 10 (50%) | 3 (15%) | 3 (15%) | 0.003[3] |
| CIN+ tumors | 9 (39.1%) | 2 (8.7%) | 6 (26.1%) | 6 (26.1%) | |
| MACS | 12 (70.6%) | 1 (5.9%) | 1 (5.9%) | 3 (17.6%) |
Data shown in parentheses represent median values. Tumors with more than 3 whole chromosomes affected were considered as CIN+; tumors with 1–3 whole chromosomes affected were considered as MACS; tumors with no whole chromosome affected were considered as CIN-. [1]Statistical comparison was performed using analysis of variance (ANOVA). [2]Statistical comparison was performed using the Kruskal-Wallis test. [3]Statistical comparison was performed using Pearson's Chi Square (χ2) test. ⁎CIN: Chromosomal instability. †CNAs: Copy number alterations. ‡GII: Genomic instability index. §MACS: Microsatellite and chromosome-stable. ᵟMb: Megabasepairs. ᵠNo.: Number. ᵜNS: Not significant. ᶘSD: Standard deviation.
Concerning aneuploidy, we observed that the vast majority of MACS (also MSS) were included in A1, whereas the vast majority of CIN- tumors were included in A2 (Table 3). As expected, the most unstable subgroup (A1) included the highest proportion of MSS tumors and the most stable subgroup (A2) included the highest proportion of MSI tumors (Tables 2 and 3); this is not surprising, given that CIN and MSI are carcinogenesis pathways traditionally considered as mutually exclusive. Moreover, we observed some aneuploid events that seemed to be more frequent in some subgroups than in others. Thus, alterations in chromosomes 19 and 22 were predominant in subcluster A1 (60%), whereas alterations in chromosomes 4, 11 and 15 were predominant in B3 (40%) and alterations in chromosome 13 were predominant in B4 (67%).
Recurrent CNAs
Among the most frequent alterations in cluster A were gains at 19p13 (72%) and 9q33-q34 (62%), and losses at 5q13 (67%) and 1q12-q21 (62%), while the most frequent alterations in cluster B were gains at 8q22-q24 and 13q13-q14 (41%), and losses at 12q24 (50%). In addition, there were 63 recurrent alterations (frequencies greater than or equal to 30%) occurring with statistically significantly different frequencies in clusters A and B (Supplementary Table S5). On the other hand, gains at 19p13-p12 and 19q13 (96%) were common in A1, but only losses at 2q12-q13 (64%) were recurrent in A2. Finally, gains at 4q31-q32, 5q14, 12p13-q13 and 13q12 (100%), and losses at 17q11 (80%) were frequent in B3, whereas gains at 1p36-p34, 2p25-p24, 2q11-q14, 2q37, 3p25, 3q21-q23, 7p14, 7q32 and 16p13-p11 (92%) were frequent in B4.
Molecular Classification
Based on MSI and CIMP status, we defined four molecular groups as described in Materials and Methods [15]. MSS/CIMP-Low/0 tumors were the most frequent whereas MSI/CIMP-High tumors were uncommon (Table 2). Moreover, MSI/CIMP-High tumors were related with LS patients and/or Amsterdam II-positive families.
The unsupervised analysis clustered the samples into four groups according to the similarity of their CNAs. These groups showed a certain degree of correspondence with the molecular classification proposed by Ogino and Goel [15], in such a way that we were able to outline an algorithm by which tumors might be categorized according to their genomic status based on CIMP. Thus, the CIMP-High tumors were mostly contained within clusters A1 or A2 depending on the CIN degree, whereas the CIMP-Low/0 tumors were mostly contained within clusters B3 or B4 depending on the presence/absence of MSI (Fig. 3). Interestingly, the genomic instability of each subcluster mostly affected particular chromosomes allowing us to define chromosomal regions more specifically affected depending on the CIMP/MSI status of the samples. The most frequent events of CIN in A1 involved chromosomes 19 and 22, whereas in B3 they involved chromosomes 4, 11 and 15, and in B4 they involved chromosome 13 (Fig. 3).
Fig. 3.
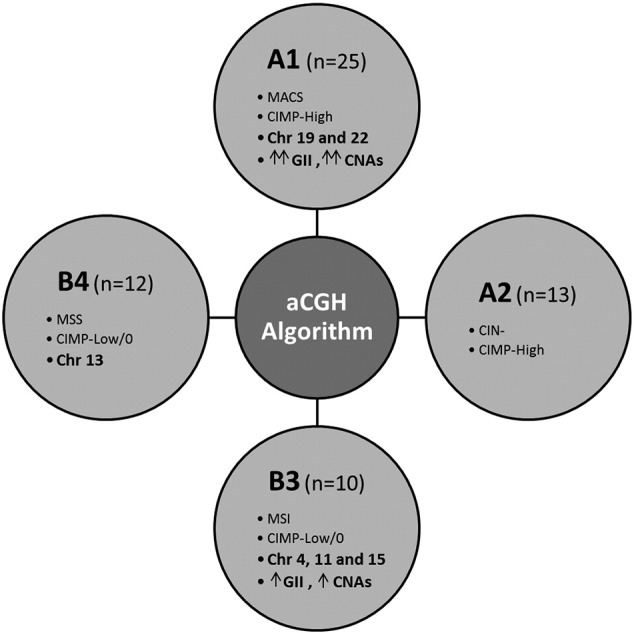
Algorithm by which tumors may be categorized according to their genomic status based on CIN, MSI and CIMP. The CIMP-High tumors were mostly contained within clusters A1 or A2 depending on the CIN degree, whereas the CIMP-Low/0 tumors were mostly contained within clusters B3 or B4 depending on the MSI status.
Discussion
In a previous study, we confirmed the existence of substantial differences between the CIN pattern of early-onset and late-onset colorectal tumors, suggesting that genomic profiles may be associated with age of onset in CRC [13]. In the present study, our aim was to investigate whether the CIN profile is also associated with the clinicopathological implications and/or with the global genomic status based on MSI and CIMP in EOCRC. For this purpose, we analyzed a cohort of 60 EOCRCs and performed an unsupervised hierarchical clustering analysis of the aCGH data.
The unsupervised analysis organized samples into two main clusters (A and B) and four secondary clusters (A1, A2, B3, B4) (Fig. 1). No significant differences were observed regarding the clinicopathological features of the tumors included in clusters A and B, and they were overall those expected considering the familial component of each one of them (Table 1). Both MSI and CIMP showed a heterogeneous distribution across the clusters, with CIMP-High tumors mostly stratified into cluster A and CIMP-Low/0 tumors mostly stratified into cluster B (Table 2). There were no differences either between the subgroups derived from the main clusters, and it was not surprising that the cluster with the highest percentage of metastasis at diagnosis (B4) exhibited the lowest values of OS (overall survival) and DFS (disease-free survival), that is, they had the worst prognosis (Table 1).
It is well known that sporadic tumors commonly develop via the CIN pathway, which is characterized by chromosomal imbalances (aneuploidy) and a high frequency of loss of heterozygosity [17]. On this basis, it would be expected that tumors contained in cluster B showed a higher degree of CIN, as has in fact been observed (Table 3). Contrary to expectations, the average number of CNAs per sample was higher in cluster A.
The average of whole chromosomes affected was significantly higher within the cluster with the lowest hereditary component (cluster B; P = .026) even though we did not observe statistical differences regarding the GII total (Table 3). Interestingly, we observed that A1 contained almost the entire instability linked to cluster A. Therefore, the lack of statistical differences in the comparison of the GII total between A and B may be understood as a consequence of the masking of the high degree of instability associated to A1, because in a normal distribution the mean of the sampling distribution is equal to the mean of the population. Despite the fact that high levels of CIN have been related to a worse prognosis in CRC [18], in our series the worst clinical outcomes were linked to the cluster with a lower average of GII total (cluster A) which showed a significantly higher proportion of tumor recurrence (P = .035; Fig. 2). Probably, this contradiction is a consequence of the high discrepancy between A1 and A2 in terms of CIN being neutralized when cluster A was evaluated as a whole entity.
Each subcluster manifested prevalence of certain events of CIN. The most frequent aneuploidies in A1 involved chromosomes 19 and 22 (60%) where some genes related to CRC are located (MMP11, GSTT2, CHEK2 and EP300). Moreover, about 96% of tumors included in A1 showed gains at 19p13-p12 and 19q13 while only losses at 2q12-q13 (64%) were recurrent in A2. This is an interesting finding because 19p13-p12 and 19q13 harbor genes previously related with the development of malignancy and growth progression of certain tumors such as STK11 (a tumor suppressor related to Peutz-Jeghers syndrome) or MADCAM1, whose relationship with inflammatory bowel disease has been reported [19]. More interesting, the region 2q12-q13 harbors BUB1, a gene encoding a mitotic checkpoint serine/threonine-protein kinase whose alterations have been related to early-onset CRC [20]. The most frequent aneuploidies observed in B3 affected chromosomes 4, 11, 15 (40%). In this respect, it is important to mention that losses of large parts of chromosome 4 (where some genes important for carcinogenesis appear to be located) are repeatedly reported in several cancers [21], [22], [23], [24], [25], [26], [27], [28]. Also supporting our results, there are many studies that identify different loci in chromosomes 11 and 15 as candidates for CRC susceptibility [29], [30], [31], [32], [33], [34]. Finally, it is worth mentioning chromosome 13 since all tumors of B3 showed alterations in 13q12. This region contains CDX2, a specific transcription factor expressed during intestinal development whose lack of expression appears to be associated with a poor prognosis in CRC [35]. In addition, chromosome 13 was the autosome most frequently affected by aneuploidy within subcluster B4 (67%), which may be important given the large clinical and molecular heterogeneity of this subgroup.
Our unsupervised analysis revealed a certain parallelism with the molecular classification proposed by Ogino and Goel [15], which on the one hand allowed us to outline an algorithm by which tumors may be classified according to their genomic instability features, and, on the other hand, allowed us to define some chromosomal alterations recurrently affected depending on the CIMP/MSI status of the samples (Fig. 3). On this basis, the CIMP-High/CIN- tumors were mainly included within the cluster A whereas the CIMP-Low/0/CIN+ tumors were mainly included within the cluster B. In the same way, the MSI tumors were predominant in A2 and B3 whereas the MSS tumors were predominant in A1 and B4. Therefore, the cluster A1 mostly included the CIMP-High/MSS/CIN- tumors, the cluster A2 mostly included the CIMP-High/MSI/CIN- tumors, the cluster B3 mostly included the CIMP-Low/0/MSI/CIN+ tumors and the cluster B4 mostly included the CIMP-Low/0/MSS/CIN+ tumors. Interestingly, the tumors contained within A1 frequently showed alterations in chromosomes 19 and 22, the tumors contained in B3 frequently showed alterations in chromosomes 4, 11 and 15, and the tumors contained in B4 frequently showed alterations in chromosome 13 (Fig. 3). Validation and improvement of this algorithm in further studies may ultimately provide a new form of classifying EOCRC according to the CNAs detected, and consequently might have a significant impact on the understanding of the still poorly characterized EOCRC.
Acknowledgements
We thank the Tumor Registry of the Pathology Department of the 12 de Octubre University Hospital for providing the paraffin-embedded tissues, and Ron Hartong for his help with the English revision of this manuscript.
Footnotes
Financial support: This work was supported by Projects PI10/0683, PI13/01741, PI13/0127 from the Spanish Ministry of Health and Consumer Affairs; FEDER; and Mari Paz Jiménez Casado Foundation.
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.neo.2016.11.006.
Appendix A. Supplementary data
Supplementary materials.
References
- 1.Jemal A., Bray F., Center M.M., Ferlay J., Ward E., Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61(2):69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Ferlay J., Shin H.R., Bray F., Forman D., Mathers C., Parkin D.M. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127(12):2893–2917. doi: 10.1002/ijc.25516. [DOI] [PubMed] [Google Scholar]
- 3.Leslie A., Carey F.A., Pratt N.R., Steele R.J. The colorectal adenoma-carcinoma sequence. Br J Surg. 2002;89(7):845–860. doi: 10.1046/j.1365-2168.2002.02120.x. [DOI] [PubMed] [Google Scholar]
- 4.Boyle P., Ferlay J. Cancer incidence and mortality in Europe, 2004. Ann Oncol. 2005;16(3):481–488. doi: 10.1093/annonc/mdi098. [DOI] [PubMed] [Google Scholar]
- 5.Siegel R.L., Jermal A., Ward E.M. Increase in incidence of colorectal cancer among young men and women in the United States. Cancer Epidemiol Biomarkers Prev. 2009;18(6):1695–1698. doi: 10.1158/1055-9965.EPI-09-0186. [DOI] [PubMed] [Google Scholar]
- 6.Sia C.S., Paul E., Wale R.J., Lynch A.C., Heriot A.G., Warrier S.K. No increase in colorectal cancer in patients under 50 years of age: a Victorian experience from the last decade. Colorectal Dis. 2014;16:690–695. doi: 10.1111/codi.12648. [DOI] [PubMed] [Google Scholar]
- 7.Ahnen D.J., Wade S.W., Jones W.F., Sifri R., Mendoza Silveiras J., Greenamyer J., Guiffre S., Axilbund J., Spiegel A., You Y.N. The increasing incidende of young-onset colorectal cancer: A call to action. Mayo Clin Proc. 2014;89:216–224. doi: 10.1016/j.mayocp.2013.09.006. [DOI] [PubMed] [Google Scholar]
- 8.Banerjea A., Hands R.E., Powar M.P., Bustin S.A., Dorudi S. Microsatellite and chromosomal stable colorectal cancers demonstrate poor immunogenicity and early disease recurrence. Colorectal Dis. 2009;11(6):601–608. doi: 10.1111/j.1463-1318.2008.01639.x. [DOI] [PubMed] [Google Scholar]
- 9.Perea J., Rueda D., Canal A., Rodríguez Y., Álvaro E., Osorio I., Alegre C., Rivera B., Martínez J., Benítez J. Age at Onset Should Be a Major Criterion for Subclassification of Colorectal Cancer. J Mol Diagn. 2014;16(1):116–126. doi: 10.1016/j.jmoldx.2013.07.010. [DOI] [PubMed] [Google Scholar]
- 10.Hawkins N.J., Tomlinson I., Meagher A., Ward R.L. Microsatellite-stable diploid carcinoma: a biologically distinct and aggressive subset of sporadic colorectal cancer. Br J Cancer. 2001;84(2):232–236. doi: 10.1054/bjoc.2000.1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oliveira C., Velho S., Moutinho C., Ferreira A., Preto A., Domingo E., Capelinha A.F., Duval A., Hamelin R., Machado J.C. KRAS and BRAF oncogenic mutations in MSS colorectal carcinoma progression. Oncogene. 2007;26(1):158–163. doi: 10.1038/sj.onc.1209758. [DOI] [PubMed] [Google Scholar]
- 12.De Angelis P.M., Clausen O.P., Schjølberg A., Stokke T. Chromosomal gains and losses in primary colorectal carcinomas detected by CGH and their associations with tumour DNA ploidy, genotypes and phenotypes. Br J Cancer. 1999;80(3–4):526–535. doi: 10.1038/sj.bjc.6690388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arriba M., García J.L., Inglada-Pérez L., Rueda D., Osorio I., Rodríguez Y., Álvaro E., Sánchez R., Fernández T., Pérez J. DNA copy number profiling reveals different patterns of chromosomal instability within colorectal cancer according to the age of onset. Mol Carcinog. 2015 doi: 10.1002/mc.22315. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 14.Ogino S., Kawasaki T., Kirkner G.J., Kraft P., Loda M., Fuchs C.S. Evaluation of markers for CpG island methylator phenotype (CIMP) in colorectal cancer by a large population-based sample. J Mol Diagn. 2007;9(3):305–314. doi: 10.2353/jmoldx.2007.060170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ogino S., Goel A. Molecular classification and correlates in colorectal cancer. J Mol Diagn. 2008;10:13–27. doi: 10.2353/jmoldx.2008.070082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.R Core Team . R Foundation for Statistical Computing; Vienna, Austria: 2012. R: A language and environment for statistical computing. [ISBN 3-900051-07-0, URL http://www.R-project.org/] [Google Scholar]
- 17.Dunican D.S., McWilliam P., Tighe O., Parle-McDermott A., Croke D.T. Gene expression differences between the microsatellite instability (MIN) and chromosomal instability (CIN) phenotypes in colorectal cancer revealed by high-density cDNA array hybridization. Oncogene. 2002;21(20):3253–3257. doi: 10.1038/sj.onc.1205431. [DOI] [PubMed] [Google Scholar]
- 18.Sinicrope F.A., Rego R.L., Halling K.C., Foster N., Sargent D.J., La Plant B., French A.J., Laurie J.A., Goldberg R.M., Thibodeau S.N. Prognostic impact of microsatellite instability and DNA ploidy in human colon carcinoma patients. Gastroenterology. 2006;131(3):729–737. doi: 10.1053/j.gastro.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 19.Arihiro S., Ohtani H., Suzuki M., Murata M., Ejima C., Oki M., Kinouchi Y., Fukushima K., Sasaki I., Nakamura S. Differential expression of mucosal addressin cell adhesion molecule-1 (MAdCAM-1) in ulcerative colitis and Crohn's disease. Pathol Int. 2002;52(5–6):367–374. doi: 10.1046/j.1440-1827.2002.01365.x. [DOI] [PubMed] [Google Scholar]
- 20.de Voer R.M., Geurts van Kessel A., Weren R.D., Ligtenberg M.J., Smeets D., Fu L., Vreede L., Kamping E.J., Verwiel E.T., Hahn M.M. Germline mutations in the spindle assembly checkpoint genes BUB1 and BUB3 are risk factors for colorectal cancer. Gastroenterology. 2013;145(3):544–547. doi: 10.1053/j.gastro.2013.06.001. [DOI] [PubMed] [Google Scholar]
- 21.Berg M., Agesen T.H., Thiis-Evensen E., INFAC-study group. Merok M.A., Teixeira M.R., Vatn M.H., Nesbakken A., Skotheim R.I., Lothe R.A. Distinct high resolution genome profiles of early onset and late onset colorectal cancer integrated with gene expression data identify candidate susceptibility loci. Mol Cancer. 2010;9:100. doi: 10.1186/1476-4598-9-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jiang L.X., Xu J., Wang Z.W., Li D.P., Peng Z.H., Gao J.J., He L., Zheng H.T. Tumor suppress genes screening analysis on 4q in sporadic colorectal carcinoma. World J Gastroenterol. 2008;14(36):5606–5611. doi: 10.3748/wjg.14.5606. [discussion 5609-10] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ried T., Knutzen R., Steinbeck R., Blegen H., Schröck E., Heselmeyer K., du Manoir S., Auer G. Comparative genomic hybridization reveals a specific pattern of chromosomal gains and losses during the genesis of colorectal tumors. Genes Chromosomes Cancer. 1996;15(4):234–245. doi: 10.1002/(SICI)1098-2264(199604)15:4<234::AID-GCC5>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 24.Polascik T.J., Cairns P., Chang W.Y., Schoenberg M.P., Sidransky D. Distinct regions of allelic loss on chromosome 4 in human primary bladder carcinoma. Cancer Res. 1995;55(22):5396–5399. [doi: Published 15 November 1995] [PubMed] [Google Scholar]
- 25.Shivapurkar N., Sood S., Wistuba I.I., Virmani A.K., Maitra A., Milchgrub S., Minna J.D., Gazdar A.F. Multiple regions of chromosome 4 demonstrating allelic losses in breast carcinomas. Cancer Res. 1999;59(15):3576–3580. [doi: Published 1 August 1999] [PubMed] [Google Scholar]
- 26.Hertz S., Rothämel T., Skawran B., Giere C., Steinemann D., Flemming P., Becker T., Flik J., Wiese B., Soudah B. Losses of chromosome arms 4q, 8p, 13q and gain of 8q are correlated with increasing chromosomal instability in hepatocellular carcinoma. Pathobiology. 2008;75(5):312–322. doi: 10.1159/000151712. [DOI] [PubMed] [Google Scholar]
- 27.Cetin E., Cengiz B., Gunduz E., Gunduz M., Nagatsuka H., Bekir-Beder L., Fukushima K., Pehlivan D., MO N., Nishizaki K. Deletion mapping of chromosome 4q22-35 and identification of four frequently deleted regions in head and neck cancers. Neoplasma. 2008;55(4):299–304. [PubMed] [Google Scholar]
- 28.Krona C., Carén H., Sjöberg R.M., Sandstedt B., Laureys G., Kogner P., Martinsson T. Analysis of neuroblastoma tumour progression; loss of PHOX2B on 4p13 and 17q gain are early events in neuroblastoma tumourigenesis. Int J Oncol. 2008;32(3):575–583. [PubMed] [Google Scholar]
- 29.Biancolella M., Fortini B.K., Tring S., Plummer S.J., Mendoza-Fandino G.A., Hartiala J., Hitchler M.J., Yan C., Schumacher F.R., Conti D.V. Identification and characterization of functional risk variants for colorectal cancer mapping to chromosome 11q23.1. Hum Mol Genet. 2014;23(8):2198–2209. doi: 10.1093/hmg/ddt584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hes F.J., Ruano D., Nieuwenhuis M., Tops C.M., Schrumpf M., Nielsen M., Huijts P.E., Wijnen J.T., Wagner A., Gómez García E.B. Colorectal cancer risk variants on 11q23 and 15q13 are associated with unexplained adenomatous polyposis. J Med Genet. 2014;51(1):55–60. doi: 10.1136/jmedgenet-2013-102000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fernandez-Rozadilla C., Cazier J.B., Tomlinson I., Brea-Fernández A., Lamas M.J., Baiget M., López-Fernández L.A., Clofent J., Bujanda L., Gonzalez D. A genome-wide association study on copy-number variation identifies a 11q11 loss as a candidate susceptibility variant for colorectal cancer. Hum Genet. 2014;133(5):525–534. doi: 10.1007/s00439-013-1390-4. [DOI] [PubMed] [Google Scholar]
- 32.Tu L., Yan B., Peng Z. Common genetic variants (rs4779584 and rs10318) at 15q13.3 contributes to colorectal adenoma and colorectal cancer susceptibility: evidence based on 22 studies. Mol Genet Genomics. 2015;290(3):901–912. doi: 10.1007/s00438-014-0970-x. [DOI] [PubMed] [Google Scholar]
- 33.Maleno I., Aptsiauri N., Cabrera T., Gallego A., Paschen A., López-Nevot M.A., Garrido F. Frequent loss of heterozygosity in the β2-microglobulin region of chromosome 15 in primary human tumors. Immunogenetics. 2011;63(2):65–71. doi: 10.1007/s00251-010-0494-4. [DOI] [PubMed] [Google Scholar]
- 34.Fitzgerald L.M., McDonnell S.K., Carlson E.E., Langeberg W., McIntosh L.M., Deutsch K., Ostrander E.A., D.J. Schaid D.J., Stanford J.L. Genome-wide linkage analyses of hereditary prostate cancer families with colon cancer provide further evidence for a susceptibility locus on 15q11-q14. Eur J Hum Genet. 2010;18(10):1141–1147. doi: 10.1038/ejhg.2010.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bae J.M., Lee T.H., Cho N.Y., Kim T.Y., Kang G.H. Loss of CDX2 expression is associated with poor prognosis in colorectal cancer patients. World J Gastroenterol. 2015;21(5):1457–1467. doi: 10.3748/wjg.v21.i5.1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary materials.