

Summary
Common variable immunodeficiency (CVID) is the most common severe adult primary immunodeficiency and is characterized by a failure to produce antibodies leading to recurrent predominantly sinopulmonary infections. Improvements in the prevention and treatment of infection with immunoglobulin replacement and antibiotics have resulted in malignancy, autoimmune, inflammatory and lymphoproliferative disorders emerging as major clinical challenges in the management of patients who have CVID. In a proportion of CVID patients, inflammation manifests as granulomas that frequently involve the lungs, lymph nodes, spleen and liver and may affect almost any organ. Granulomatous lymphocytic interstitial lung disease (GLILD) is associated with a worse outcome. Its underlying pathogenic mechanisms are poorly understood and there is limited evidence to inform how best to monitor, treat or select patients to treat. We describe the use of combined 2‐[(18)F]‐fluoro‐2‐deoxy‐d‐glucose positron emission tomography and computed tomography (FDG PET‐CT) scanning for the assessment and monitoring of response to treatment in a patient with GLILD. This enabled a synergistic combination of functional and anatomical imaging in GLILD and demonstrated a widespread and high level of metabolic activity in the lungs and lymph nodes. Following treatment with rituximab and mycophenolate there was almost complete resolution of the previously identified high metabolic activity alongside significant normalization in lymph node size and lung architecture. The results support the view that GLILD represents one facet of a multi‐systemic metabolically highly active lymphoproliferative disorder and suggests potential utility of this imaging modality in this subset of patients with CVID.
Keywords: common variable immunodeficiency, fluorodeoxyglucose positron emission tomography, granulomatous lymphocytic interstitial lung disease, rituximab
Introduction
Common variable immunodeficiency (CVID) is the most common serious immunodeficiency of adults, and represents a heterogeneous group of disorders which present not only with acute and chronic infections but also with a range of inflammatory and autoimmune disorders. Patients with CVID have an increased incidence of lymphoma and other malignancies, gastrointestinal in particular 1.
Sinopulmonary infection is the most frequent infectious complication in CVID, and there has been much improvement in the prevention of serious infections such as pneumonia with immunoglobulin replacement and antibiotic therapy 1. With reduced infection burden, non‐infectious complications, including lymphoproliferative disease, pulmonary complications, hepatic and gastrointestinal disease, have become increasingly important causes of morbidity and mortality 6, 7, 8, 9, 10, 11, 12, 13. This is underscored by the finding that the presence of one or more non‐infectious complications in CVID was associated with an almost 11‐fold increase in mortality during follow‐up 2, 3. Patients with no other disease‐related complications had a long‐term survival of 95 versus 42% in those with non‐infectious complications 3. Approximately 5–15% of patients with CVID develop granulomatous lymphocytic interstitial lung disease (GLILD), which is accompanied frequently by splenomegaly, diffuse adenopathy, autoimmune cytopenias, gastrointestinal and hepatic disease 4, 13, 14, 15, 16, 17, 18, 19, 20. More recent revised definitions for CVID aim to recognize the granulomatous, lymphoproliferative and autoimmune manifestations alongside antibody failure and the infectious sequelae 5, 6, 7.
GLILD is a useful term, although it does not encompass the multi‐systemic nature of the underlying pathology and is a less clearly defined entity from a histopathological and radiological perspective where patterns may overlap.
Histologically, GLILD is described as pulmonary tissue containing both granulomatous and lymphoproliferative patterns [i.e. lymphocytic interstitial pneumonitis (LIP), follicular bronchiolitis and/or lymphoid hyperplasia]. Radiographically, GLILD consists of scattered nodular disease (macronodular disease) with areas of consolidation and ground glass, often more prominent on the lower zones 8. It is almost always associated with splenomegaly and diffuse adenopathy, and the differential diagnosis for GLILD includes lymphoma as well as infection, cryptogenic organizing pneumonia and other interstitial lung diseases.
As patients with CVID and GLILD often have poorer outcomes 9, 10 a number of therapeutic interventions have been tried in small numbers of patients with variable effect, and there is an impression that combination therapy may be more successful 11, 12, 13. A recent report describes the use of rituximab and azathioprine used in a small series of patients with GLILD with promising early results 14.
Integrated positron emission tomography/computed tomography (PET‐CT) with glucose analogue, 2‐[(18)F]‐fluoro‐2‐deoxy‐d‐glucose (FDG) can not only identify the source of infection or inflammation before morphological changes on conventional anatomical imaging techniques, such as computed tomography (CT) and magnetic resonance imaging (MRI), but also map the extent and severity of disease, identify sites for tissue sampling and assess therapy response 15. With a rapidly expanding body of evidence, it is recognized increasingly that, in addition to its established role in oncological imaging, FDG PET‐CT also has clinical utility in a range of suspected infectious and inflammatory conditions. These include fever of unknown origin, sarcoidosis, large‐vessel vasculitis, musculoskeletal infections, joint prosthesis or implant‐related complications, human immunodeficiency virus (HIV)‐related infections and immunoglobulin (Ig)G4‐related systemic disease.
It is also possible to make semiquantitative measurements of the level of FDG uptake using the ratio between observed and expected tracer uptake in a particular volume, this being expressed as standardized uptake value (SUV).
In our patient with GLILD, the effect of treatment on lung structure and metabolic activity is assessed using combined FDGPET‐CT scanning.
Materials and methods
The patient was fasted for at least 6 h prior to tracer administration. Serum glucose levels were confirmed to be < 7·0 mmol/l prior to each imaging visit. The patient received an activity of 4 MBq 18F FDG per kilogram body weight. Uptake time was 90 min. FDG PET‐CT imaging was performed with a GE 690 PET CT system (GE Healthcare, Pollards Wood, UK). CT images were acquired in a helical acquisition with a pitch of 0·98 and a tube rotation speed of 0·5 s. Tube output was 120 kVp with output modulation between 20 and 200 mA. Matrix size for the CT acquisition was 512 × 512 pixels with a 50‐cm field of view. No oral or intravenous contrast medium was administered. PET images were acquired at 3 min per field of view. The length of the axial field of view was 15·7 cm. Images were reconstructed with the ordered subset expectation maximization algorithm, with 24 subsets and two iterations. Matrix size was 256 × 256 pixels, using the VUE point of time‐of‐flight algorithm. Image analysis was performed on a GE Xeleris workstation running version 2.1704 software (GE Healthcare).
The standardized uptake value is a semi‐quantitative measurement of tracer uptake calculated using the formula:
SUV is usually considered to be a unitless measurement representing the ratio of observed activity in a volume/expected if the tracer is distributed evenly throughout the patient. For each of the measurements a 3‐cm diameter spherical region of interest was used. The SUVmax represents the highest SUV within this spherical region of interest.
In many oncology applications, SUV measurements are compared to the SUV of liver as an internal standard to reduce variations in serial measurements and reduce the effect of variations in metabolic state at the time of acquisition as well as minor differences in uptake time 16.
The five largest measurable lymph nodes were selected for more detailed analysis, as well as the two largest lung nodules. The SUVmax and maximum linear dimension was recorded on both scans for each of these lesions. FDG uptake was also recorded in the pharyngeal tonsils and liver on both occasions. Comparisons were made for all of the lesions pre‐ and post‐immunosuppression as well as an overall change of size of the lesions and FDG uptake expressed as a percentage change compared to baseline.
The response to treatment was captured using a fitness tracker application called the ‘Health app’, which is loaded into all iPhones™ with iOS 8™. The pedometer function records the number of steps taken on a daily basis (when the phone is carried on the person) and is an indication of fitness and mobility. The data was collected retrospectively, as the patient had been using the iPhone™ ‘Health app’ routinely and agreed to share the information allowing weekly total steps to be calculated.
Immunoglobulins and flow cytometry were undertaken in the Immunology Laboratory at University Hospital of Wales, which is both Clinical Pathology Accreditation (CPA) and United Kingdom Accreditation Service (UKAS) accredited.
Case and results
A 51‐year‐old female presented in her 20s with immune thrombocytopenia (ITP) requiring 6 months of steroid therapy and a second episode following swine flu vaccination. She recalled the onset of respiratory tract infections requiring multiple courses of antibiotics in her late 30s. Four years later she was found to be antibody‐deficient, diagnosed with common variable immunodeficiency (CVID) and commenced on replacement intravenous immunoglobulin with significant decrease in infection frequency.
She then moved to our centre in 2010. On examination there were bi‐basal crepitations and she was noted to have verrucas on the right foot. She had splenomegaly confirmed on ultrasound. Her previous CT scan (2007) was reported as showing mild bronchiectasis and findings consistent with lymphoid interstitial pneumonitis. Blood results on immunoglobulin replacement therapy showed trough IgG 7·76 g/l (6·00–15·50), IgA < 0·22 g/l (0·80–4·00) and an elevated IgM 6·67 g/l (0·48–1·90) with normal serum electrophoresis. Levels of β‐2‐microglobulin were elevated at 4·2 mg/l (1·2–2·4). T and natural killer (NK) cell numbers were slightly low: CD3+ 772 × 106 cells/l (800–2500), CD3CD4+ 662 × 106 cells/l (400–1500), CD3CD8+ 111 ×106 cells/l (200–1100), B cells CD19+ 57 × 106 cells/l (50–500) and NK cells CD56+ 30 × 106 cells/l (80–650). Complement studies were normal and reduced class switched memory B cells were noted at 0·7% (6·5–29·1) of CD19 cells with EUROclass phenotype SmB–, Trnorm, CD21low.
In 2011 she developed autoimmune haemolytic anaemia (AIHA) and was treated with high‐dose intravenous immunoglobulin and oral steroids and switched from subcutaneous to intravenous immunoglobulin replacement. In 2012 she underwent splenectomy for AHA and increasing splenomegaly (15–23 cm with multiple small hypoechoic foci consistent with granulomas) with improvement in her AHA and abdominal discomfort. Histology showed granulomatous splenitis and a bone marrow aspiration and peripheral lymphoma panel found no evidence of lymphoma following review at the All Wales Lymphoma Panel. Post‐splenectomy, an increase in lymphocyte numbers was observed.
In 2015 she became increasingly tired and short of breath on exertion. A repeat CT scan showed mild bronchiectasis and a predominantly peri‐bronchial infiltrate with a basal predominance and lymphadenopathy within the thorax and abdomen. Comparisons with previous imaging showed no major change since 2010. The CRP was 8 mg/l (< 6 mg/l) and the echocardiogram was normal. Lung function tests compared with values in 2012 showed a reduction in the forced vital capacity (FVC) (95% from 108% predicted), total lung capacity (TLC) (73% from 104%) and gas transfer (53% from 62% predicted). Transbronchial lung biopsy demonstrated a variable histiocytic and lymphocytic population infiltrating the interstitium and lamina propria with possible lymphoepithelial lesions present. These findings, alongside the supportive previous finding of granulomatous splenitis, were consistent with GLILD. In November 2015 an FDGPET‐CT scan was performed and she commenced treatment with two doses of (1 g) of rituximab and mycophenolate mofetil, as she did not tolerate azathioprine (thiopurine methyltransferase (TPMT) was normal). At the time of treatment, she was unable to climb a single flight of stairs without stopping due to shortness of breath. Two months later she could run up five flights of stairs, and was able to return to walking her daughter to school and said she felt she was able ‘to take a deep breath again’. She underwent repeat FDGPET‐CT scan (Fig. 1) and lung function testing. Repeat lung function tests performed in February and August 2016 and cumulative lung function tests are shown in Fig. 2, there being an improvement in her FVC (107.5%) and gas transfer (63.9% predicted). Table 1 shows the changes in the standardized uptake values before and after treatment. Overall, there was a reduction of 77% in the liver‐corrected SUVmax of her measured sites of disease. The total anatomical size of the lesions also reduced, although to a lesser extent (47%).
Figure 1.
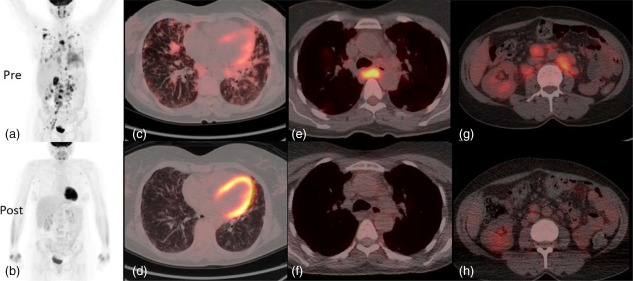
2‐[(18)F]‐fluoro‐2‐deoxy‐d‐glucose positron emission tomography and computed tomography (FDG PET‐CT) pre‐ and 3 months post‐rituximab and mycophenolate treatment. Whole body images acquired at 90 min following 285 MBq 18‐F fluorodeoxyglucose. Maximum intensity projection (MIP) whole body images pretreatment showing widespread abnormal uptake of tracer in the lung parenchyma and lymph nodes above and below the diaphragm (a). MIP images following treatment show near resolution of all the areas of abnormal tracer uptake (b). (c,d) Axial fused PET/CT images at the mid‐thoracic level. The level of FDG uptake is represented by the intensity of colour superimposed upon the CT image. There is a combination of interstitial septal thickening and ill‐defined FDG avid peri‐bronchovascular nodules (c). Post‐treatment images at the same level shows improvement of the nodularity with near resolution of abnormal FDG uptake within them (d). Images at the level of the carina show an enlarged lymph node exhibiting intense abnormal FDG uptake prior to treatment (e). Following treatment the lymph node at this site normalizes in size and shows no abnormal tracer uptake (f). Finally, pretreatment images through the abdomen at the level of the right renal hilum demonstrate numerous enlarged FDG avid retroperitoneal lymph nodes (g) which reduce in size and FDG uptake following treatment (h). [Colour figure can be viewed at wileyonlinelibrary.com.]
Figure 2.
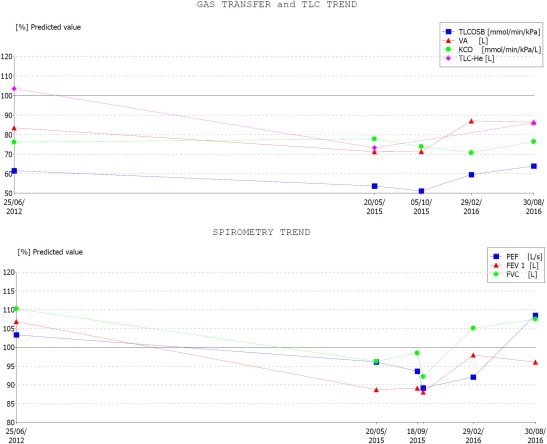
Lung function pre‐ and post‐rituximab and mycophenolate treatment. Changes in lung function as a percentage of predicted are demonstrated over time. Initially there is a reduction in lung volume [forced vital capacity (FVC), total lung capacity (TLC)], airflow [forced expiratory volume in 1 s (FEV1) and peak expiratory flow (PEF)] and gas transfer [total lung capacity for carbon monoxide (TLCO)] from 2012 to 2015. Following initiation of treatment (end 2015) there has been an improvement in these measurements. [Colour figure can be viewed at wileyonlinelibrary.com.]
Table 1.
Measurements and standardized uptake values (SUV) pre‐ and post‐therapy for granulomatous lymphocytic interstitial lung disease (GLILD)
| Area | Preimmunosuppression SUVmax | Post‐immunosuppression SUVmax | Preimmunosuppression linear measurement (cm) | Post‐immunosuppression linear measurement (cm) |
|---|---|---|---|---|
| L pharyngeal tonsil | 9·2 | 4·7 | n.a. | n.a. |
| R pharyngeal tonsil | 8·9 | 5·4 | n.a. | n.a. |
| R axilla LN | 13·4 | 1·4 | 24 | 13 |
| Subcarinal LN | 14·8 | 2·6 | 26 | 14 |
| RML lung nodule | 6·1 | 1·5 | 9 | 7 |
| RLL lung nodule | 6·2 | 2·3 | 26 | 10 |
| Inter‐aortocaval node | 12·3 | 3 | 26 | 12 |
| Left para‐aortic node | 9·2 | 1·9 | 32 | 17 |
| Right comm. iliac node | 12·5 | 2·3 | 33 | 21 |
| Liver | 2·66 | 3·1 | n.a. | n.a. |
| Mean disease SUVmax | 10·29 | 2·79 | n.a. | n.a. |
| Disease/liver ratio | 3·87 | 0·9 | n.a. | n.a. |
| Sum of measurable disease length | n.a. | n.a. | 176 | 94 |
| % Reduction | n.a. | 77% | n.a. | 47% |
Table 1 sets out changes in the largest measurable lesions on the positron emission tomography (PET) and computed tomography (CT) components pre‐ and post‐treatment. SUV measurements are given for each pharyngeal tonsil, target lymph nodes in the right axilla, subcarinal space, inter‐aortocaval space, left para‐aortic space and right common iliac chain. Also measured are the two largest lung nodules in the right middle and lower lobes. 2‐[(18)F]‐fluoro‐2‐deoxy‐d‐glucose (FDG) uptake in the liver is given as an internal reference point for the two scans. The average SUVmax over the disease sites is given as well as the ratio of disease to liver activity both pre‐ and post‐treatment. The dimensions of all the anatomically measurable lesions are given in centimetres both pre‐ and post‐treatment. Following treatment the average SUV max has fallen by 73 and 77% when expressed as a ratio to liver uptake. The linear size of the lesions over the same period has reduced by only 47%. The lesions have reduced in size and metabolic activity, although reductions in metabolic activity are more marked.
Lymphocyte subset analysis reflected the higher overall values post‐splenectomy and absent B cells following rituximab. There was also a decline in the β‐2‐microglobulin concentration from 4·2 to 2·7 mg/l (1·2–2·4) and a reduction in the serum IgM concentration from 6·67 to 1·48 g/l (0·5‐2). Data was extracted retrospectively from the patient's fitness tracker app. and is shown in Fig. 3.
Figure 3.
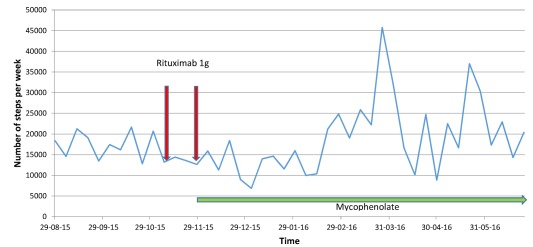
Number of steps per week pre‐ and post‐rituximab and mycophenolate treatment. This figure indicates the weekly total of steps taken as recorded by the ‘Health app’ present on the patient's own iPhone™ prior to and following treatment. The markers (arrows) indicate the administration of rituximab and initiation of mycophenolate which after a delay of approximately 8 weeks is followed by an increase in the weekly steps taken. [Colour figure can be viewed at wileyonlinelibrary.com.]
Discussion
FDG PET‐CT is used primarily in oncological disease and combines anatomical imaging with CT with functional images reflecting levels of glucose uptake and utilization 17. Increased levels of FDG uptake are also seen in a wide variety of systemic infective and inflammatory conditions, and there is increasing interest in the use of FDG PET‐CT in the assessment and monitoring of these disorders. Here, we show that combined modality FDG PET‐CT is able to demonstrate both anatomical and functional improvements following immunosuppressant therapy. Of note, the metabolic changes are much more marked than anatomical improvements, and this is concordant with observations of treatment response in both oncology and inflammatory applications 15.
GLILD represents a major assessment and therapeutic challenge in CVID, with many unanswered questions. This is, to our knowledge, the first report of the use of FDG PET‐CT to delineate the extent of disease both anatomically and functionally and to monitor treatment response. It demonstrated a high rate of metabolic activity within structurally abnormal tissue and we were able to show reversibility of much of the metabolic and structural features following combination therapy. The pretreatment images underscore the multi‐systemic metabolically active lymphoproliferative nature of this condition, where GLILD represents only the pulmonary manifestation of a more widespread disorder. The images also demonstrate abnormal metabolic activity within lymph nodes which are not enlarged significantly and might be missed using conventional CT or MRI imaging. Conversely, the appearances may suggest a diagnosis of lymphoma, reinforcing the need for full clinicopathological correlation where clinical immunologists may need to draw upon the expertise of a multi‐disciplinary team including respiratory, radiology, pathology and haematology.
It is important to recognize that conventional inflammatory markers such as C‐reactive protein (CRP) may not be elevated significantly and that care also needs to be taken in the interpretation of lung function tests. Normal results for the latter do not necessarily rule out the diagnosis, as the baseline lung function prior to disease onset may not be available and time of onset of GLILD may not be known.
Lung function tests, especially basic spirometric measurements, have a low sensitivity for detecting GLILD 14. This patient had normal spirometry and lung volume, with only a modest reduction in gas transfer.
This was against a background of a long duration of relatively stable clinical symptoms, signs and CT abnormalities before a more rapid decline in symptoms. This raises the question of the usefulness of serial CT monitoring imaging in primary immunodeficiency (PID) performed typically at 5‐yearly intervals. This frequency of monitoring may miss the clinically relevant period of acceleration of progression and fail to guide the timing of treatment initiation. It seems appropriate to perform a risk assessment at cohort entry (Fig. 4) in order to help individualize monitoring, and new strategies may be needed for non‐invasive monitoring at much shorter intervals than 5‐yearly. In addition, patients deemed high risk may benefit from targeted information and education in guiding the reporting of specific symptoms associated with disease progression.
Figure 4.
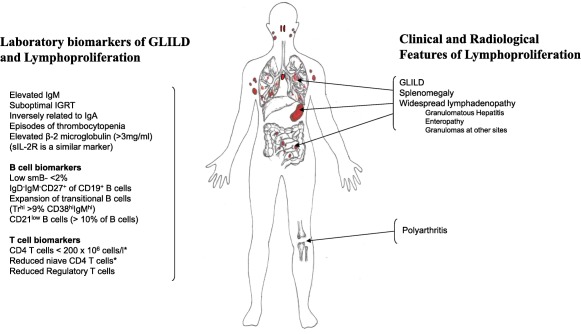
Clinical and laboratory risk factors for granulomatous lymphocytic interstitial lung disease (GLILD). The figure shows the major findings involved in this multi‐systemic lymphoproliferative disorder in terms of clinical and imaging findings on the right and associated laboratory biomarkers on the left. The reduced T cell and naive T cell abnormalities, if sufficiently pronounced, would exclude these patients from the revised common variable immunodeficiency (CVID) classifications 5, 6, 7. [Colour figure can be viewed at wileyonlinelibrary.com.]
Once a change in symptoms is identified, it is important to consider carefully the combination of clinical features, laboratory markers, lung function measurements and radiological investigations that inform management decision‐making in GLILD. FDG PET‐CT helps to identify active disease that may respond to treatment.
The optimal treatment regimen for GLILD and the wider multi‐systemic lymphoproliferation has not been defined. Although steroids have been used as first‐line treatment, combination therapy using rituximab and a second‐line agent such as mycophenolate may offer more promise 12, 13, 14, 18. The duration of benefit from treatment needs to be determined in larger studies with longer periods of follow‐up, but retreatment is likely to be needed in many cases. Lung transplantation, aside from the associated immunosuppression, may not influence the underlying systemic nature of the condition and results have been variable and granulomas have been reported to recur 19, 20.
It is also not known which patients with GLILD will develop progressive disease. In a small retrospective study, nine of 15 patients had physiological worsening of their interstitial lung disease 21. A number of potential biomarkers for GLILD have been identified, and these include suboptimal immunoglobulin replacement, elevated IgM, thrombocytopenic episodes and polyarthritis alongside cellular markers shown in Fig. 4 2, 3, 18, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32. It is not established whether there might be a relationship between the amount and volume of functional activity detected and measured semi‐quantitatively by FDG PET‐CT and risk of progressive disease.
While there are no guidelines to define when further treatment would be appropriate, a number of potential clinical, radiological, lung function and laboratory parameters may help, including exercise tolerance, lung function tests including gas exchange, radiological deterioration, the return of peripheral B cells, β‐2‐microglobulin and IgM levels. There is some evidence to support MRI scanning in reducing the overall radiation exposure in patients who may require multiple monitoring imaging studies 33, 34.
It is not yet clear whether improved molecular characterization of this group of disorders will help to define those patients at greatest risk. Patients with X‐linked agammaglobulinaemia (XLA) are not affected by GLILD, suggesting a potential role for interaction between B and T cells in the pathogenesis. The revised CVID classifications aim to exclude patients with severe T cell immunodeficiency and late‐onset combined immunodeficiency (LOCID) 5, 6, 7 while accepting that multiple T cell abnormalities have been described in CVID 35.
There remain numerous unresolved questions, such as how to predict which patients are most at risk of developing GLILD and the timing of progression of disease. Large observational studies such as the Study of Interstitial Lung Disease in Primary Antibody Deficiency (STILPAD) 36 may help to address these questions. The risk of developing lymphoma due to the widespread and high‐volume ongoing functionally metabolically active lymphoproliferative processes within the lungs, lymph nodes and spleen has not been determined, nor has the potential benefit of debulking this with early rituximab therapy for early GLILD. The multi‐systemic nature of the lymphoproliferation also presents a mechanistic challenge for the separate non‐overlapping clinical classification of CVID 2. The clinically defined subtypes of CVID other than ‘infection only’ are likely to be linked, given that lymphoproliferation may increase the risk of autoimmunity (due to the breakdown of tolerance) 37 as well as the development of lymphoid malignancy.
In the light of the difficulties in assessment, treatment and monitoring of this multi‐systemic lymphoproliferative condition, combined with the poor outcomes for GLILD, there may be a role in selected patients for FDG PET‐CT imaging in view of the sensitivity of the combined anatomical and functional information obtained. Further studies will be needed to define the role of this imaging modality in the management of GLILD.
Disclosure
The authors have no disclosures.
Author contributions
S. J. wrote the paper with contributions and data interpretation from E. C., T. E.‐S., P. F., P. W., M. B. and C. M. S. J., T. E.‐S., M. B. and E. C. were involved in the clinical care and P. F. and C. M. undertook and interpreted the FDG PET‐CT studies. Pippa Jolles drew Fig. 4.
Acknowledgements
Many thanks to Jenny Hughes for editorial review of the manuscript.
References
- 1. Jolles S. The variable in common variable immunodeficiency: a disease of complex phenotypes. J Allergy Clin Immunol Pract 2013; 1:545–56. [DOI] [PubMed] [Google Scholar]
- 2. Chapel H, Lucas M, Lee M et al Common variable immunodeficiency disorders: division into distinct clinical phenotypes. Blood 2008; 112:277–86. [DOI] [PubMed] [Google Scholar]
- 3. Resnick ES, Moshier EL, Godbold JH, Cunningham‐Rundles C. Morbidity and mortality in common variable immune deficiency over 4 decades. Blood 2012; 119:1650–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Prasse A, Kayser G, Warnatz K. Common variable immunodeficiency‐associated granulomatous and interstitial lung disease. Curr Opin Pulm Med 2013; 19:503–9. [DOI] [PubMed] [Google Scholar]
- 5. Ameratunga R, Woon ST, Gillis D, Koopmans W, Steele R. New diagnostic criteria for common variable immune deficiency (CVID), which may assist with decisions to treat with intravenous or subcutaneous immunoglobulin. Clin Exp Immunol 2013; 174:203–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bonilla FA, Barlan I, Chapel H et al International consensus document (ICON): common variable immunodeficiency disorders. J Allergy Clin Immunol Pract 2016; 4:38–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Thon V, Martinez N, Kanariou M, Warnatz K, Quinti I, Chapel H. Common variable immunodeficiency disorders (CVID) – clinical diagnostic criteria: esid.org/Working‐Parties/Registry/Diagnosis‐criteria, 2014. Accessed August 2016.
- 8. Park JE, Beal I, Dilworth JP, Tormey V, Haddock J. The HRCT appearances of granulomatous pulmonary disease in common variable immune deficiency. Eur J Radiol 2005; 54:359–64. [DOI] [PubMed] [Google Scholar]
- 9. Bouvry D, Mouthon L, Brillet PY et al Granulomatosis‐associated common variable immunodeficiency disorder: a case–control study versus sarcoidosis. Eur Respir J 2013; 41:115–22. [DOI] [PubMed] [Google Scholar]
- 10. Bates CA, Ellison MC, Lynch DA, Cool CD, Brown KK, Routes JM. Granulomatous‐lymphocytic lung disease shortens survival in common variable immunodeficiency. J Allergy Clin Immunol 2004; 114:415–21. [DOI] [PubMed] [Google Scholar]
- 11. Pathria M, Urbine D, Zumberg MS, Guarderas J. Management of granulomatous lymphocytic interstitial lung disease in a patient with common variable immune deficiency. BMJ Case Rep 2016; 2016. doi:10.1136/bcr-2016-215624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tashtoush B, Memarpour R, Ramirez J, Bejarano P, Mehta J. Granulomatous‐lymphocytic interstitial lung disease as the first manifestation of common variable immunodeficiency. Clin Respir J 2016. doi: 10.1111/crj.12511. [DOI] [PubMed] [Google Scholar]
- 13. Verma N, Grimbacher B, Hurst JR. Lung disease in primary antibody deficiency. Lancet Respir Med 2015; 3:651–60. [DOI] [PubMed] [Google Scholar]
- 14. Chase NM, Verbsky JW, Hintermeyer MK et al Use of combination chemotherapy for treatment of granulomatous and lymphocytic interstitial lung disease (GLILD) in patients with common variable immunodeficiency (CVID). J Clin Immunol 2013; 33:30–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vaidyanathan S, Patel CN, Scarsbrook AF, Chowdhury FU. FDG PET/CT in infection and inflammation–current and emerging clinical applications. Clin Radiol 2015; 70:787–800. [DOI] [PubMed] [Google Scholar]
- 16. Cheson BD, Fisher RI, Barrington SF et al Recommendations for initial evaluation, staging, and response assessment of Hodgkin and non‐Hodgkin lymphoma: the Lugano classification. J Clin Oncol 2014; 32:3059–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Scarsbrook AF, Barrington SF. PET‐CT in the UK: current status and future directions. Clin Radiol 2016; 71:673–90. [DOI] [PubMed] [Google Scholar]
- 18. Mannina A, Chung JH, Swigris JJ et al Clinical predictors of a diagnosis of common variable immunodeficiency‐related granulomatous‐lymphocytic interstitial lung disease. Ann Am Thorac Soc 2016; 13:1042–9. [DOI] [PubMed] [Google Scholar]
- 19. Burton CM, Milman N, Andersen CB, Marquart H, Iversen M. Common variable immune deficiency and lung transplantation. Scand J Infect Dis 2007; 39:362–7. [DOI] [PubMed] [Google Scholar]
- 20. Thickett KM, Kumararatne DS, Banerjee AK, Dudley R, Stableforth DE. Common variable immune deficiency: respiratory manifestations, pulmonary function and high‐resolution CT scan findings. Q J Med 2002; 95:655–62. [DOI] [PubMed] [Google Scholar]
- 21. Maglione PJ, Overbey JR, Cunningham‐Rundles C. Progression of common variable immunodeficiency interstitial lung disease accompanies distinct pulmonary and laboratory findings. J Allergy Clin Immunol Pract 2015; 3:941–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Arumugakani G, Wood PM, Carter CR. Frequency of Treg cells is reduced in CVID patients with autoimmunity and splenomegaly and is associated with expanded CD21lo B lymphocytes. J Clin Immunol 2010; 30:292–300. [DOI] [PubMed] [Google Scholar]
- 23. Bateman EA, Ayers L, Sadler R et al T cell phenotypes in patients with common variable immunodeficiency disorders: associations with clinical phenotypes in comparison with other groups with recurrent infections. Clin Exp Immunol 2012; 170:202–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Carter CR, Aravind G, Smalle NL, Cole JY, Savic S, Wood PM. CVID patients with autoimmunity have elevated T cell expression of granzyme B and HLA‐DR and reduced levels of Treg cells. J Clin Pathol 2013; 66:146–50. [DOI] [PubMed] [Google Scholar]
- 25. Chapel H, Lucas M, Patel S et al Confirmation and improvement of criteria for clinical phenotyping in common variable immunodeficiency disorders in replicate cohorts. J Allergy Clin Immunol 2012; 130:1197–98. [DOI] [PubMed] [Google Scholar]
- 26. Giovannetti A, Pierdominici M, Mazzetta F et al Unravelling the complexity of T cell abnormalities in common variable immunodeficiency. J Immunol 2007; 178:3932–43. [DOI] [PubMed] [Google Scholar]
- 27. Gregersen S, Aalokken TM, Mynarek G et al Development of pulmonary abnormalities in patients with common variable immunodeficiency: associations with clinical and immunologic factors. Ann Allergy Asthma Immunol 2010; 104:503–10. [DOI] [PubMed] [Google Scholar]
- 28. Horn J, Manguiat A, Berglund LJ et al Decrease in phenotypic regulatory T cells in subsets of patients with common variable immunodeficiency. Clin Exp Immunol 2009; 156:446–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Malphettes M, Gerard L, Carmagnat M et al Late‐onset combined immune deficiency: a subset of common variable immunodeficiency with severe T cell defect. Clin Infect Dis 2009; 49:1329–38. [DOI] [PubMed] [Google Scholar]
- 30. Melo KM, Carvalho KI, Bruno FR et al A decreased frequency of regulatory T cells in patients with common variable immunodeficiency. PLOS ONE 2009; 4:e6269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Quinti I, Soresina A, Guerra A et al Effectiveness of immunoglobulin replacement therapy on clinical outcome in patients with primary antibody deficiencies: results from a multicenter prospective cohort study. J Clin Immunol 2011; 31:315–22. [DOI] [PubMed] [Google Scholar]
- 32. Wehr C, Kivioja T, Schmitt C et al The EUROclass trial: defining subgroups in common variable immunodeficiency. Blood 2008; 111:77–85. [DOI] [PubMed] [Google Scholar]
- 33. Milito C, Pulvirenti F, Serra G et al Lung magnetic resonance imaging with diffusion weighted imaging provides regional structural as well as functional information without radiation exposure in primary antibody deficiencies. J Clin Immunol 2015; 35:491–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Serra G, Milito C, Mitrevski M et al Lung MRI as a possible alternative to CT scan for patients with primary immune deficiencies and increased radiosensitivity. Chest 2011; 140:1581–9. [DOI] [PubMed] [Google Scholar]
- 35. Wong GK, Huissoon AP. T‐cell abnormalities in common variable immunodeficiency: the hidden defect. J Clin Pathol 2016; 69:672–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.STILPAD Observational Study. Available at: https://www.uniklinik-freiburg.de/cci/studien/stilpad.html. Accessed August 2016.
- 37. van de Ven AA, Warnatz K. The autoimmune conundrum in common variable immunodeficiency disorders. Curr Opin Allergy Clin Immunol 2015; 15:514–24. [DOI] [PubMed] [Google Scholar]