

Abstract
The first ring-forming thioboration reaction of C–C π bonds is reported. This catalyst-free method proceeds in the presence of a commercially available external electrophilic boron source (B-chlorocatecholborane) in good to high yields. The method is scalable and tolerates a variety of functional groups that are intolerant of other major borylation methods. The resulting borylated benzothiophenes participate in a variety of in situ derivatization reactions, showcasing that these borylated intermediates do not need to be isolated prior to downstream functionalization. This methodology has been extended to the synthesis of borylated dihydrothiophenes. Mechanistic experiments suggest that the operative mechanistic pathway is through boron-induced activation of the alkyne followed by electrophilic cyclization, as opposed to S–B σ bond formation, providing a mechanistically distinct pathway to the thioboration of C–C π bonds.
Keywords: Boron, C–C Activation, Sulfur Heterocycles, Sulfur, Cyclization
Borylative Cyclizations
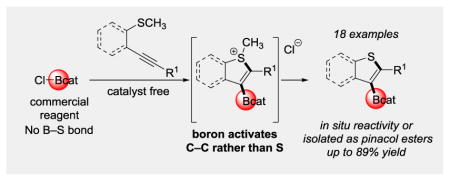
The first ring-forming thioboration reaction of C–C π bonds is reported. The resulting borylated benzothiophenes participate in a variety of in situ derivatization reactions, showcasing that these borylated intermediates do not need to be isolated prior to downstream functionalization. Mechanistic experiments suggest that the operative mechanistic pathway is through boron-induced activation of the alkyne followed by electrophilic cyclization.
Thioboration, the addition of sulfur and boron across C–C π bonds, holds promise as an efficient route to synthesize functionalized thioethers.[1] This area of research has focused on reagents containing B–S σ bonds that are capable of adding in a direct fashion to π systems. In 2015, Bo, Fernández, and Westcott demonstrated the ability of B–S σ bonds from in-house synthesized reagents to add across Michael acceptors through boron activation of the carbonyl oxygen (Figure 1a, top), but without generation of a B–C bond for downstream functionalization.[2] In 1993, Miyaura and Suzuki developed a thioboration reaction of B–S σ bonds across alkynes.[3,4] This method similarly employed in-house synthesized reagents containing B–S σ bonds, however it used a carbophilic palladium catalyst to activate the C–C π bond. Protodeboration and in situ Suzuki cross-coupling reactions of these thioboration products were demonstrated, establishing the utility of such synthetic intermediates (Figure 1a, bottom).
Figure 1.

(a) Previously reported thioboration methods. (b) This work demonstrating formal thioboration. ClBcat = B-chlorocatecholborane.
In contrast, formal thioboration, wherein the equivalents of boron and sulfur add across a C–C π bond, is underexplored, despite the potential advantages of employing commercially available boron reagents as opposed to the thioboration reagents requiring synthesis, and the plausibility of avoiding a palladium catalyst as previously required in the direct thioboration of alkynes.[3,4] Although little is known about the thiophilicity versus carbophilicity of boron reagents in synthesis, such knowledge would facilitate the development of thioboration reactions by indicating when B–S σ bonds are necessary and when such bonds can be avoided, aiming instead for previously unknown carbophilic activation of the C–C π bond by boron with simultaneous attack by sulfur via an AdE3 or AdE2 reaction mechanism (Figure 1b).[5,6] Herein the first formal thioboration of C–C π bonds is reported, concurrently developing fundamental knowledge about guiding principles of relative carbophilicity and thiophilicity. The experiments were motivated by a broader study on gold-catalyzed and catalyst-free oxyboration and aminoboration (B–O and B–N addition) reactions in our research group.[7–10] This catalyst-free thioboration method generates borylated benzothiophene derivatives, a heterocyclic scaffold found in a variety of bioactive molecules and pharmaceuticals, such as raloxifene and sertaconazole (Figure 2).[11–13] These borylated benzothiophenes can then be further elaborated using the wide range of established boron functionalization chemistry.[14–16] This reaction employs a commercial boron reagent, B-chlorocatecholborane (ClBcat), removing the need for B–S σ bond formation in starting materials or in intermediates and making the reaction mechanistically distinct for thioboration.
Figure 2.

Two bioactive molecules that contain a benzothiophene core (in red). Raloxifene is used in the treatment of osteoporosis, and sertoconazole is used to treat skin infection.
Primary competing strategies for the synthesis of borylated benzothiophenes include lithiation/electrophilic trapping[17,18] and transition metal-catalyzed borylation of the benzothiophene core.[19,20] The formal thioboration strategy described herein provides complementary functional group tolerance to these other borylation methods, and also furnishes the benzothiophene core in the same synthetic step. Alternative routes to borylated benzothiophenes, in contrast, require separate steps for borylation and generation of the benzothiophene core.
We hypothesized that 2-alkynylthioanisoles (1) would react upon treatment with ClBcat to yield thioboration products 2 (Table 1). After initial identification of successful reactivity, reaction conditions were optimized. Examination of the equiv of ClBcat (1.0–1.4 equiv) identified 1.4 equiv as the optimal value at 1.3 M concentration in substrate 1 as determined by 1H NMR spectroscopy relative to 1,3,5-triisopropylbenzene as an internal standard. Transesterification of 2 to the more air and moisture stable pinacolboronic ester (3) provided bench-stable organoboron building blocks. The use of ClBpin as an alternative electrophilic boron reagent, which would theoretically provide direct access to the desired pinacolboronic ester 3 from 1, was not evaluated because of its instability above −35 °C and its difficulty of synthesis.[21]
Table 1.
Synthesis of Borylated Benzothiophenes via the Formal Thioboration Reaction.

|
Yield is that of the isolated product.
H NMR yields were determined using mesitylene as an internal standard in d8-toluene, and are listed in parantheses. [a] required 24 h.
The functional group compatibility of the thioboration reaction was next examined (Table 1). Esters, aryl and alkyl halides, amines and cyano groups, an O-silyl protecting group, and several heterocycles tolerated the thioboration reaction conditions in good yields. Functional groups that were incompatible with the thioboration reaction included pyridinyl and alcohol (in both cases, only starting material was observed by 1H NMR spectroscopy, consistent with reaction inhibition by the heteroatom lone pairs). Consistent with the need to favor carbophilicity and avoid competing heteroatomphilicity of boron in the formal thioboration reaction, amide-containing compound 3p required 24 h rather than the standard 4 h to reach complete conversion (70% isolated yield at 24 h vs 25% isolated yield at 4 h). The slower reactivity was attributed to competitive coordination of the amide to boron. Notably, functional groups that cannot be tolerated by existing methods of borylation of benzothiophenes (i.e., lithiation/electrophilic trapping or Pd-catalyzed Miyaura borylation)[17–20] were tolerated by these thioboration reaction conditions (e.g., substrates 3f–3h, 3j, 3m, 3n). Thus, this thioboration reaction provided access to borylated benzothiophenes that had limited accessibility through traditional methods. A crystal structure confirmed the regioselectivity of the thioboration method (Figure 3).
Figure 3.
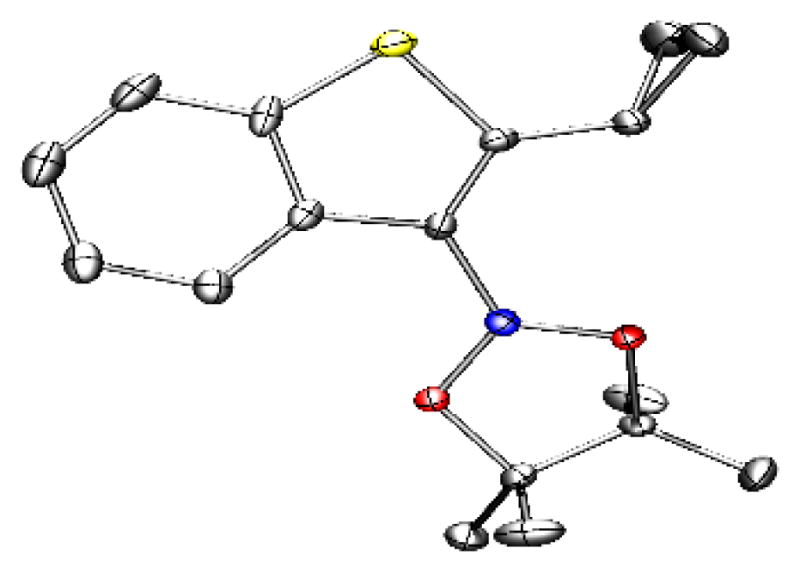
X-ray crystallographic structure of 3d, with the thermal ellipsoids shown at 50% probability (B, blue; C, gray; S, yellow; O, red).
Scale up
In addition to its good functional group tolerance, the thioboration reaction was scalable. Alkynylthioanisole 1d underwent smooth thioboration at the 2.0 g scale to generate 3d in 69% yield (Table 1).
In situ functionalizations
We hypothesized that the catalyst-free conditions of the formal thioboration reaction would provide minimal interference to downstream functionalization conditions due to the absence of residual metal salts. Indeed, synthetic intermediate 2a participated in a wide range of C–B σ-bond functionalization reactions without the need for the additional synthetic manipulations of boron ligand exchange from catechol to pinacol or the requirement to isolate any boron-containing compound (Scheme 1).
Scheme 1.

In Situ Functionalization without Isolation of Organoboron Intermediates: Direct Access to Downstream Products.
Oxidative workup of the C–B bond furnished 1-benzothiophene-3(2H)-one derivative 4, a heterocyclic motif that has been examined as a donor-acceptor chromophore,[22] in 73% yield from 1a. Rhodium-catalyzed conjugate addition to methyl vinyl ketone furnished product 5 in 71% yield over two steps.[23,24] Subjecting intermediate 2a to two different Suzuki conditions[15,25] produced products 6 and 7 in 65% and 56% yield over two steps, respectively. Trifluoromethylation using a modification of a procedure developed by Sanford[26] furnished 8 in 37% yield over two steps. Albeit in low yield, this reaction provided access to 3-trifluoromethylated benzothiophenes, which have limited alternative synthetic routes.[27,28] These in situ reactions illustrate strategies for efficiently generating the heterocyclic core and functionalizing at the 3-position in one pot.
Mechanistic studies: boron as a thiophilic or carbophilic Lewis acid
Three main mechanistic pathways were considered for the thioboration reaction (Scheme 2). The first route was through thiophilic activation of 1 via coordination of ClBcat to the sulfur rather than the C–C π bond, forming activated intermediate 9 (Scheme 2, top). Demethylation furnishes thioboric ester 10; subsequent B–S bond addition across the alkyne yields product 2, which is a known pathway for several other B–X σ bond addition reactions.[29–32] In order to examine the thiophilicity of ClBcat toward 1a at ambient temperature, the initial reaction mixture was evaluated by NMR spectroscopy in d8-toluene. 1H and 11B NMR spectra obtained at t = 0 at ambient temperature showed no evidence of sulfur coordination to boron as judged by persistence resonances corresponding to starting material 1a and ClBcat and the absence of other resonances. This result provided an early indication of the lack of thiophilicity of this reagent, but did not rule out sulfur–boron coordination or activation leading to possible reaction intermediates, which was next investigated.
Scheme 2.

Three Possible Mechanistic Pathways: B-S σ Bond Formation (top), Haloboration (middle), and Alkyne Activation by ClBcat (bottom).
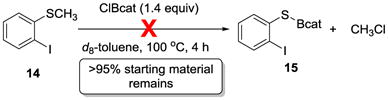
If demethylation were occurring first, through activated sulfonium intermediate 9 and B–S bond containing 10, then a no-alkyne control would demethylate at the same rate (or faster, but not slower) than the thioboration reaction proceeds (4 h at 100 °C). Treatment of o-iodothioanisole 14, however, under these conditions resulted in no reaction: >95% of the starting material remained after 4 h as determined by 1H NMR spectroscopy using mesitylene as an internal standard, with no demethylated product 15 observed (eq 1). Moreover, by 11B NMR spectroscopy, only the ClBcat peak at δ = 28.6 ppm was detected, suggesting that the sulfur was not significantly coordinating to ClBcat, even after extended reaction times. This lack of chemical shift change in the 11B NMR spectrum also ruled out formation of detectable amounts of sulfur-based borenium species.[33,34] This mechanistic control reaction demonstrated that demethylation of 1 is too slow relative to the timescale of the overall thioboration reaction (4 h) to be a step in the operative pathway and therefore ruled out the thiophilic activation pathway.
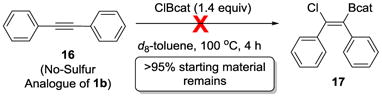
Next, two pathways were considered in which the ClBcat acts as a carbophilic Lewis acid by activating the C–C π system.[34] The first is through a haloboration/cyclization pathway proceeding through a chloroboration reaction analogous to that reported for alkynes,[30] generating intermediate 11 (Scheme 2, middle). This chloroboration product then undergoes cyclization to form sulfonium 12. This mechanism was probed by using a substrate without sulfur (eq 2). Chloroboration of alkynes with other reagents containing B–Cl bonds is thermodynamically downhill;[30, 35] we therefore hypothesize that the chloroborated products in this system would be observable. Treatment of diphenylacetylene 16, which is the no-sulfur analogue of substrate 1b, under the otherwise standard thioboration conditions resulted in no reaction by 1H (>95% 16 remaining using mesitylene as an internal standard) or 11B NMR spectroscopy (only signal detected at δ = 28.6 ppm, corresponding to unreacted ClBcat) in 4 h. This demonstrated that chloroboration product 17 did not form, and thus is an unlikely operative pathway in this thioboration reaction.
On the basis of these mechanistic experiments, a role for boron as a carbophilic Lewis acid in this cyclization is proposed, plausibly through an AdE2/AdE3 mechanism[5,6] (Scheme 2, bottom). Subsequent attack by the sulfur via transition state 13 generates sulfonium intermediate 12. Demethylation furnishes borylated benzothiophene 2. Notably, this proposed pathway has no productive B–S coordination.
 |
(1) |
 |
(2) |
Extension of the mechanistic concept to other substrate classes
Having established the feasibility of this thioboration reaction, we hypothesized that this method could be extended towards the synthesis of dihydrothiophenes, a class of compounds that are useful toward anti-HIV therapeutics[36] and in agricultural products.[37] Subjecting alkynyl thioether 18 to the standard thioboration reaction conditions furnished the desired cyclic thioether 20a, which was transesterified to the bench-stable pinacolboronic ester 21a in 60% overall yield (eq 3). This additional substrate class established that the thioboration reaction did not require the entropic assistance of a rigid backbone or the enthalpic assistance of a gain of aromaticity to proceed.
The formal thioboration reaction also proceeded from thioacetate 22, expanding the reactivity concept from dealkylation to deacylation of sulfur. Subjecting thioacetate 22 to ClBcat at 100 °C for 2 h furnished the desired cyclic thioether 20b, plausibly via the analogous sulfonium intermediate 23 (eq 4). Compound 20b was transesterified to the bench-stable pinacolboronic ester 21b in 51% overall yield.
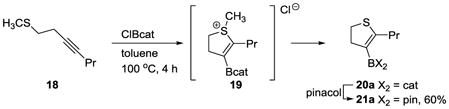 |
(3) |
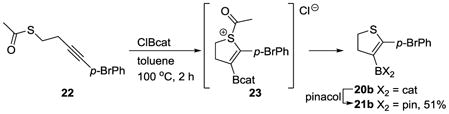 |
(4) |
In conclusion, the first formal thioboration of C–C π bonds is reported. This scalable method efficiently generates both the benzothiophene core and a C–B functional group handle in one synthetic step. These borylated products are primed for downstream in situ functionalization reactions or for isolation as bench-stable building blocks. The mechanistic concept of this thioboration reaction was extended to the synthesis of borylated dihydrothiophenes via both demethylation and deacylation pathways. Mechanistic studies documented an unusual pathway for thioboration reactions in which an S–B σ bond is not formed. This thioboration reaction demonstrates a strategy for harnessing the carbophilic reactivity of boron without concurrent thiophilicity. We envision that this knowledge gained about the thiophilic versus carbophilic reactivity available to boron reagents can be used as a guiding principle for the design of catalyst-free direct or formal boron–element addition reactions.
Supplementary Material
Acknowledgments
This work was supported by a grant from the NIH (1R01GM098512-01), by the University of California, Irvine, and by an Allergan Foundation Graduate Fellowship to D.J.F.
References
- 1.Hall DG. Medicine, and Materials. Wiley-VCH; Weinheim, Germany: 2011. Boronic Acids: Preparation and Applications in Organic Synthesis. [Google Scholar]
- 2.Civit MG, Sanz X, Vogels CM, Webb JD, Geier SJ, Decken A, Bo C, Westcott SA, Fernández E. J Org Chem. 2015;80:2148. doi: 10.1021/jo5026354. [DOI] [PubMed] [Google Scholar]
- 3.Ishiyama T, Nishijima K, Miyaura N, Suzuki A. J Am Chem Soc. 1993;115:7219. [Google Scholar]
- 4.Cui Q, Musaev DG, Morokuma K. Organometallics. 1998;17:1383. [Google Scholar]
- 5.Ashtekar KD, Vetticatt M, Yousefi R, Jackson JE, Borhan B. J Am Chem Soc. 2016;138:8114. doi: 10.1021/jacs.6b02877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hansmann MM, Melen RL, Rominger F, Hashmi ASK, Stephan DW. Chem Commun. 2014;50:7243. doi: 10.1039/c4cc01370k. [DOI] [PubMed] [Google Scholar]
- 7.Hirner JJ, Faizi DJ, Blum SA. J Am Chem Soc. 2014;136:4740. doi: 10.1021/ja500463p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chong E, Blum SA. J Am Chem Soc. 2015;137:10144. doi: 10.1021/jacs.5b06678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Faizi DJ, Issaian A, Davis AJ, Blum SA. J Am Chem Soc. 2016;138:2126. doi: 10.1021/jacs.5b12989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tu KN, Hirner JJ, Blum SA. Org Lett. 2016;18:480. doi: 10.1021/acs.orglett.5b03530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li L, Berthelette C, Chateauneuf A, Ouellet M, Sturino CF, Wang Z. Biorg Med Chem Lett. 2010;20:7440. doi: 10.1016/j.bmcl.2010.10.024. [DOI] [PubMed] [Google Scholar]
- 12.Muchmore D. The Oncologist. 2000;5:388. doi: 10.1634/theoncologist.5-5-388. [DOI] [PubMed] [Google Scholar]
- 13.Croxtall JD, Plosker GL. Drugs. 2009;69:339. doi: 10.2165/00003495-200969030-00009. [DOI] [PubMed] [Google Scholar]
- 14.Sakai M, Hayashi T, Miyaura N. Organometallics. 1997;16:4229. [Google Scholar]
- 15.Suzuki A. In: Modern Arene Chemistry. Astruc D, editor. Wiley-VCH; Weinheim, Germany: 2002. [Google Scholar]
- 16.Ley SV, Thomas AW. Angew Chem Int Ed. 2003;42:5400. doi: 10.1002/anie.200300594. [DOI] [PubMed] [Google Scholar]
- 17.Nakagawa H, Shigekazu K, Nakashima T, Kawai T. Org Lett. 2009;11:1475. doi: 10.1021/ol802969b. [DOI] [PubMed] [Google Scholar]
- 18.Kawai S, Nakashima T, Kutsunugi Y, Nakagawa H, Nakano H, Kawai T. J Mater Chem. 2009:3606. [Google Scholar]
- 19.Kawamorita S, Ohmiya H, Sawamura M. J Org Chem. 2010;75:3855. doi: 10.1021/jo100352b. [DOI] [PubMed] [Google Scholar]
- 20.Guerrand HDS, Vaultier M, Pinet S, Pucheault M. Adv Synth Catal. 2015;357:1167. [Google Scholar]
- 21.Bettinger HF, Filthaus M, Bornemann H, Oppel IM. Angew Chem Int Ed. 2008;47:4744. doi: 10.1002/anie.200705936. [DOI] [PubMed] [Google Scholar]
- 22.Nakazumi H, Watanabe S, Maeda K, Kitao T. Chem Lett. 1990:679. [Google Scholar]
- 23.Takaya Y, Ogasawara M, Hayashi T. Tetrahedron Lett. 1998;39:8479. [Google Scholar]
- 24.Takaya Y, Senda T, Kurushima H, Ogasawara M, Hayashi T. Tetrahedron: Asymmetry. 1999;10:4047. [Google Scholar]
- 25.Billingsley K, Buchwald SL. J Am Chem Soc. 2007;129:3358. doi: 10.1021/ja068577p. [DOI] [PubMed] [Google Scholar]
- 26.Ye Y, Künzi SA, Sanford M. Org Lett. 2012;14:4979. doi: 10.1021/ol3022726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Owton WM. Tetrahedron Lett. 2003;44:7147. [Google Scholar]
- 28.Yoshida S, Yorimitsu H, Oshima K. Org Lett. 2007;9:5573. doi: 10.1021/ol702643j. [DOI] [PubMed] [Google Scholar]
- 29.Brown HC. Tetrahedron. 1961;12:117. [Google Scholar]
- 30.Hara S, Dojo H, Takinami S, Suzuki A. Tetrahedron Lett. 1983;24:731. [Google Scholar]
- 31.Satoh Y, Serizawa H, Hara S, Suzuki A. J Am Chem Soc. 1985;107:5225. [Google Scholar]
- 32.Yang CH, Zhang YS, Fan WW, Liu GQ, Li YM. Angew Chem Int Ed. 2015;54:12636. doi: 10.1002/anie.201505489. [DOI] [PubMed] [Google Scholar]
- 33.Stahl T, Müther K, Ohki Y, Tatsumi K, Oestreich M. J Am Chem Soc. 2013;135:10978. doi: 10.1021/ja405925w. [DOI] [PubMed] [Google Scholar]
- 34.Lawson JR, Clark ER, Cade IA, Solomon SA, Ingleson MJ. Angew Chem Int Ed. 2013;52:7518. doi: 10.1002/anie.201302609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cragg RH, Lappert MF, Tilley BP. J Orgmet Chem. 1964;1:384. [Google Scholar]
- 36.Zhu W, Chong Y, Choo H, Mathews J, Schinazi RF, Chu CK. J Med Chem. 2004;47:1631. doi: 10.1021/jm0303148. [DOI] [PubMed] [Google Scholar]
- 37.Bindschadler P, Von Deyn W, Braun FJ. Dihydrothiophene Compounds for Controlling Invertebrate Pests. WO2015104422. International Patent Application. 2015 Jul 16;
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.