

Abstract
Objective
Despite widespread use of antibiotics for the treatment of life-threatening infections and for research on the role of commensal microbiota, our understanding of their effects on the host is still very limited.
Design
Using a popular mouse model of microbiota depletion by a cocktail of antibiotics, we analysed the effects of antibiotics by combining intestinal transcriptome together with metagenomic analysis of the gut microbiota. In order to identify specific microbes and microbial genes that influence the host phenotype in antibiotic-treated mice, we developed and applied analysis of the transkingdom network.
Results
We found that most antibiotic-induced alterations in the gut can be explained by three factors: depletion of the microbiota; direct effects of antibiotics on host tissues and the effects of remaining antibiotic-resistant microbes. Normal microbiota depletion mostly led to downregulation of different aspects of immunity. The two other factors (antibiotic direct effects on host tissues and antibiotic-resistant microbes) primarily inhibited mitochondrial gene expression and amounts of active mitochondria, increasing epithelial cell death. By reconstructing and analysing the transkingdom network, we discovered that these toxic effects were mediated by virulence/quorum sensing in antibiotic-resistant bacteria, a finding further validated using in vitro experiments.
Conclusions
In addition to revealing mechanisms of antibiotic-induced alterations, this study also describes a new bioinformatics approach that predicts microbial components that regulate host functions and establishes a comprehensive resource on what, why and how antibiotics affect the gut in a widely used mouse model of microbiota depletion by antibiotics.
Introduction
Antibiotics are widely used in people and animals, with estimates suggesting that 4 out of 10 adults and 7 out of 10 children receive antibiotics each year,1,2 along with billions of food animals.3 Although antibiotics are essential for the treatment of many life-threatening bacterial infections and have significantly increased the life expectancy of human populations, more than 10% of people who receive these medications suffer from adverse effects.4–6 Some of these side effects are known to be related to the perturbation in host resident microorganisms (microbiota) because along with elimination of pathogens antibiotics can lead to long-lasting disturbances in the commensal micro-biota.7–12 Antibiotic-induced changes in the micro-biota have been implicated in the development of such pathologies as diarrhoea, colitis, sepsis and an increased risk of inflammatory bowel disease, obesity and allergies.13–21 In experimental models and in human patients, the growth of opportunistic Clostridium difficile and infection with Salmonella are related to susceptibility to colitis caused by antibiotic use.12,22 Administration of antibiotics can also lead to deficit of the innate antimicrobial protein Reg3γ that decreases resistance against antibiotic-resistant pathogens.23 These effects have led to the emerging concept that the use of antibiotics may negatively affect host physiology by altering the composition of the microbiota; however, a comprehensive understanding of these alterations is not clear.
Antibiotics also have a long history in research using animal models, and a recent surge in investigations on the physiological role of the normal microbiota has resulted in a number of studies that use mixtures of antibiotics as an alternative to germ-free experimental animals (whose production is expensive and time-consuming).24 Although this strategy has been successful in providing evidence for the involvement of the microbiota in a particular physiological or pathophysiological process (such as the role of some commensal bacteria in tumour susceptibility to chemotherapy),25 antibiotic-induced depletion can also be misleading.26 For example, in a study on the interaction between intestinal B cells and the microbiota, the use of germ-free animals allowed us to uncover links that were not apparent using antibiotic-treated animals.27 This discrepancy suggested that the effect(s) of antibiotics can be more complex than mere depletion of microbiota.
We aimed to comprehensively understand potential differences between the absence of microbiota (as in germ-free animals) and the effects of antibiotics using a popular cocktail of antibiotics.24,28–32 We took a top-down systems biology approach to examine antibiotic-induced changes in the intestinal transcriptome and microbiome of mice. We found that intestinal alterations resulting from antibiotic treatment could be explained by three major influences: (1) depletion of microbiota by antibiotics; (2) direct effects of antibiotics on host tissues; (3) effects of microbes surviving after the antibiotics treatment (denoted antibiotic-resistant in this study). The effect of microbe depletion mostly resulted in depression of immunity in the gut, while the two other influences were surprisingly similar, mediating repression of mitochondrial function and death of the intestinal epithelium. Finally, we created a novel analytic tool, combining microbiome gene abundances and intestinal transcriptome data, to reveal ‘transkingdom’ gene networks that drive intestinal epithelial function.
Materials and Methods
Mice and antibiotics
C57BL/6 mice were obtained from the Jackson Labs, Swiss-Webster, B10A and BALB/c mice were obtained from Taconic Farms. Germ-free Swiss Webster mice were obtained from Taconic farms Gnotobiotic Center. C57BL/6 germ-free mice were obtained from the National Gnotobiotic Rodent Resource Center at the University of North Carolina. The study was approved by the NIAID, NIH and Oregon State University Animal Care and Use Committees. Mice used in the experiments were of both sexes and 2–4 months old. Antibiotics were administered in drinking water for 4–5 weeks in the following concentrations: ampicillin (1 g/L), vancomycin (0.5 g/L), neomycin trisulfate (1 g/L), metronidazole (1 g/L). Mouse small intestines (ileum) were either snap frozen for RNA isolations or processed for epithelial cell isolations as described in online supplementary methods.
Gene expression microarrays
RNA from ileum samples was isolated, labelled, hybridised to Agilent arrays and scanned as described in ref. 27. All the analyses were performed using BRB Array Tools developed by the Biometric Research Branch of the National Cancer Institute under the direction of R. Simon (http://linus.nci.nih.gov/BRB-ArrayTools.html). Full description of the analyses is provided in online supplementary material. Raw and processed data files are available under GSE60568 super-series in the Gene Expression Omnibus (GEO) data repository.
Microbiota analysis
DNA from caecum content was isolated using QIAamp DNA Stool Mini Kit (Qiagen) following the protocol with optional 10 min 90°C incubation for better lysis of bacteria. For Phylochip microarrays, DNA was amplified and hybridised according to manufacturer's protocols (Affymetrix). Phylotrac33 was used in order to retrieve normalised probe results. All further analyses including ‘class comparison’ between different groups and NMF sample clustering were performed using BRB Array Tools.
For microbiome sequencing, DNA libraries were prepared using Illumina Nextera sample preparation kit and Illumina HiSeq2000 was used for sequencing libraries. The sequencing resulted in 2 816 120 546 sequences for 32 samples. Because microbiota in normal mice had higher diversity than in antibiotic (ABx)-treated mice, we performed deeper sequencing for control mice (4 samples per lane) than for ABx-treated (12 samples per lane) with averages (±SD) of 113 248 484 (±37 769 496) reads and 38 102 424 (±12 613 196) reads per sample, respectively. Quality filtering, alignments, differential abundance and network analysis were performed as described in online supplementary material.
Network reconstruction and interrogation
Detailed description of the procedures is provided in ref 64 and online supplementary material.
Confocal microscopy, laser microdissection, evaluation of mitochondria and cell viability
Detailed description of the procedures is provided in online supplementary material.
Statistical analysis of data (except for microarrays and sequencing data)
For paired comparisons we used paired t test or Wilcoxon matched-paired signed-ranked test. For non-paired comparisons we used t test or Mann-Whitney test. We used non-parametric tests in case the data does not pass normality test (p<0.05). The outliers were identified as values outside of mean±3SD and removed from the further analysis.
Results
We started with a cocktail of four antibiotics (ampicillin, vancomycin, neomycin and metronidazole) that were originally designed to deplete cultivable microbes,24 and investigated the global effects of this cocktail on the microbiota and on host intestinal tissues of treated mice. Because the commensal micro-biota differ in mice from different vendors,34 and because host genetics can also influence the microbial makeup, we compared antibiotic-treated and conventional mice on two different genetic backgrounds obtained from two different vendors.
Changes in microbial populations after treatment with antibiotics
We first evaluated the effect of the antibiotic cocktail on the microbiota. As expected, ABx treatment induced a pronounced drop in total bacterial mass per gram of intestinal content (see online supplementary figure S1). Analysis of bacterial 16S ribosomal RNA (rRNA) genes by Phylochip demonstrated that ∼5500 taxonomic probes were significantly decreased (figure 1A). However, there was also a relative increase in about 320 probes, mostly corresponding to bacteria of the class Gammaproteobacteria (order Enterobacteriales and a few other orders such as Pseudomonadales) and class Betaproteobacteria.
Figure 1.
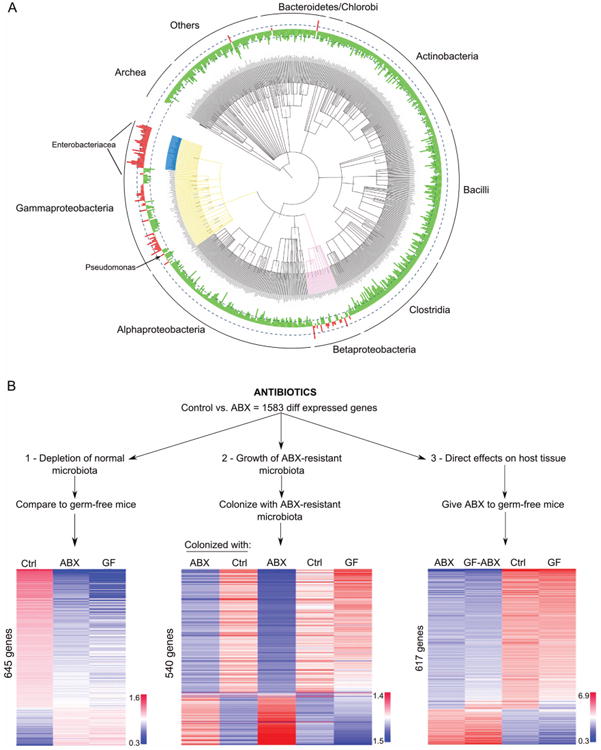
(A) Taxonomic distribution (class level) of gut microbiota after ABx treatment created, using the Tree of Life Web Project http://tolweb. org. Green are depleted and red are taxa enriched by antibiotics, respectively; height of columns corresponds to the fold change in abundance; dashed lines denote 10 times fold change; main bacterial taxa enriched in antibiotics-treated mice are highlighted in blue (Enterobacteriacea), yellow (Gammaproteobacteria) and pink (Betaproteobacteria). See also online supplementary figures S1 and S2; (B) design of the study and heatmaps for host gene expression in the ileum across the groups. Each column corresponds to median value of the group; rows of genes are ranked by fold change in Ctrl versus ABx; colours represent relative log intensities of expression of genes normalised to a reference sample (n=7–16 mice per group); Ctrl, control; ABX, antibiotic-treated; GF, germ-free mice.
Antibiotic-induced expression changes in the host can be attributed to three major factors
We next inquired about the effects of the cocktail on the host's intestinal tissues by comparing global gene expression of ileum samples taken from ABx-treated versus untreated conventional mice. We found a set of 1583 genes that were differentially expressed (see online supplementary table S1; FDR<10%) between the two groups, the majority of which (1180 genes) showed lower expression in ABx-treated than in control mice.
To determine the impact of the three potential influencing factors (depletion of microbes and their functions, direct effect of antibiotics on host tissues, and effects of remaining antibiotic-resistant microbes), we designed three different sets of experiments (figure 1B).
First, to reveal which of these 1583 antibiotic-induced gene expression changes might be affected by depletion of normal microbiota, we compared their expression levels in mice that completely lack microbes (germ-free mice) with expression in ABx-treated and conventional mice. We found that 645 of the 1583 genes were similarly regulated in germ-free and ABx-treated animals (figure 1B, left panel). Therefore, although it is commonly thought that the majority of antibiotic-induced effects are due to depletion of commensal microbiota, our results indicate that this is true for only about one-third of the ABx-induced gene expression changes in the gut.
Second, to reveal the potential effects of ABx-resistant microbes (in the absence of antibiotics), we colonised germ-free mice with microbes from antibiotic-treated or conventional mice, and assessed gene expression and bacterial content in the gut 3 and 7 days later (at which time the bacterial distribution in the intestine of colonised mice resembled that of their respective (conventional or antibiotic-treated) donors (see online supplemental figure S2)). We observed that changes in expression of 540 of the 1583 ABx-regulated genes were similar between ABx-treated mice and germ-free mice colonised with antibiotic-resistant microbes (figure 1B, middle panel), but different from those colonised with normal control microbiota.
Third, to assess the direct effect of antibiotics on mouse tissues (in the absence of potential microbial effects), we treated germ-free mice with the antibiotic cocktail, and found that 617 genes out of the 1583 were similarly affected by antibiotics in both germ-free and conventional mice (figure 1B, right panel). Thus, about a third of the antibiotic-induced changes in gene expression are regulated by the antibiotics directly, and not through the depletion of the microbiota or the outgrowth of antibiotic-resistant organisms.
Altogether, almost 90% of the changes in gene expression (1380 genes out of 1583) could be clearly attributed to one of these three factors (figure 2A). Some were influenced by more than one. For example, a group of 135 genes that were regulated in germ-free mice (ie, effect of the absence of normal microbiota) were also concordantly regulated by antibiotics in the absence of microbes (108/135 repressed). The largest, and most surprising, overlap was between the genes directly regulated by antibiotics and those regulated by antibiotic-resistant microbes (258 genes; figure 2A). Genes in this intersection were similarly regulated by both factors: that is, the 182/258 genes directly repressed by ABx were also repressed in the presence of ABx-resistant microbes (see online supplementary figure S3).
Figure 2.
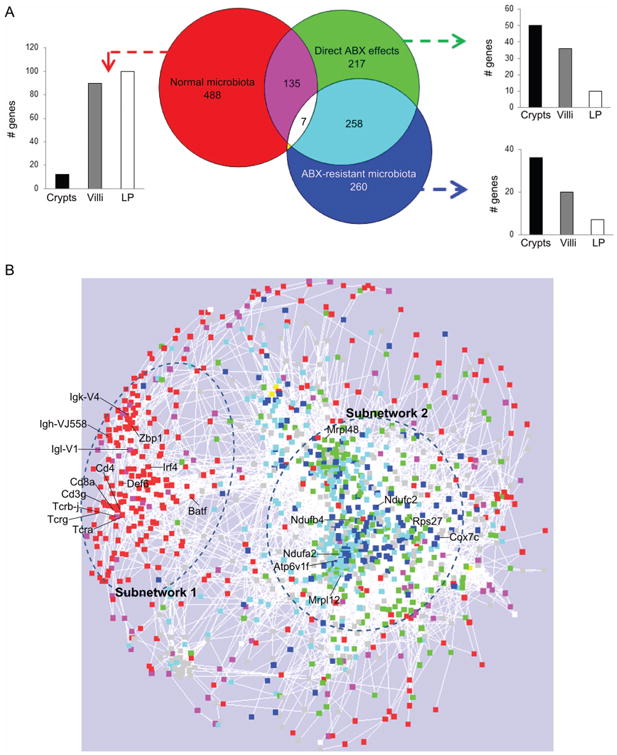
(A) Venn diagram analysis of host differentially expressed genes affected by each factor (normal microbiota, direct antibiotic (ABX) effects and ABX-resistant microbiota; and assignment of the genes to crypts, villous epithelium (villi), lamina propria (LP). See also online supplementary figure S3; (B) gene expression network of 1583 differentially expressed genes; colours correspond to the Venn diagram analysis, grey corresponds to genes not attributed to any of the three factors; symbols are genes; circles define the subnetworks #1 (left) and #2 (right); see also online supplementary figure S4.
The differentially expressed genes form a network with two functionally and geographically distinct subnetworks
In order to understand whether these different gene groupings were related to different functions, we first asked if they were expressed in different intestinal compartments (villous epithelium, lamina propria, crypts). Using laser-microdissection of the small intestine (see online supplementary movie), we found that genes that were regulated by depletion of the normal microbiota were expressed predominantly in the lamina propria, whereas genes regulated by ABx directly, or by ABx-resistant bacteria, as well as the 258-gene overlapping set, were mostly expressed in crypts and villous epithelium (figure 2A). Thus, the depletion of microbes appeared to affect a distinct subset of genes and tissues compared with the direct effects of antibiotics and the effects of antibiotic-resistant microbes.
To uncover regulatory relationships among the three regulated gene groups, we reconstructed a gene expression network of all of the ABx-regulated genes, using an approach that we previously applied in other models to gain insights into regulatory relationships between differentially expressed genes.27,35 Briefly, we analysed pairwise correlations between expression levels of the 1583 genes in control mice, allowing natural covariation in gene expression to reveal connections in the network. Using correlations that were consistent across four groups of mice on different genetic backgrounds, we found a network consisting of 1275 genes that contained two distinct subnetworks (figure 2B). Subnetwork #1 was largely composed of the microbiota-dependent genes (figure 2B, red and magenta) while subnetwork #2 contained similar numbers of genes that were regulated directly by antibiotics and those regulated by antibiotic-resistant microbes (including their overlap; figure 2B, turquoise, green, blue). This high connectedness indicates that both antibiotics and the antibiotic-resistant microbiota might influence the same biological functions (ie, a group of coordinately regulated genes) rather than affect functionally unrelated biological processes.
Functional analysis of subnetwork #1 shows its role in mucosal immunity
Gene ontology (GO) enrichment analysis (figure 3A) demonstrated that subnetwork #1 was highly enriched for functions of adaptive and innate immunity. Among the most enriched GO categories were several T and B lymphocyte processes, antigen presentation and innate defense responses (figure 3A). Many components of mucosal immunity were downregulated after ABx treatment, including immunoglobulin genes (plasma cells), transcripts for T-cell receptor (TCR) γ, CD8 α and granzymes (intraepithelial T lymphocytes); TCR β, CD8 β (lamina propria T lymphocytes), along with transcription factors such as Irf4 and Batf, which are critical for CD4 and CD8T cell differentiation. Interestingly, antibiotics brought down expression of T cell transcripts to a greater extent than those of plasma cells (see online supplementary figure S5). Genes expressed by myeloid cells were also repressed, including CD11c, Fc γ receptors, dectin 2 and other C-type lectins. In addition, immune activation of epithelial cells was also reduced, as evidenced by decreased expression of defensin 6, Zbp1, Duoxa2, serum amyloids and interferon-induced genes (see online supplementary table S1).
Figure 3.
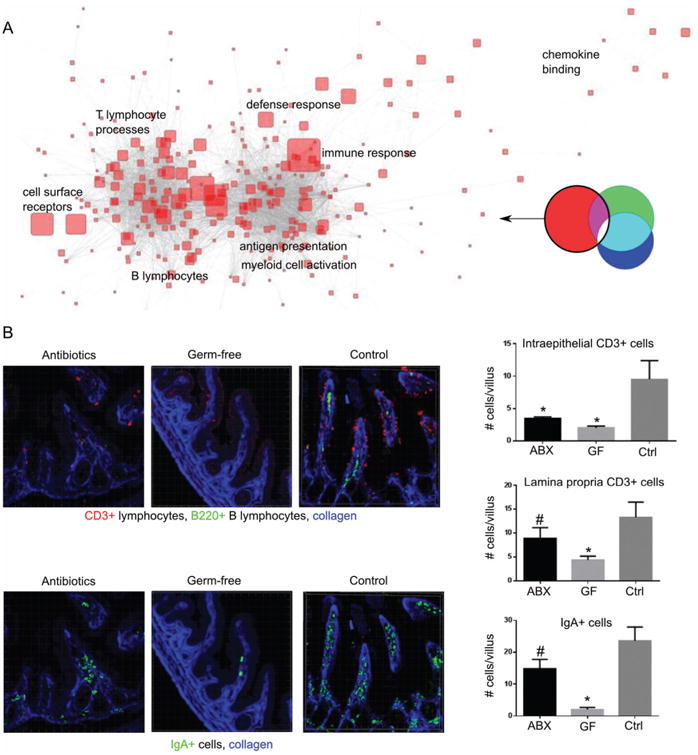
Effects dependent on normal microbiota depletion: (A) gene ontology categories over-represented in the genes influenced by normal microbiota. Size of symbols represents enrichment fold; only most enriched categories are indicated. (B) immunofluorescent staining for T cells (CD3+) and IgA+ plasma cells in ABx-treated, germ-free and control mice; n=4–6 mice per group; counted ∼6 fields/mouse containing 2–3 villi; *p<0.01; #p<0.1 compared to control; see also online supplementary figure S5. Ctrl, control; ABx, antibiotic-treated; GF, germ-free mice.
Because immune cells traffic through tissues, these changes in gene expression could reflect alterations of immune cell composition and/or changes in cellular gene expression. To discriminate between these possibilities, we measured the numbers of T lymphocytes and plasma cells (the two types of cells most clearly identified by the transcriptional network). Using immunofluorescent confocal microscopy, we observed a marked decrease in the number of intraepithelial T and B lymphocytes after antibiotic administration, and few IgA-producing cells remained (figure 3B). Thus, the ABx-induced drop in the normal microbiota induced some changes in gene expression that could be assigned to alterations in the cellular composition of the gut-associated immune system (such as intraepithelial and lamina propria T lymphocytes, plasma cells and myeloid cells), and other expression changes more likely to be due to modifications of individual cell function, such as adjustments in the innate immune properties of intestinal epithelial cells.
Analysis of subnetwork #2 points to alterations in mitochondria and increased cell death
The network analysis (figure 2B) indicated that the genes regulated directly by antibiotics and those regulated by antibiotic-resistant microbes are involved in one coregulated subnetwork, 70% of which were repressed by direct ABx effects and by ABx-resistant microbes (see online supplementary figure S3). GO enrichment analysis of the downregulated genes comprising subnetwork #2 pointed to several ribosomal and mitochondrial gene categories as being over-represented in this subnetwork (figure 4A). The most enriched categories included genes coding for electron transport chain, oxidation-reduction, ATP biosynthesis, and cellular and mitochondrial ribosomes. Strikingly, both antibiotics and antibiotic-resistant microbes induced repression of genes coding for proteins constituting all five complexes of the mitochondrial respiratory chain (see online supplementary figure S6).
Figure 4.
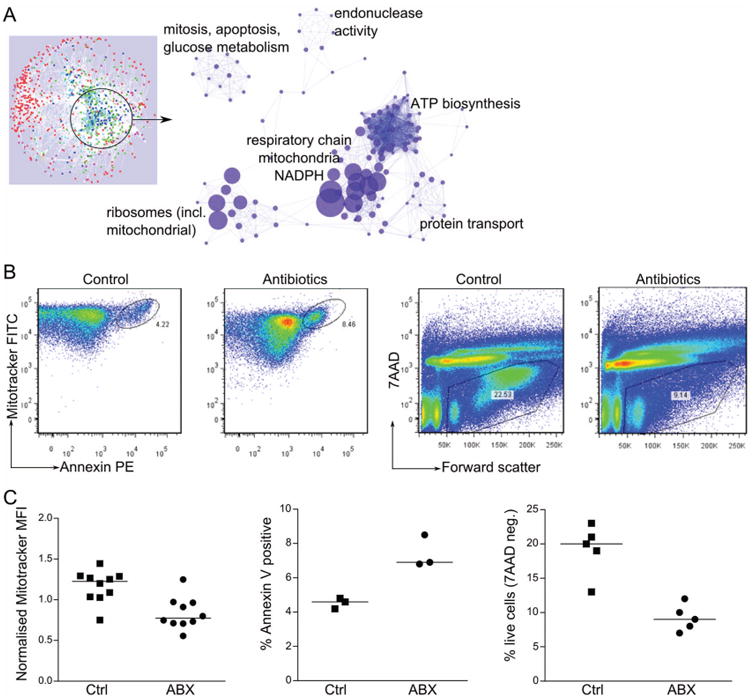
Effects of ABx and of ABx-resistant microbes on the host: (A) gene ontology categories over-represented in the genes of the subnetwork directly affected by ABx and by ABx-resistant microbiota. Size of the symbol represents enrichment fold; only most enriched categories are indicated. See also online supplementary figure S6; (B and C) mitochondrial staining (Mitotracker MFI normalised to the average MFI for each experiment), apoptosis (Annexin V) and cell viability (7AAD), in gut epithelial cells isolated from ABx-treated and control mice; (B) a single representative experiment; (C) summary graphs of 3–5 experiments; each symbol represents one mouse; median values are represented by horizontal lines; all comparisons significant at p<0.01. Ctrl, control; ABx, antibiotic-treated; MFI, mean fluorescent intensity.
Because genes from this subnetwork were predominantly assigned to the crypts and villous epithelium (figure 2A), we assessed the consequences of the impaired mitochondrial gene expression at the cellular level in epithelial cells. Mitotracker green staining of intestinal epithelial cells demonstrated that the numbers of mitochondria per epithelial cell were reduced in antibiotic-treated mice (figure 4B, C). As mitochondria play a major role in apoptosis induction, we also evaluated cell viability and apoptosis. Consistent with the general mitochondrial perturbation, we observed more apoptotic (annexinpos) and dead (7AADpos) cells in the epithelium of treated animals (figure 4B, C).
Metagenomics and transkingdom network reconstruction between genes of antibiotic-resistant microbiota and host genes
Although it has recently been shown36 that antibiotics can have off target effects on mitochondria, we were surprised to find that antibiotic-resistant microbes could do the same, in the complete absence of antibiotics. In retrospect, however, this result is consistent with the finding that mitochondria are descended from bacteria,37 and that bacteria compete with each other via a wide range of mechanisms, including the secretion of antibiotics.38
To identify potential mechanisms by which the antibiotic resistant microbes might modify gut mitochondria and gut epithelium, we performed a ‘transkingdom’ analysis, integrating information on the abundance of various bacterial genes with information on intestinal gene expression, to reveal regulatory relationships between the microbiota and the host. Similar integration of DNA copy numbers and gene expression was highly informative in our previous work for elucidating key gene drivers of cervical carcinogenesis.35
Shotgun sequencing of the microbiota demonstrated 4523 bacterial genes (FDR<0.07) with differential abundance between ABx and controls with 1689 genes enriched by ABx (see online supplementary table S2). Importantly, several well-known antibiotic-resistance genes had higher abundance in ABx-treated mice supporting the notion of antibiotic resistance in the microbiota surviving the treatment (see online supplementary table S2, category “Resistance to antibiotics and toxic compounds”). We then integrated abundances of 1689 ABx-enriched microbial genes in each individual mouse with mRNA levels of the host's genes assigned to the effect of the ABx-resistant microbiota (figure 2A). The resulting transking-dom network simultaneously reflects relationships within and between the host and microbial kingdoms (figure 5). Nodes in this network represent microbial and host genes and edges denote correlations between and within each group of genes. Of the 1689 bacterial genes enriched by antibiotics, 412 were connected to the host portion of the network.
Figure 5.
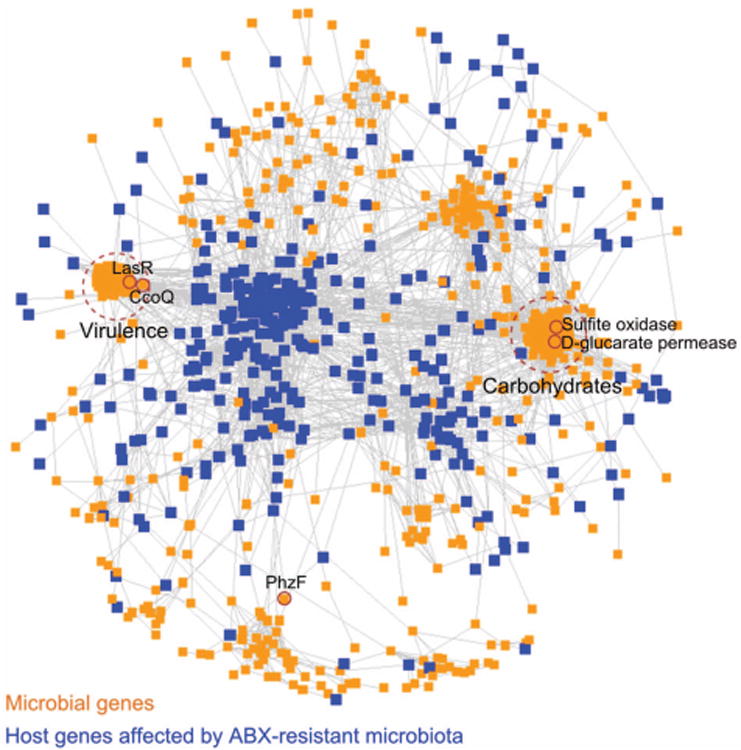
Transkingdom network—the network reconstructed from host gene expression corresponding to those driven by direct effect of antibiotics and antibiotic-resistant bacteria (figure 2) and DNA abundance for microbial genes enriched in ABx-treated mice. Microbial genes—orange; host genes—blue; edges denote correlations between genes; two major microbial subnetworks defined by dashed circles; see also online supplementary figure S7; microbial genes with top 1% value of betweenness centrality between host and microbial compartments are indicated by small solid circles and gene symbols.
Interrogation of the microbial portion of this transkingdom network revealed two major subnetworks containing the highest number of genes connected to the mammalian expression levels (see online supplementary figure S7). One subnetwork was enriched for virulence genes and the other subnetwork had over-representation of carbohydrate metabolism according to the first level functional annotation from SEED database 39,40 (see online supplementary table S3). Deeper investigation of the ‘virulence’ subnetwork (ie, SEED second level) demonstrated enrichment for type III secretion system, iron scavenging mechanisms, quorum sensing and biofilm formation while the ‘carbohydrate’ subnetwork was over-represented for monosaccharides, disaccharides, oligosaccharides, and some metabolic pathways (see online supplementary table S4).
Although the ABx-resistant microbes were much less diverse than the conventional microbiota (figure 1A), they nevertheless represented more than a hundred different taxa. Therefore, it was not clear whether the observed effects in ABx-treated mice were due to complex changes in microbial communities or to the outgrowth of specific microbes. To discriminate between these possibilities, we asked how many different taxa might be contributing to the two major microbial subnetworks. While the ‘virulence’ subnetwork was comprised of genes from a small number of taxa (median of 17 taxa per gene), the taxonomic diversity for the ‘carbohydrate’ subnetwork, and for other bacterial genes in the transkingdom network, was significantly higher (medians of 72 and 110 taxa per gene, respectively; figure 6A). The few taxa that mapped to the genes of the first subnetwork were dominated by various types of Pseudomonas aeruginosa (figure 6B), while most of the taxa from the second subnetwork were from the Enterobacteriaceae family (see online supplementary figure S8). To identify candidate microbial genes that may have critical roles in the effects of bacteria on the host, we searched the transkingdom network for microbial genes that represent ‘bottlenecks’ in the connection between microbial and mouse genes, that is, those with high betweenness centrality (see M&M). This approach has been useful in revealing key disease genes in gene/protein networks.41,42 Among the top 1% of genes with the highest betweenness centrality, there were two (ccoQ, lasR) in the ‘virulence’ subnetwork enriched for P. aeruginosa, two others (sulfite oxidase, D-glucarate permease) in the ‘carbohydrate’ subnetwork, and the phzF gene not assigned to any subnetwork (figures 5 and 6C). These microbial genes were therefore the top candidates to be involved in the repression of mitochondrial function in the gut epithelium by ABx-resistant microbiota.
Figure 6.
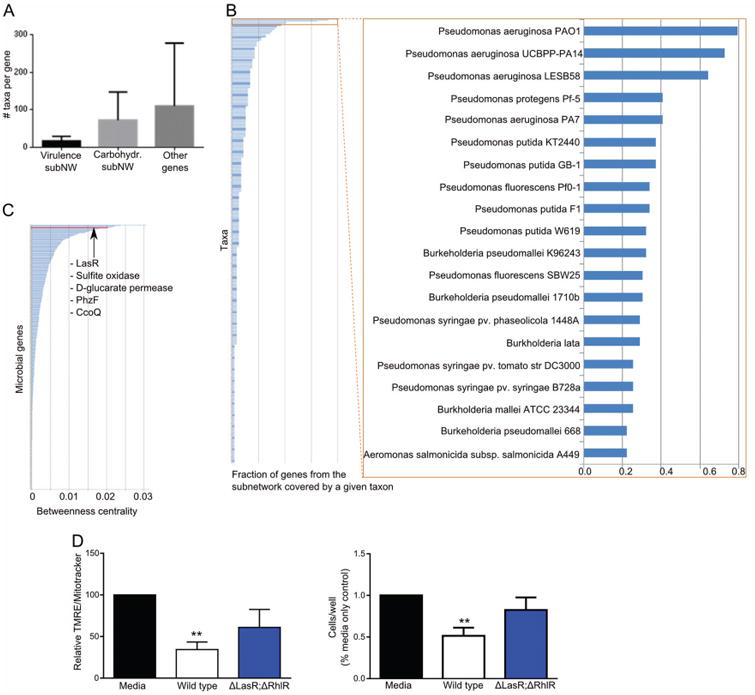
Predicting and validating a key microbial gene driving changes in the host mitochondria. (A) Number of microbial taxa to which genes from two microbial subnetworks (subNW) or all other microbial genes from the transkingdom network (figure 5) can be assigned; (B) fraction of genes from virulence subnetwork (figure 5) that is found in the genome of a particular bacterial strain; see also online supplementary figure S8; (C) distribution of values of betweenness centrality for microbial genes calculated for connections between host and microbial compartments of the transkingdom network; (D) mitochondrial activity (left) and cell viability (right) of MODE-K epithelial cells of control (untreated cells, black column), cells treated with supernatants from wild type Pseudomonas aeruginosa PAO1 (white column), or with quorum sensing mutant of P. aeruginosa (lasR/rhlR knockouts, blue column); n=3 independent experiments.
P. aeruginosa is a human commensal that can become pathogenic, develop multidrug resistance and secrete a variety of extracellular enzymes and toxins that damage host tissue.43–47 Considering these aspects as well as the fact that the ‘virulence’ subnetwork is enriched for P. aeruginosa genes, we studied the possibility that P. aeruginosa might be one of the key members of the ABx-associated microbiota that cause depression of host mitochondria and cell death. Focusing on candidate genes from the ‘virulence’ subnetwork, we found that, whereas cytochrome c oxidase (ccoQ) is encountered in the genomes of a wide variety of microbes, LasR is almost uniquely present in P. aeruginosa. This gene codes for the major quorum-sensing transcription factor in P. aeruginosa, and is known to regulate numerous secreted virulence factors in a cell density-dependent manner.48 Thus, the reconstruction and analysis of the transkingdom network associated with ABx treatment allowed us to hone in on a candidate microbe (P. aeruginosa) and a candidate gene (LasR) as potential mediators of the mitochondrial depression and host cell death caused by antibiotic-resistant microbes.
In order to test these predictions, we coincubated an epithelial cell line with supernatant from in vitro cultured P. aeruginosa and saw a decrease in mitochondrial membrane potential and viability (figure 6). To test the effect of quorum sensing in our system, we used a P. aeruginosa quorum-sensing mutant with a lasR knockout. The mutant was also deficient for a second, subordinate quorum-sensing transcription factor, RhlR. We chose the double knockout because these mutations together block expression of all QS-dependent secreted virulence factors. None of the mutations affect bacterial growth in culture. Epithelial cells treated with supernatant from quorum-sensing-deficient P. aeruginosa cultures showed improved mitochondrial membrane potential and cell survival compared to those treated with supernatants from wild type bacteria (figure 6D). Thus, quorum-sensing-controlled factors secreted by P. aeruginosa have an inhibitory effect on the mitochondria and viability of mouse gut cells.
Discussion
Although it is generally thought that antibiotics mediate their major effects by killing bacteria, we found that this accounts for only about one-third of the changes seen in intestinal gene expression of mice treated with a popular cocktail of antibiotics. The other two-thirds were due to direct effects of the antibiotics on host tissues, and to the effects of the outgrowth of antibiotic-resistant bacteria. Analysing germ-free mice, germ-free mice treated with antibiotics and germ-free mice colonised with microbiota from antibiotic-treated or normal mice, and using a novel systems biology approach to construct transkingdom networks, we were able to hone in on some of the mechanisms by which these three factors influenced intestinal cell physiology, and to create a resource that should be valuable for future work by our laboratory and others.
Microbiota depletion leads to local immunodeficiency
Previous studies have shown that antibiotic treatment induces a decrease in immunity in the gut including decreased numbers of Th17-producing and IFNγ-producing lymphocytes, reduced expression of pro-IL1b, pro-IL-18 and of the antimicrobial peptides resistin-like β, Reg3b and Reg3 g.28,49,50 None of these investigations, however, addressed whether the observed effects resulted from the depletion of normal microbiota or from other factors operating in antibiotic-treated animals. We demonstrated that several aspects of mucosal immunity were indeed perturbed in antibiotic-treated mice because of depletion of commensal microbiota, as they were also perturbed in germ-free mice. Laser-capture microdissection indicated that these genes mostly mapped to the lamina propria and villous epithelium, reflecting the normally high abundance of immune cells in these two compartments that disappear in the absence of microbes. Interestingly, IgA-producing plasma cells were also reduced in antibiotic-treated mice, but to a significantly lesser extent than in germ-free animals and not as much as T cells (see figure 3B and online supplementary figure S5) indicating a long life span of mucosal plasma cells in the absence of antigen. Consistent with this interpretation, it was shown that plasma cells can persist in the gut for up to 16 weeks after encounter with microbes, even though the bacteria have long disappeared.51
Differential loss of T cells compared with plasma cells could be due to their high dependence on the presence of antigen, differences in life spans and/or to reduction in homing of T cells after microbiota removal by antibiotics.52 In this regard, we observed a decreased expression of several chemokines including CCL5 after antibiotic treatment (see online supplementary table S1). It was previously demonstrated that CCL5/CCR5 interaction contributes to the mucosal recruitment of T cells.53,54
Antibiotics have direct toxic effects on host tissues
The direct toxic effects of antibiotic treatment are commonly missed, most likely because of difficulty in discriminating between different mechanisms in the gut when multiple factors can contribute to the final phenotype. However, in systems where a microbial factor is not present, such as in vitro cultures, antibiotics have been shown to elicit mitochondrial damage by targeting various mitochondrial functions,55 and a recent study observed that antibiotics were toxic to all five complexes of mitochondrial respiratory chain leading to oxidative damage in mammalian cells.36 The most plausible hypothesis is that these mitochondrion-associated side effects are related to the fact that mitochondria evolved from ancient bacteria and, although very distant, they seem to carry the key features enabling toxic effects of antibiotics. In our study, in addition to the mitochondrial changes, we also found that antibiotic-treated germ-free mice exhibited suppression of gene expression related to both cellular and mitochondrial ribosomes. At least two non-mutually exclusive mechanisms may explain this observation: the ABx might inhibit RNA polymerase I that transcribes rRNA, or a deficit in ATP can lead to repression rRNA promoters as their activation requires ATP-dependent chromatin remodelling.56
Transkingdom network as a model of interactions between host and antibiotic-resistant pathobionts
The most surprising finding in our study was the discovery that ABx-resistant microbes had toxic effects that were highly overlapping with those of the antibiotics themselves. Functional enrichment and network analysis revealed that, like the antibiotics, the antibiotic-resistant microbes affected mitochondrial respiration and mitochondrial ribosomes. Thus, the intriguing question was which bacteria and which bacterial genes were responsible for this mitochondrial inhibition.
To address this problem, we developed a new approach that models host-microbe interactions. This approach consisted in metagenomics analysis and reconstruction of a transkingdom network integrating the abundances of microbial genes and host gene expression in one network. A similar strategy was successful in our recent work elucidating the mechanisms underlying cervical cancer35 where gene copy numbers for cancer gene regulators were correlated with expression levels of the mRNA for both regulator and target genes. A recent study of human micro- biota reported a correlation between gene copy number and transcript level for a considerable fraction of genes indicating that metagenomics information can reflect the functional state of microbiota.57 Thus, we reasoned that copy numbers for bacterial genes in the intestine might reflect activity of various bacterial functions, some of which would affect host mitochondria. By correlating these bacterial gene copy numbers with the levels of intestinal gene transcripts, we constructed a ‘transkingdom’ network that delineated a comprehensive model of the key processes at play in the antibiotic-resistant microbial community, and how those influence host gene expression. We revealed that the virulence-enriched subnetwork and P. aeruginosa were the most prominent candidates for mitochondrial inhibition in the host cells. Focusing on ‘bottleneck’ microbial genes (genes with high betweenness centrality), we identified LasR as one of putative key genes responsible for the effect of virulence associated genes from P. aeruginosa. Although multiple groups (reviewed in refs. 58 and 59), including ours,27,35,60,64 have widely used network analysis for uncovering underlying biological processes and master regulators within single organisms, this study is the first to integrate information about microbial genomes with host gene expression into one regulatory transkingdom network to reveal microbial gene candidates that regulate host functions.
P. aeruginosa induces mitochondrial depression and death of gut epithelium
It has not previously been demonstrated that P. aeruginosa is involved in intestinal antibiotic-associated disorders; however, the bacterium has been shown to be able to kill worms61 and mammalian cells in vitro62 by mechanisms involving oxidative stress61 and mitochondrial dysfunction.62 In our study, death of the intestinal epithelium might be a result of mitochondrial repression by factors regulated by the master regulator of quorum sensing, LasR. One such factors might be cyanide, whose synthase was over-represented in the antibiotic-resistant microbiota (see online supplementary figure S9).
Taking all this together, we can speculate how Pseudomonas-induced gut epithelial cell death occurs in vivo. Treatment with antibiotics results in depletion of the majority of commensals, and decreases gut immunity, providing an opportunity for Pseudomonas and other antibiotic-resistant bacteria to grow and increase in density. The increased density, in turn, allows for the activation of quorum-sensing-controlled virulence factors.48 These factors, including cyanide, directly stimulate mitochondria-dependent apoptosis in the intestinal epithelium.
It might seem unexpected that antibiotic-resistant bacteria have very similar effects on intestinal tissue as do the antibiotics themselves. However, the original antibiotics were molecules produced by microbes to compete with each other in their natural environment. Given that mitochondria are descended from α-proteobacteria,37 and still maintain many bacterial features, it could be anticipated that they would be targets for both natural antibiotics made by bacteria, and synthetic antibiotics administered by humans.
Practical considerations and data resource
The cocktail of antibiotics in this study has been used in hundreds of other studies and continues to be widely employed not only in microbiology and immunology, but also in more distant fields like neuroscience63 because of the rise in research on the role of the microbiome. However, an important practical consideration from our findings is that, despite the fact that antibiotics can substitute for germ-free mice in some specific instances, they are far from identical situations, and the absence of an effect after ABX treatment can lead to the false conclusion of no involvement of microbiota in the given phenotype. Thus, it would be useful, for any specific set of experiments, to be able to make the decision between using germ-free animals or antibiotics in advance.
To this end, we offer here a user-friendly tool for making an informed decision about this choice (see online supplementary table S1). This resource provides information on: (1) which genes in the intestine are affected by the cocktail of antibiotics; (2) why they change their expression (ie, microbiota depletion, direct effect or rise of ABx-resistant bacteria); (3) in which compartment specific changes occur (crypt, lamina propria, villous epithelium) and (4) which group of bacteria (or sometimes which specific bacterium) seem to be responsible for the changes in the host. The use of this resource can help predict if the gene or phenotype of interest that is known (or suspected) to be modulated by the microbiota is also modulated directly by antibiotic administration, thus allowing the decision of using germ-free animals or antibiotics for a specific set of experiments to be made in advance.
Conclusions
In this study, we have dissected the effects of antibiotics on the mouse gut, revealing microbiome-dependent and independent mechanisms. Depletion of commensals results in local immunodeficiency, whereas both direct effects of the antibiotics and proliferation of antibiotic-resistant microbes induce mitochondria-dependent cell death in the intestine, a phenomenon that has not been previously appreciated. We also developed a new bioinformatics approach that pinpoints microbes and microbial genes regulating host physiological functions, and established a comprehensive data set to allow other investigators to examine the effects of antibiotics.
Supplementary Material
Significance of this study.
What is already known on this subject?
Use of antibiotics is essential in clinical practice but is associated with several adverse side effects related to the perturbation of microbiota.
In research, antibiotics are frequently employed to examine the role of microbiota.
No comprehensive evaluation of the effects of antibiotics on the host tissues and microbiota in a popular model of microbiota depletion by a cocktail of antibiotics that is intended to mimic germ-free situation.
What are the new findings?
Depletion of normal microbiota by antibiotics led to a decline in immune gene expression and decrease in T lymphocyte numbers in the gut but IgA-producing plasma cells were less affected.
There was a repression in mitochondrial gene expression and mitochondrial functions as a direct effect of antibiotics on mammalian cells and as an effect of microbiota-resistant microbiota.
Transkingdom gene networks uncovered that virulence/quorum sensing genes of Pseudomonas aeruginosa were critical in inducing mitochondrial dysfunction in the host cells.
How might it impact on clinical practice in the foreseeable future?
Although not used as a cocktail in clinical practice, individual antibiotics constituting the mix are prescribed to patients. Therefore, our findings may be considered for the management of patients treated with long courses of antibiotics by including strategies to enhance immunity and to protect mitochondria.
Acknowledgments
This research was supported by startup funds for AM and NS from Oregon State University (OSU), USA, Medical Research Foundation of Oregon New Investigator Award (NS) and by the Intramural Research Program of the NIAID, NIH. The authors thank Ekaterina Peremyslova for the library preps for sequencing; OSU Center for Genome Research and Biocomputing; Maureen Bower from National Gnotobiotic Rodent Resource Center, University of North Carolina supported by P40RR018603.
Funding Division of Intramural Research, National Institute of Allergy and Infectious Diseases. Oregon State University Medical Research Foundation of Oregon
Footnotes
Contributors AM and NS conceived, led and supervised the study; performed most of animal and microarray experiments; analysed and/or supervised analyses of all data and wrote the manuscript. AD participated in study design and analyses, performed confocal microscopy and laser microdissection. XD performed metagenomic and network analyses, drafted respective parts of the manuscript. RLG participated in design and performed in vitro experiments. DJS and MS participated in design of experiments with Pseudomonas aeruginosa and prepared bacterial cultures. WH participated in design and coguided analysis of shotgun sequencing for metagenomics. PM participated in design of the study, interpretation of results and writing of manuscript. JR performed Phylochip microarray experiments.
Competing interests None
Provenance and peer review Not commissioned; externally peer reviewed
Data sharing statement Raw and processed data files are available under GSE60568 super-series in the Gene Expression Omnibus (GEO) data repository. Microbiome sequencing data are available at NCBI (SRA), BioProject ID: PRJNA258166.
References
- 1.Sharland M SACAR Paediatric Subgroup. The use of antibacterials in children: a report of the Specialist Advisory Committee on Antimicrobial Resistance (SACAR) Paediatric Subgroup. J Antimicrob Chemother. 2007;60(Suppl 1):i15–26. doi: 10.1093/jac/dkm153. [DOI] [PubMed] [Google Scholar]
- 2.European Commission. Antimicrobial resistance. Brussels: 2010. [Google Scholar]
- 3.Gustafson RH, Bowen RE. Antibiotic use in animal agriculture. J Appl Microbiol. 1997;83:531–41. doi: 10.1046/j.1365-2672.1997.00280.x. [DOI] [PubMed] [Google Scholar]
- 4.Hempel S, Newberry SJ, Maher AR, et al. Probiotics for the prevention and treatment of antibiotic-associated diarrhea: a systematic review and meta-analysis. JAMA. 2012;307:1959–69. doi: 10.1001/jama.2012.3507. [DOI] [PubMed] [Google Scholar]
- 5.Dancer SJ. How antibiotics can make us sick: the less obvious adverse effects of antimicrobial chemotherapy. Lancet Infect Dis. 2004;4:611–19. doi: 10.1016/S1473-3099(04)01145-4. [DOI] [PubMed] [Google Scholar]
- 6.Power SE, O'Toole PW, Stanton C, et al. Intestinal microbiota, diet and health. Br J Nutr. 2014;111:387–402. doi: 10.1017/S0007114513002560. [DOI] [PubMed] [Google Scholar]
- 7.Cho I, Yamanishi S, Cox L, et al. Antibiotics in early life alter the murine colonic microbiome and adiposity. Nature. 2012;488:621–6. doi: 10.1038/nature11400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jernberg C, Lofmark S, Edlund C, et al. Long-term ecological impacts of antibiotic administration on the human intestinal microbiota. ISME J. 2007;1:56–66. doi: 10.1038/ismej.2007.3. [DOI] [PubMed] [Google Scholar]
- 9.Dethlefsen L, Huse S, Sogin ML, et al. The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol. 2008;6:e280. doi: 10.1371/journal.pbio.0060280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Antonopoulos DA, Huse SM, Morrison HG, et al. Reproducible community dynamics of the gastrointestinal microbiota following antibiotic perturbation. Infect Immun. 2009;77:2367–75. doi: 10.1128/IAI.01520-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hill DA, Hoffmann C, Abt MC, et al. Metagenomic analyses reveal antibiotic-induced temporal and spatial changes in intestinal microbiota with associated alterations in immune cell homeostasis. Mucosal Immunol. 2010;3:148–58. doi: 10.1038/mi.2009.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ubeda C, Taur Y, Jenq RR, et al. Vancomycin-resistant Enterococcus domination of intestinal microbiota is enabled by antibiotic treatment in mice and precedes bloodstream invasion in humans. J Clin Invest. 2010;120:4332–41. doi: 10.1172/JCI43918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Greer RL, Morgun A, Shulzhenko N. Bridging immunity and lipid metabolism by gut microbiota. J Allergy Clin Immunol. 2013;132:253–62. doi: 10.1016/j.jaci.2013.06.025. quiz 63. [DOI] [PubMed] [Google Scholar]
- 14.Bartlett JG. Clinical practice. Antibiotic-associated diarrhea. N Engl J Med. 2002;346:334–9. doi: 10.1056/NEJMcp011603. [DOI] [PubMed] [Google Scholar]
- 15.Cunha BA. Antibiotic side effects. Med Clin North Am. 2001;85:149–85. doi: 10.1016/s0025-7125(05)70309-6. [DOI] [PubMed] [Google Scholar]
- 16.Hviid A, Svanstrom H, Frisch M. Antibiotic use and inflammatory bowel diseases in childhood. Gut. 2011;60:49–54. doi: 10.1136/gut.2010.219683. [DOI] [PubMed] [Google Scholar]
- 17.Mogg GA, Keighley MR, Burdon DW, et al. Antibiotic-associated colitis—a review of 66 cases. Br J Surg. 1979;66:738–42. doi: 10.1002/bjs.1800661017. [DOI] [PubMed] [Google Scholar]
- 18.Hill DA, Siracusa MC, Abt MC, et al. Commensal bacteria-derived signals regulate basophil hematopoiesis and allergic inflammation. Nat Med. 2012;18:538–46. doi: 10.1038/nm.2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cho I, Blaser MJ. The human microbiome: at the interface of health and disease. Nat Rev Genet. 2012;13:260–70. doi: 10.1038/nrg3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Russell SL, Gold MJ, Hartmann M, et al. Early life antibiotic-driven changes in microbiota enhance susceptibility to allergic asthma. EMBO Rep. 2012;13:440–7. doi: 10.1038/embor.2012.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cox LM, Yamanishi S, Sohn J, et al. Altering the intestinal microbiota during a critical developmental window has lasting metabolic consequences. Cell. 2014;158:705–21. doi: 10.1016/j.cell.2014.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reeves AE, Theriot CM, Bergin IL, et al. The interplay between microbiome dynamics and pathogen dynamics in a murine model of Clostridium difficile Infection. Gut Microbes. 2011;2:145–58. doi: 10.4161/gmic.2.3.16333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brandl K, Plitas G, Mihu CN, et al. Vancomycin-resistant enterococci exploit antibiotic-induced innate immune deficits. Nature. 2008;455:804–7. doi: 10.1038/nature07250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, et al. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell. 2004;118:229–41. doi: 10.1016/j.cell.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 25.Iida N, Dzutsev A, Stewart CA, et al. Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science. 2013;342:967–70. doi: 10.1126/science.1240527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ridley EV, Wong AC, Douglas AE. Microbe-dependent and nonspecific effects of procedures to eliminate the resident microbiota from Drosophila melanogaster. Appl Environ Microbiol. 2013;79:3209–14. doi: 10.1128/AEM.00206-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shulzhenko N, Morgun A, Hsiao W, et al. Crosstalk between B lymphocytes, microbiota and the intestinal epithelium governs immunity versus metabolism in the gut. Nat Med. 2011;17:1585–93. doi: 10.1038/nm.2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hall JA, Bouladoux N, Sun CM, et al. Commensal DNA limits regulatory T cell conversion and is a natural adjuvant of intestinal immune responses. Immunity. 2008;29:637–49. doi: 10.1016/j.immuni.2008.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chappert P, Bouladoux N, Naik S, et al. Specific gut commensal flora locally alters T cell tuning to endogenous ligands. Immunity. 2013;38:1198–210. doi: 10.1016/j.immuni.2013.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kuss SK, Best GT, Etheredge CA, et al. Intestinal microbiota promote enteric virus replication and systemic pathogenesis. Science. 2011;334:249–52. doi: 10.1126/science.1211057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ayres JS, Trinidad NJ, Vance RE. Lethal inflammasome activation by a multidrug-resistant pathobiont upon antibiotic disruption of the microbiota. Nat Med. 2012;18:799–806. doi: 10.1038/nm.2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kirkland D, Benson A, Mirpuri J, et al. B cell-intrinsic MyD88 signaling prevents the lethal dissemination of commensal bacteria during colonic damage. Immunity. 2012;36:228–38. doi: 10.1016/j.immuni.2011.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schatz MC, Phillippy AM, Gajer P, et al. Integrated microbial survey analysis of prokaryotic communities for the PhyloChip microarray. Appl Environ Microbiol. 2010;76:5636–8. doi: 10.1128/AEM.00303-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ivanov II, Atarashi K, Manel N, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. 2009;139:485–98. doi: 10.1016/j.cell.2009.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mine KL, Shulzhenko N, Yambartsev A, et al. Gene network reconstruction reveals cell cycle and antiviral genes as major drivers of cervical cancer. Nat Commun. 2013;4:1806. doi: 10.1038/ncomms2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kalghatgi S, Spina CS, Costello JC, et al. Bactericidal antibiotics induce mitochondrial dysfunction and oxidative damage in mammalian cells. Sci Transl Med. 2013;5:192ra85. doi: 10.1126/scitranslmed.3006055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Williams TA, Foster PG, Cox CJ, et al. An archaeal origin of eukaryotes supports only two primary domains of life. Nature. 2013;504:231–6. doi: 10.1038/nature12779. [DOI] [PubMed] [Google Scholar]
- 38.Hibbing ME, Fuqua C, Parsek MR, et al. Bacterial competition: surviving and thriving in the microbial jungle. Nat Rev Microbiol. 2010;8:15–25. doi: 10.1038/nrmicro2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Overbeek R, Begley T, Butler RM, et al. The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res. 2005;33:5691–702. doi: 10.1093/nar/gki866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aziz RK, Bartels D, Best AA, et al. The RAST Server: rapid annotations using subsystems technology. BMC Genomics. 2008;9:75. doi: 10.1186/1471-2164-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Prifti E, Zucker JD, Clement K, et al. Interactional and functional centrality in transcriptional co-expression networks. Bioinformatics. 2010;26:3083–9. doi: 10.1093/bioinformatics/btq591. [DOI] [PubMed] [Google Scholar]
- 42.McDermott JE, Diamond DL, Corley C, et al. Topological analysis of protein co-abundance networks identifies novel host targets important for HCV infection and pathogenesis. BMC Syst Biol. 2012;6:28. doi: 10.1186/1752-0509-6-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sadikot RT, Blackwell TS, Christman JW, et al. Pathogen-host interactions in Pseudomonas aeruginosa pneumonia. Am J Respir Crit Care Med. 2005;171:1209–23. doi: 10.1164/rccm.200408-1044SO. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Babrowski T, Romanowski K, Fink D, et al. The intestinal environment of surgical injury transforms Pseudomonas aeruginosa into a discrete hypervirulent morphotype capable of causing lethal peritonitis. Surgery. 2013;153:36–43. doi: 10.1016/j.surg.2012.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rampioni G, Schuster M, Greenberg EP, et al. Contribution of the RsaL global regulator to Pseudomonas aeruginosa virulence and biofilm formation. FEMS Microbiol Lett. 2009;301:210–17. doi: 10.1111/j.1574-6968.2009.01817.x. [DOI] [PubMed] [Google Scholar]
- 46.Schuster M, Sexton DJ, Diggle SP, et al. Acyl-homoserine lactone quorum sensing: from evolution to application. Annu Rev Microbiol. 2013;67:43–63. doi: 10.1146/annurev-micro-092412-155635. [DOI] [PubMed] [Google Scholar]
- 47.Mellbye B, Schuster M. Physiological framework for the regulation of quorum sensing-dependent public goods in Pseudomonas aeruginosa. J Bacteriol. 2014;196:1155–64. doi: 10.1128/JB.01223-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jimenez PN, Koch G, Thompson JA, et al. The multiple signaling systems regulating virulence in Pseudomonas aeruginosa. Microbiol Mol Biol Rev. 2012;76:46–65. doi: 10.1128/MMBR.05007-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ubeda C, Pamer EG. Antibiotics, microbiota, and immune defense. Trends Immunol. 2012;33:459–66. doi: 10.1016/j.it.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Reikvam DH, Erofeev A, Sandvik A, et al. Depletion of murine intestinal microbiota: effects on gut mucosa and epithelial gene expression. PLoS ONE. 2011;6:e17996. doi: 10.1371/journal.pone.0017996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hapfelmeier S, Lawson MA, Slack E, et al. Reversible microbial colonization of germ-free mice reveals the dynamics of IgA immune responses. Science. 2010;328:1705–9. doi: 10.1126/science.1188454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cauley LS, Lefrancois L. Guarding the perimeter: protection of the mucosa by tissue-resident memory T cells. Mucosal Immunol. 2013;6:14–23. doi: 10.1038/mi.2012.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mazzucchelli L, Hauser C, Zgraggen K, et al. Differential in situ expression of the genes encoding the chemokines MCP-1 and RANTES in human inflammatory bowel disease. J Pathol. 1996;178:201–6. doi: 10.1002/(SICI)1096-9896(199602)178:2<201::AID-PATH440>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 54.Agace WW, Roberts AI, Wu L, et al. Human intestinal lamina propria and intraepithelial lymphocytes express receptors specific for chemokines induced by inflammation. Eur J Immunol. 2000;30:819–26. doi: 10.1002/1521-4141(200003)30:3<819::AID-IMMU819>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 55.Barnhill AE, Brewer MT, Carlson SA. Adverse effects of antimicrobials via predictable or idiosyncratic inhibition of host mitochondrial components. Antimicrob Agents Chemother. 2012;56:4046–51. doi: 10.1128/AAC.00678-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Murano K, Okuwaki M, Hisaoka M, et al. Transcription regulation of the rRNA gene by a multifunctional nucleolar protein, B23/nucleophosmin, through its histone chaperone activity. Mol Cell Biol. 2008;28:3114–26. doi: 10.1128/MCB.02078-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Franzosa EA, Morgan XC, Segata N, et al. Relating the metatranscriptome and metagenome of the human gut. Proc Natl Acad Sci U S A. 2014;111:E2329–38. doi: 10.1073/pnas.1319284111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pe'er D, Hacohen N. Principles and strategies for developing network models in cancer. Cell. 2011;144:864–73. doi: 10.1016/j.cell.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lefebvre C, Rieckhof G, Califano A. Reverse-engineering human regulatory networks. Wiley Interdiscip Rev Syst Biol Med. 2012;4:311–25. doi: 10.1002/wsbm.1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Skinner J, Kotliarov Y, Varma S, et al. Construct and compare gene coexpression networks with DAPfinder and DAPview. BMC Bioinformatics. 2011;12:286. doi: 10.1186/1471-2105-12-286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mahajan-Miklos S, Tan MW, Rahme LG, et al. Molecular mechanisms of bacterial virulence elucidated using a Pseudomonas aeruginosa-Caenorhabditis elegans pathogenesis model. Cell. 1999;96:47–56. doi: 10.1016/s0092-8674(00)80958-7. [DOI] [PubMed] [Google Scholar]
- 62.Jendrossek V, Grassme H, Mueller I, et al. Pseudomonas aeruginosa-induced apoptosis involves mitochondria and stress-activated protein kinases. Infect Immun. 2001;69:2675–83. doi: 10.1128/IAI.69.4.2675-2683.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ochoa-Reparaz J, Mielcarz DW, Ditrio LE, et al. Role of gut commensal microflora in the development of experimental autoimmune encephalomyelitis. J Immunol. 2009;183:6041–50. doi: 10.4049/jimmunol.0900747. [DOI] [PubMed] [Google Scholar]
- 64.Dong X, Yambartsev A, Ramsey S, et al. Reverse enGENEering of regulatory networks from Big Data: a simple guide for biologist. Bioinform Biol Insights. 2015 doi: 10.4137/BBI.S12467. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.