

Summary
Tolerogenic dendritic cells (tolDC) are a new immunotherapeutic tool for the treatment of rheumatoid arthritis (RA) and other autoimmune disorders. We have established a method to generate stable tolDC by pharmacological modulation of human monocyte‐derived DC. These tolDC exert potent pro‐tolerogenic actions on CD4+ T cells. Lack of interleukin (IL)−12p70 production is a key immunoregulatory attribute of tolDC but does not explain their action fully. Here we show that tolDC express transforming growth factor (TGF)‐β1 at both mRNA and protein levels, and that expression of this immunoregulatory cytokine is significantly higher in tolDC than in mature monocyte‐derived DC. By inhibiting TGF‐β1 signalling we demonstrate that tolDC regulate CD4+ T cell responses in a manner that is at least partly dependent upon this cytokine. Crucially, we also show that while there is no significant difference in expression of TGF‐βRII on CD4+ T cells from RA patients and healthy controls, RA patient CD4+ T cells are measurably less responsive to TGF‐β1 than healthy control CD4+ T cells [reduced TGF‐β‐induced mothers against decapentaplegic homologue (Smad)2/3 phosphorylation, forkhead box protein 3 (FoxP3) expression and suppression of (IFN)‐γ secretion]. However, CD4+ T cells from RA patients can, nonetheless, be regulated efficiently by tolDC in a TGF‐β1‐dependent manner. This work is important for the design and development of future studies investigating the potential use of tolDC as a novel immunotherapy for the treatment of RA.
Keywords: regulation, rheumatoid arthritis, TGF‐β1, tolerogenic dendritic cells
Introduction
Rheumatoid arthritis (RA) is a debilitating, chronic autoimmune disease resulting in joint inflammation and progressive joint destruction. It is thought that RA arises from a breakdown of immunological self‐tolerance leading to aberrant immune responses to autoantigens. CD4+ T cells, specifically T helper type 1 (Th1) and Th17 cells, are thought to be the main contributors to this response via the production of proinflammatory cytokines, such as interferon (IFN)‐γ and interleukin (IL)−17A 1. There is currently no known cure for RA, and current treatments involve chronic, non‐antigen‐specific global immunosuppression, which can be associated with numerous side effects. New paradigms for the treatment of RA are focused on the reinstatement of self‐tolerance in an antigen‐specific manner, leaving immunogenic responses to pathogen‐derived antigens and cancer immunity intact.
We have developed a novel, immunotherapeutic tool for inhibiting T cell‐mediated pathology, namely tolerogenic dendritic cells (tolDC) 2, 3, 4, 5, 6. These cells are generated by treating monocyte‐derived DC with the immunosuppressive glucocorticoid, dexamethasone (Dex); the vitamin D receptor agonist, vitamin D3 (VitD3); and the Toll‐like receptor 4 ligand, lipopolysaccharide (LPS). The phenotype of these cells is stable and is characterized by intermediate levels of the T cell co‐stimulatory molecules, CD80 and CD86, low levels of the proinflammatory cytokines, IL‐12 and tumour necrosis factor (TNF)‐α, and high levels of the anti‐inflammatory cytokine, IL‐10. We have demonstrated that equivalent murine tolDC can inhibit the destructive immune response in a collagen‐induced arthritis (CIA) mouse model of RA 5, and we have recently completed a Phase I clinical trial demonstrating the safety and feasibility of autologous tolDC therapy in inflammatory arthritis patients 7. tolDC have low immunostimulatory capacity for CD4+ T cells. Specifically, they skew naive CD4+ T cells responses to an anti‐inflammatory profile, while they render memory CD4+ T cells hyporesponsive 2, 3, 4. We have demonstrated previously that, despite the production of high levels of IL‐10 by tolDC, this cytokine appears not to be essential for their regulatory action on CD4+ T cells, whereas deficient IL‐12p70 production is essential 2.
Transforming growth factor (TGF)‐β is a pleiotropic cytokine, produced by a wide variety of cells. Its most immunologically relevant and potent isoform is TGF‐β1, which plays an important role in immunoregulation via direct suppression of T cell proliferation and cytokine production 8 and the induction of forkhead box protein 3 (FoxP3)+ regulatory T cells (Tregs) 9. TGF‐β1 is produced in a latent inactive form through non‐covalent linkage to latency‐associated peptide (LAP) 10. LAP (TGF‐β1) can bind to the surface of cells, components of the extracellular matrix, or can exist in a soluble form. LAP (TGF‐β1) is activated (released from LAP) by a number of mechanisms, such as cleavage by thrombospondin. Activated TGF‐β1 binds to TGF‐β receptor II (TGF‐βRII) followed by recruitment and activation of the primary signal transducer, TGF‐βRI (ALK5). This leads to phosphorylation of the receptor‐regulated mothers against decapentaplegic homologues (Smads), Smad2 and Smad3, that regulate target gene transcription 11. Negative feedback of the pathway is mediated by the inhibitory Smad, Smad7, which can either block or degrade TGF‐βRI 12.
TGF‐β1 is abundant in the affected joints of RA patients 13, 14, 15, 16, 17, 18, 19 and increases with disease duration 20. However, this high level of TGF‐β1 does not regulate the pathogenic T cell response adequately in RA. This may be due to the overall proinflammatory cytokine environment in the synovium and the high level of T cell activation, which are known to down‐regulate TGF‐βRII expression 21, 22 and/or may be due to induction of inhibitory molecules, such as Smad7 23.
We aimed to determine a potential role for TGF‐β1 in the regulatory function of tolDC, and more specifically to investigate the ability of tolDC to regulate CD4+ T cells from RA patients in a TGF‐β1‐dependent manner. A more complete understanding of the molecular mechanisms by which tolDC regulate T cell responses is essential for the development of future studies on tolDC immunotherapy for RA.
Materials and methods
The minimum information about the tolerogenic antigen‐presenting cells (MITAP) checklist was followed for the preparation of this paper 24. See http://w3id.org/ontolink/mitap for MITAP document and checklist.
Isolation of cells from peripheral blood and synovial fluid
Human samples were obtained from healthy controls (HC) and patients with RA (disease duration over 1 year) with informed consent and following a favourable ethical opinion from South West 3 Research Ethics Committee. All samples were transported at room temperature and were processed within 3 h of sample retrieval. Peripheral blood mononuclear cells (PBMC) were isolated from 18 ml fresh ethylenediamine tetraacetic acid (EDTA) anti‐coagulated peripheral blood (PB) or 5 ml leucocyte reduction system cones by density centrifugation on Lymphoprep (Axis‐Shield Diagnostics, Dundee, UK). Synovial fluid mononuclear cells (SFMC) were isolated from 10–50 ml synovial fluid (SF) by incubation with heparin (1 U/ml) and hyaluronidase (10 U/ml) for 30 min at 37°C before density centrifugation on Lymphoprep. CD14+ monocytes were isolated from HC PBMC by positive magnetic selection using anti‐CD14 magnetic microbeads and a VarioMACS (both from Miltenyi Biotec, Bergisch Gladbach, Germany). CD4+ T cells were isolated from HC or RA PBMC with anti‐CD4 magnetic microbeads (Miltenyi Biotec) or with the RosetteSep CD4+ T cell enrichment kit (StemCell Technologies, Vancouver, BC, Canada). Purities of CD14+ monocytes and CD4+ T cells were routinely >90%, as determined by flow cytometry (data not shown).
Generation of DC populations
Freshly isolated monocytes were cultured immediately in RPMI‐1640 (Sigma, Poole, UK) supplemented with 10% fetal calf serum (FCS) (Lonza, Basel, Switzerland), 2 mM glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin (all from Sigma) at 0·5 × 106 cells/ml in a 1 ml volume in a 24‐well culture plate (Costar, Corning, NY, USA) in the presence of IL‐4 and granulocyte–macrophage colony‐stimulating factor (GM‐CSF) (50 ng/ml each; Immunotools, Friesoythe, Germany) for 7 days at 37°C with 5% CO2. Fresh pre‐warmed medium and cytokines were added on day 3. Mature LPS‐activated dendritic cells (matDC) were matured on day 6 by addition of LPS from Escherichia coli (0·1 μg/ml; Sigma) for 24 h. tolDC were generated as above, but with the addition of Dex (1 × 10−6 M; Sigma) at day 3 and Dex (1 × 10−6 M), the active form of vitD3, 1α,25‐dihydroxyvitamin D3 (1 × 10−10 M; Leo‐Pharma, Ballerup, Denmark) and LPS (0·1 μg/ml) at day 6 for 24 h. On day 7 tolDC and matDC morphology was checked using an inverted microscope – tolDC were slightly elongated and adhered to the culture plates, whereas matDC were more rounded, had visible dendrites, and did not adhere to the culture plates. All DC populations were washed extensively before using them in functional assays. DC phenotype was checked using flow cytometry and was consistent with tolDC exhibiting a semi‐mature phenotype, expressing low levels of CD83, intermediate levels of CD80 and CD86 and high levels of human leucocyte antigen D‐related (HLA‐DR) (data not shown).
Micro fluidic cards
RNA was extracted from DC using an RNeasy kit (Qiagen, Crawley, UK). RNA was reverse‐transcribed to cDNA using random hexamers and SuperScript II RT (Invitrogen, Paisley, UK). cDNA samples were run on a custom Micro Fluidic Card (Applied Biosystems, Foster City, CA, USA) using an ABI Prism 7900HT system (Applied Biosystems). TGF‐β1 mRNA expression was normalized to that of human glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) by subtracting the comparative threshold (CT) value of GAPDH from the CT value of TGF‐β1 (ΔCT). Results are expressed as 2‐ΔCT.
Flow cytometry
Anti‐human LAP (TGF‐β1)‐phycoerythrin (PE) antibody (27232; R&D Systems, Abingdon, UK) was used for cell surface marker analysis of DC. Anti‐human CD3‐allophycocyanin (APC) (HIT3a; BD Bioscience, San Jose, CA, USA), CD4‐fluorescein isothiocyanate (FITC) (RPA‐T4; eBioscience Ltd, Hatfield, UK), and TGF‐βRII‐PE (25508; R&D Systems) antibodies were used for cell surface marker analysis of PBMC and SFMC. Briefly, cells were centrifuged and resuspended in flow cytometry buffer [phosphate‐buffered saline (PBS; Lonza) supplemented with 0·5% bovine serum albumin (BSA; Sigma), 1 mM EDTA (Fisher Scientific, Fair Lawn, New York, NY, USA) and 0·01% sodium azide (Sigma)]. 200 μg/ml human immunoglobulin (Ig)G (Grifols, Los Angeles, CA, USA) was added with antibodies to prevent Fc receptor binding. Cells were incubated on ice for 30 min, centrifuged and resuspended in flow cytometry buffer. Intracellular FoxP3 was detected using a FoxP3‐APC staining kit (PCH101; eBioscience).
Intracellular pSmad2/3 was detected using a Phosflow assay by serum starving PBMC overnight by culture in serum‐free X‐VIVO 15 (Lonza) at 37°C with 5% CO2. PBMC were stimulated with 10 ng/ml TGF‐β1 (PeproTech, EC Ltd, London, UK) for 30 min at 37°C. Untreated control samples were set up in parallel. PBMC were fixed using 1× BD Phosflow Lyse/Fix Buffer (BD Bioscience) and then permeabilized using BD Perm Buffer III (BD Bioscience). To reduce background staining the cells were blocked with 2% mouse serum (Sigma) for 15 min prior to addition of anti‐human CD3‐Pacific Blue (UCHT 1; BD Bioscience), Smad2 (pS465/pS467)/Smad3 (pS423/pS425)‐PE (pSmad2/3; O72‐670; BD Bioscience) and CD4‐APC‐eFluor780 (SK3; eBioscience) antibodies. PBMC were incubated at room temperature for 1 h, centrifuged and resuspended in stain buffer (PBS with Ca2+ and Mg2+ (Lonza) supplemented with 0·2% BSA and 0·09% sodium azide). Data were collected on a BD FACSCanto II (BD Biosciences) and analysed using FlowJo (Tree Star Inc., Ashland, OR, USA). Results are shown as either the median fluorescent intensity (MFI) of the marker of interest or as a percentage of cells expressing the marker of interest.
Stimulation of cells by αCD3αCD28 expander beads and TGF‐β1
PBMC, SFMC and CD4+ T cells were stimulated with αCD3αCD28 expander beads (10 : 1 ratio, Dyna; Invitrogen) in the absence or presence of 10 ng/ml TGF‐β1 in RPMI‐1640 supplemented with 10% FCS, 2 mM glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin. Supernatants were harvested after 3 days and assayed for IFN‐γ by sandwich enzyme‐linked immunosorbent assay (ELISA; BD Bioscience). Percentage suppression was calculated as follows: [(amount of cytokine in absence of TGF‐β – amount of cytokine in presence of TGF‐β)/amount of cytokine in absence of TGF‐β] × 100. The percentage of CD4+FoxP3+ cells was determined by flow cytometry.
DC‐T cell co‐cultures
DC (1 × 104) were cultured with 1 × 105 allogeneic CD4+ T cells (1 : 10 ratio) in 200 μl serum‐free X‐VIVO 15 medium (Lonza). TGF‐βRI (ALK5) inhibitor (SB‐505124; Sigma) or recombinant human LAP (R&D Systems) was added where indicated. Supernatants were harvested after 6 days and assayed for IFN‐γ and IL‐17A by sandwich ELISA and other cytokines by an immunoassay (Meso Scale Discovery, Rockville, MD, USA). Proliferation was assessed by incorporation of [3H]‐thymidine for the last 8 h of culture by scintillation counting (Microbeta TriLux; Perkin Elmer, Waltham, MA, USA).
T cell restimulation
Allogeneic CD4+ T cells were primed with DC (10 : 1) in the presence or absence of 1 μM SB‐505124 for 6 days and rested for 4 days with 10 IU/ml IL‐2 (Proleukin; Novartis Pharmaceuticals UK Ltd, Surrey, UK). No residual DC were observed in the T cell lines. Primed T cells were washed and restimulated with matDC from the original DC donor (10 : 1) in serum‐free X‐VIVO 15 medium (Lonza). Supernatants were harvested after 72 h and assayed for IL‐10 (BD Bioscience) and IFN‐γ by sandwich ELISA. Cultures were pulsed with [3H]‐thymidine for a further 8 h to determine proliferation.
Statistics
Wilcoxon signed‐rank tests or Mann–Whitney tests were performed using Prism version 4.0 (GraphPad Software, San Diego, CA, USA). All P‐values are two‐tailed.
Results
tolDC express TGF‐β1
To investigate the role of TGF‐β1 in tolDC function we began by measuring the expression of TGF‐β1 in tolDC and matDC populations. tolDC expressed more TGFB1 mRNA than matDC (Fig. 1a). To confirm that TGFB1 transcripts were translated, we used flow cytometry to assess TGF‐β1 protein expression on the cell surface using an anti‐LAP (TGF‐β1) antibody. Mirroring the transcript data, LAP (TGF‐β1) was expressed at a higher level on the surface of tolDC compared to matDC (Fig. 1b). Together, these data show that tolDC express a higher level of TGF‐β1 than matDC at both mRNA and protein level.
Figure 1.
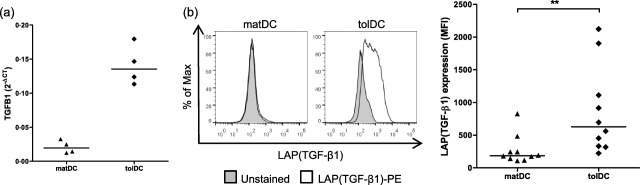
Tolerogenic dendritic cells (tolDC) express transforming growth factor (TGF)‐β. (a) Expression of the TGF‐β1 gene by mature lipopolysaccharide (LPS)‐activated dendritic cells (matDC) and tolDC was measured using a custom Micro Fluidic Card (Applied Biosystems). mRNA expression was normalized to that of human glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) by subtracting the CT value of the human GAPDH gene from the CT value of the gene of interest (ΔCT). Results are expressed as 2–ΔCT for four independent experiments. Horizontal lines represent median values. (b) matDC and tolDC were stained with anti‐human latency‐associated peptide (LAP) (TGF‐β1) antibody and were assessed by flow cytometry. Debris and dead cells were excluded on the basis of forward‐ and side‐scatter. Left panel shows one representative plot of 10 independent donors. The right panel shows the mean fluorescence intensity (MFI) for LAP (TGF‐β1) expression. Horizontal lines represent median values; n = 10. **P < 0·01 calculated with Wilcoxon signed‐rank test.
TGF‐β1 is involved in tolDC regulatory function
We next determined the role of TGF‐β1 in the regulatory action of tolDC. Allogeneic HC CD4+ T cells were co‐cultured with DC in the presence or absence of SB‐505124, a small molecule inhibitor of TGF‐βRI. In keeping with our previous finding 4, tolDC showed a reduced ability to induce T cell proliferation and IFN‐γ production in CD4+ T cells compared with matDC (Fig. 2a,b). Importantly, addition of SB‐505124 had no effect on T cell proliferation but enhanced the production of IFN‐γ by tolDC‐primed T cells in a dose‐dependent manner, restoring levels to those produced by control cells (matDC‐primed T cells) (Fig. 2a,b). The effect of blocking TGF‐β1 signalling in a primary mixed lymphocyte reaction (MLR) using 1 μM SB‐505124 resulted in a 2·7‐fold increase in IFN‐γ production from matDC‐CD4+ T cell cultures (data not shown) and a 46·5‐fold increase in IFN‐γ production from tolDC‐CD4+ T cell cultures (summary of data shown in Fig. 2b). Blocking TGF‐β signalling with SB‐505124 also enhanced IL‐4 and IL‐10 cytokine production, with minimal effect on IL‐2, TNF‐α, IL‐17A, IL‐6, IL‐1β, IL‐8 and IL‐13 production, by tolDC‐primed T cells, although this was not as pronounced as the effect seen for IFN‐γ production (Fig. 2c). We repeated the experiment using a different TGF‐β signalling inhibitor, recombinant LAP, which binds and sequesters active TGF‐β1. Although LAP was less potent, similar effects were demonstrated (Fig. 2d). The effects of blocking TGF‐β signalling with SB‐505124 were also observed at a lower DC : T cell ratio of 1 : 20 and 1 : 40 (Supporting information, Fig. S1).
Figure 2.
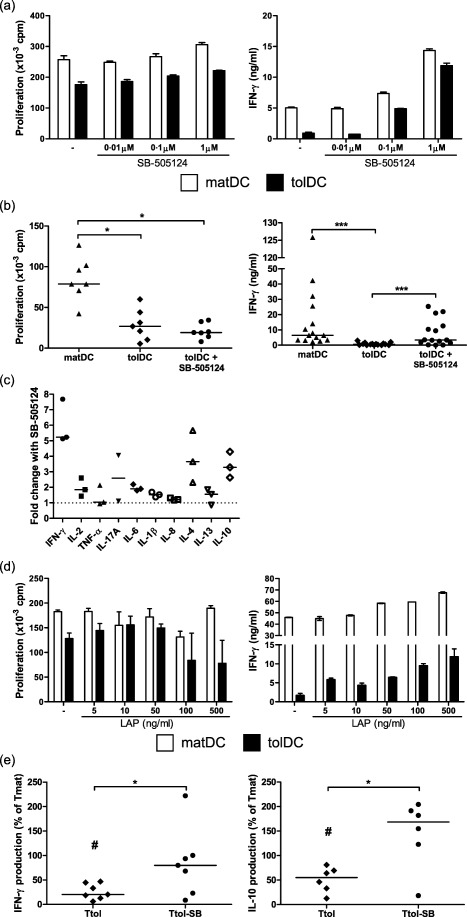
Transforming growth factor (TGF)‐β1 diminishes the ability of tolerogenic dendritic cells (tolDC) to stimulate healthy control (HC) T cells and is involved in their regulatory function. (a–d) Mature lipopolysaccharide (LPS)‐activated dendritic cells (matDC) or tolDC (1 × 104 cells/well) were co‐cultured with allogeneic HC CD4+ T cells (1 × 105 cells/well) in the absence or presence of increasing concentrations of SB‐505124, a small molecule inhibitor of TGF‐βRI (a) or 1 μM SB‐505124 (b,c) or increasing concentrations of recombinant latency‐associated peptide (LAP) (d). Proliferation (a,b,d, left panels), measured using [3H]‐thymidine uptake, and interferon (IFN)‐γ production (a,b,d, right panels), measured using enzyme‐linked immunosorbent assay (ELISA), were assessed at day 6. (c) Additional cytokines were detected in day 6 culture supernatants from tolDC‐CD4+ T‐cell cultures using an ELISA [interleukin (IL)−17A] or Meso Scale Discovery (MSD) immunoassay (all others). The fold change in cytokine production following inhibition of TGF‐β1 signalling was calculated by: concentration of cytokine produced in the presence of 1μM SB‐505124 ÷ concentration of cytokine produced in the absence of 1 μM SB‐505124. IL‐17A was undetectable in one experiment. Error bars in (a) and (d) represent standard error of the mean (s.e.m.) of triplicates (proliferation) or duplicates (IFN‐γ production). Horizontal lines in (b) and (c) represent median values, (b) left panel (proliferation) n = 7; (b) right panel (IFN‐γ production) n = 15; (c) (cytokines) n = 3. (e) Allogeneic healthy control (HC) CD4+ T cells were primed with DC (10 : 1) for 6 days and rested for 4 days with 10 IU/ml IL‐2. T cell lines primed by matDC (Tmat), tolDC (Ttol) and tolDC + SB‐505124 (Ttol‐SB) were restimulated with matDC and IFN‐γ (left panel) and IL‐10 (right panel) production, measured using ELISA, was assessed on day 3. Results are depicted as the percentage cytokine production of Tmat cell lines. Cytokine concentrations range: IFN‐γ in Ttol = 0.9–20·6 ng/ml and in Ttol‐SB = 4·4–80·7 ng/ml; IL‐10 in Ttol = 0·4–5·1 ng/ml and in Ttol‐SB = 0·9–31·7 ng/ml. Horizontal lines represent median values, n (IFN‐γ production) = 7; n (IL‐10 production) = 6. *P < 0·05 and ***P < 0·0001 calculated with Wilcoxon signed‐rank test. #Significant differences (P < 0·05) between Ttol and Tmat cells.
We further investigated whether blockade of the TGF‐β signalling pathway could overcome the regulatory effects of tolDC on CD4+ T cells that are evident on T cell restimulation. As we have shown previously 4, HC CD4+ T cells primed by tolDC (Ttol) have significantly reduced cytokine production upon restimulation compared to matDC‐primed CD4+ T cells (Tmat) (Fig. 2e). Importantly, addition of 1 μM SB‐505124 during priming significantly reversed the inhibition of IFN‐γ and IL‐10 production by tolDC to control levels (Fig. 2e and Supporting information, Fig. S2). These data indicate that part of the regulatory activity of tolDC, specifically the ability of tolDC to suppress cytokine production by CD4+ T cells, is dependent upon TGF‐β1 signalling.
HC and RA CD4+ T cells express TGF‐βRII but RA CD4+ T cells are less responsive to TGF‐β1
Proinflammatory cytokines and high levels of T cell activation, both features of RA, can induce down‐regulation of TGF‐βRII expression 21, 22 and induce expression of inhibitory molecules, such as Smad7 23. These may render RA T cells resistant to regulation by TGF‐β1. To investigate this, we first determined the expression of TGF‐βRII on the surface of HC T cells from PB and RA T cells from both PB and SF. We found no significant difference in the percentage of CD4+ T cells expressing TGF‐βRII between HC PB T cells and RA PB or SF T cells, although the trend was for a reduced proportion of TGF‐βRII+ CD4+ T cells in RA T cells (Fig. 3a).
Figure 3.
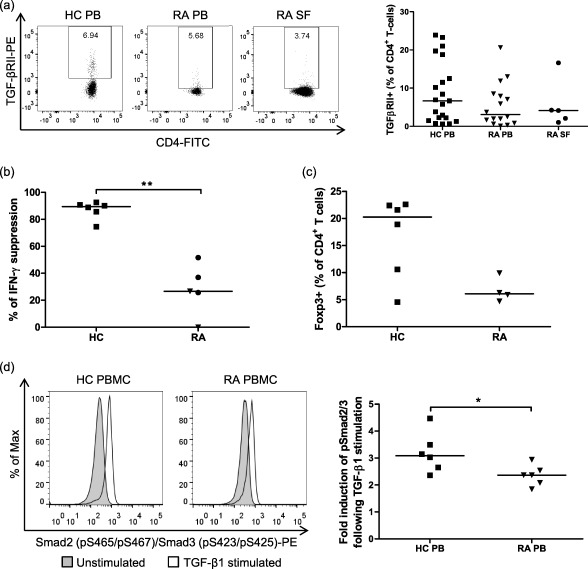
Healthy control (HC) and rheumatoid arthritis (RA) CD4+ T cells express transforming growth factor (TGF)‐βRII but RA CD4+ T cells have a reduced response to TGF‐β1 stimulation. Peripheral blood monuclear cells (PBMC) or CD4+ T cells were isolated from the peripheral blood (PB) of HC or RA patients and synovial fluid mononuclear cells (SFMC) were isolated from SF of RA patients. (a) Cells were stained with anti‐human CD3, anti‐human CD4 and anti‐human TGF‐βRII antibodies and were assessed by flow cytometry. Debris and dead cells were excluded on the basis of forward‐ and side‐scatter. A CD3+CD4+ gate was used to identify CD4+ T cells. Representative plots of TGF‐βRII expression on CD4+ T cells (left panel). The gate shows the percentage of CD4+ T cells expressing TGF‐βRII. The percentage of CD4+ T cells expressing TGF‐βRII (right panel). Horizontal lines represent median values; n (HC PB) = 21, n (RA PB) = 16, n (RA SF) = 5. (b) PBMC from HC (n = 6) and PBMC (▼) and SFMC (•) from RA patients (n = 5) were stimulated with αCD3αCD28 beads in the absence or presence of 10 ng/ml TGF‐β1 for 3 days. IFN‐γ was measured by enzyme‐linked immunosorbent assay (ELISA) and percentage suppression was calculated as follows: [(cytokine in absence of TGF‐β – cytokine in presence of TGF‐β)/cytokine in absence of TGF‐β × 100]. IFN‐γ concentrations range in cultures without TGF‐β1: HC = 1·1–32·4 ng/ml and RA patients = 0·7–32·2 ng/ml; and in cultures with TGF‐β1: HC = 0·1–5·3 ng/ml and RA patients = 0·5–79·1 ng/ml. (c) CD4+ T cells from HC PB (n = 6) and RA patient PB (n = 4) were stimulated with αCD3αCD28 beads in the presence of 10 ng/ml TGF‐β1 for 3 days. The percentage of CD4+forkhead box protein 3 (FoxP3)+ was determined by flow cytometry. (d) PBMC were left unstimulated or were stimulated with 10 ng/ml TGF‐β1 for 30 min before being fixed and permeabilized. PBMC were blocked with 2% mouse serum and stained with anti‐human CD3, anti‐human CD4 and anti‐human phospho mothers against decapentaplegic homologue (pSmad)2/3 antibodies before assessment by flow cytometry. Debris and dead cells were excluded on the basis of forward‐ and side‐scatter. A CD3+CD4+ gate was used to identify CD4+ T cells. Representative plots of pSmad2/3 expression on CD4+ T cells in unstimulated and stimulated conditions (left panel). Fold induction of pSmad2/3 in CD4+ T cells following TGF‐β1 stimulation calculated by dividing the mean fluorescence intensity (MFI) of stimulated cells by the MFI of unstimulated cells (right panel). Horizontal lines represent median values; n = 6. *P < 0·05 and **P < 0·01 calculated using a Mann‐Whitney U test.
We next examined whether HC and RA PBMC responded differently to TGF‐β1 by testing the ability of TGF‐β1 to suppress production of IFN‐γ by the cells. The extent of suppression of IFN‐γ by TGF‐β1 in PBMC and SFMC from RA patients was lower than in PBMC from HC (Fig. 3b and Supporting information, Fig. S3). We also demonstrated a trend for fewer FoxP3‐expressing CD4+ T cells from RA patients than from HC following stimulation of T cells in the presence of TGF‐β (Fig. 3c). This suggests that PBMC and SFMC from RA patients have a reduced regulatory response to TGF‐β. To explore further the ability of RA T cells to respond to TGF‐β1 we used a Phosflow assay to measure the level of phospho‐Smad2/3 (pSmad2/3) in CD4+ T cells following TGF‐β1 stimulation. CD4+ T cells from RA PB had significantly lower pSmad2/3 levels following TGF‐β1 stimulation compared to HC CD4+ T cells from PB (Fig. 3d). These data suggest that CD4+ T cells from RA patients have a reduced ability to respond to TGF‐β1.
RA CD4+ T cells can be regulated by tolDC in a TGF‐β‐dependent manner
Our work demonstrates that TGF‐β1 is involved in the regulatory function of tolDC and is responsible for inducing a hyporesponsive phenotype in CD4+ T cells. As RA T cells appear to have a reduced response to TGF‐β1, we next investigated whether these cells can be tolerized by tolDC. Similar to the findings in HC, tolDC showed a reduced ability to induce T cell proliferation and production of IFN‐γ in RA CD4+ T cells compared with matDC (Fig. 4). Unlike the result seen in HC CD4+ T cells (Fig. 2b), the addition of SB‐505124 to RA CD4+ T cell‐tolDC co‐cultures induced a small increase in T cell proliferation, although this was not restored to the levels of the control cells (matDC‐primed T cells) and the effect of SB‐505124 was very subtle. Interestingly, addition of SB‐505124 to these cultures enhanced the production of IFN‐γ by tolDC‐primed T cells, restoring levels to those of the control cells (matDC‐primed T cells). These data suggest that CD4+ T cells from RA patients can be regulated by tolDC partly via a TGF‐β1‐dependent mechanism. Interestingly, Smad7 was found to be expressed in RA patient CD4+ T cells; however it was also expressed in CD4+ T cells from a proportion of HC (Supporting information, Fig. S4). In addition, there was a trend for increased TGF‐βRII expression on CD4+ T cells cultured with tolDC (Supporting information, Fig. S5).
Figure 4.
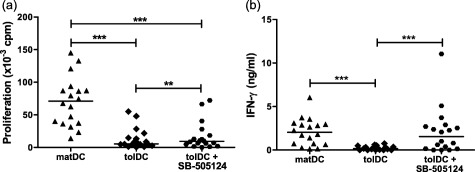
Rheumatoid arthritis (RA) CD4+ T cells can be regulated by tolerogenic dendritic cells (tolDC) in a transforming growth factor (TGF)‐β‐dependent manner. CD4+ T cells (1 × 105 cells/well) from RA patients were co‐cultured with allogenic mature lipopolysaccharide (LPS)‐activated dendritic cells (matDC) or tolDC (1 × 104 cells/well) from a healthy control in the absence or presence of 1 μM SB‐505124, a small molecule inhibitor of TGF‐βRI. Proliferation (left panel), measured using [3H]‐thymidine uptake, and interferon (IFN)‐γ production (right panel), measured using enzyme‐linked immunosorbent assay (ELISA), were assessed at day 6; n = 18. Horizontal lines represent median values. **P < 0·01 and ***P < 0·0005 calculated with Wilcoxon signed‐rank test.
Discussion
This study focused on the role of TGF‐β in the regulatory function of tolDC. We have found that tolDC express TGF‐β1 and this cytokine is involved in their ability to regulate T cells. TGF‐β1 expression by tolDC is an important functional characteristic for the regulation of cytokine production by T cells and plays a role in reducing both IFN‐γ and IL‐10 production in tolDC‐primed CD4+ T cells. However, TGF‐β1 does not seem to play a major role in the inhibition of T cell proliferation. Importantly, despite the reduced ability of CD4+ T cells from RA patients to respond to TGF‐β1, these cells are still regulated by tolDC in a TGF‐β1‐dependent manner.
We have investigated previously the effect of blocking TGF‐β1 signalling in tolDC‐T cell co‐cultures and demonstrated that the use of a TGF‐β1 neutralizing antibody (1D11; R&D Systems) had no effect on the regulatory action of tolDC 2. The discrepancy between the results with the neutralizing antibody and those shown here using the ALK5 inhibitor SB‐505124, or LAP, may be explained by the concentration of neutralizing antibody used being insufficient to inhibit the amount of TGF‐β1 produced by tolDC. In addition, FCS, which contains high levels of TGF‐β1, was added to the culture medium in the original experiments. This may have led to sequestration of the neutralizing antibody, thus interfering with efforts to neutralize TGF‐β1 in those assays.
We report that TGF‐β1 has differential effects on CD4+ T cell proliferation and cytokine production: there was no effect on proliferation but potent inhibition of cytokine production. TGF‐β1 can have both stimulatory and inhibitory effects on T cell proliferation and differentiation, depending on the differentiation stage of the cells and the cytokine microenvironment. It has been reported that TGF‐β1 can inhibit resting CD4+ T cell proliferation and cytokine production but has no inhibitory effect on activated T cells, due to down‐regulation of TGF‐βRII 22. Addition of IL‐10 enhanced TGF‐βRII expression and restored TGF‐β1 responsiveness in activated T cells, although we have been unable to reproduce this result (data not shown). Interestingly, tolDC increase TGF‐βRII expression on CD4+ T cells, although the mechanism inducing this up‐regulation requires further investigation. Other factors involved in the proliferative response of T cells to TGF‐β1 include CD28 co‐stimulation 25. In the absence of CD28 co‐stimulation, TGF‐β1 inhibits proliferation of naive T cells, but in the presence of CD28 co‐stimulation it enhances proliferation. A similar effect of CD28 co‐stimulation was not observed in memory/effector cells, as TGF‐β1 enhanced their proliferation, whether or not co‐stimulation was present. Our tolDC express intermediate levels of the co‐stimulatory molecules CD80 and CD86, which ligate CD28 on T cells. Therefore, the differential effect of TGF‐β1 on CD4+ T cell proliferation and cytokine production reported here could be due to a combination of the cytokine microenvironment and the co‐stimulatory capacity of our tolDC.
The importance of intact TGF‐β signalling in RA T cells is demonstrated by the fact that disruption of TGF‐β signalling in T cells in a CIA murine model leads to exacerbation of arthritis, which is associated with increased Th1 cytokines, IFN‐γ and TNF‐α 26. We have shown that RA PBMC and SFMC have a reduced ability to respond to TGF‐β1, as demonstrated by a reduced level of suppression of IFN‐γ production following stimulation in the presence of TGF‐β1. This is in keeping with the published literature, that exogenous TGF‐β1 cannot inhibit the spontaneous cytokine production of RA synovial mononuclear cells in culture 27 and cannot reduce IFN‐γ production by RA synovial T cells 28. In addition, we have shown that stimulation of RA CD4+ T cells with TGF‐β1 induced less FoxP3 expression and less pSmad2/3 induction compared to cells from HC. A similar result has been reported in RA PBMC, which had deficient TGF‐β1 signalling, evidenced by reduced levels of pSmad2/3 29.
This reduced response to TGF‐β1 does not appear to be linked to a reduced level of TGF‐βRII expression. The immune environment found in RA may explain the diminished response of RA T cells to TGF‐β1. For example, the proinflammatory cytokine TNF‐α, which is found in high levels in RA PB and SF, can regulate TGF‐β signalling via the induction of inhibitory Smad7 23 and via AP‐1 components that interfere with Smad signalling 30. Indeed, we found expression of Smad7 in RA CD4+ T cells. Thus, the defective response to TGF‐β1 in these cells may be due to increased negative feedback via Smad7. Defective TGF‐β signalling has also been demonstrated in T cells from other inflammatory conditions, including systemic lupus erythematosus and Crohn's disease 31, 32. In the latter case, defective TGF‐β1 signalling was due to expression of high levels of Smad7, which could be reversed with treatments targeting this molecule 32, 33. Whether tolDC can override the negative regulatory Smad7 loop by, for example, increasing the expression of TGF‐βRII on CD4+ T cells (Supporting information, Fig. S5) and enhancing the level of Smad2/3‐mediated signalling will require further investigation.
In conclusion, tolDC regulate T cell cytokine production via expression of TGF‐β1. Despite RA T cells having a reduced response to TGF‐β1 regulation, tolDC are still capable of regulating the cytokine response of RA T cells. Thus tolDC may be a promising tool for the restoration of immune tolerance in RA.
Disclosure
The authors declare no disclosures.
Author contributions
A. E. A., J. D. I. and C. M. U. H. conceived and designed the study; J. A. K. advised on experimental design; A. E. A., D. J. S., O. Y. W., M. B. and A. M. P. conducted experiments and data analysis; A. G. P., G. R. and J. D. recruited patients; A. E. A. wrote the manuscript and D. J. S., J. A. K., J. D. I. and C. M. U. H. reviewed and revised the manuscript.
Supporting information
Additional Supporting information may be found in the online version of this article at the publisher's web‐site:
Fig. S1. Transforming growth factor (TGF)‐β1 diminishes the ability of tolerogenic dendritic cells (tolDC) to stimulate healthy control (HC) T cells at different dendritic cell (DC) : T cell ratios. Mature lipopolysaccharide (LPS)‐activated dendritic cells (matDC) or tolDC were co‐cultured with allogeneic HC CD4+ T cells (1 × 105 cells/well) at different DC : T cell ratios in the absence or presence of 1 μM SB‐505124, a small molecule inhibitor of transforming growth factor (TGF)‐βRI. Proliferation (a), measured using [3H]‐thymidine uptake, and interferon (IFN)‐γ production (b), measured using a Meso Scale Discovery (MSD) immunoassay, were assessed at day 6. Columns represent median values for n = 3.
Fig. S2. Cytokine concentrations in restimulation assays. Allogeneic healthy control (HC) CD4+ T cells were primed with dendritic cells (DC) (10 : 1) for 6 days and rested for 4 days with 10 IU/ml interleukin (IL)−2. T cell lines primed by mature lipopolysaccharide (LPS)‐activated dendritic cells (matDC) (Tmat), tolerogenic dendritic cells (tolDC) (Ttol) and tolDC + SB‐505124 (Ttol‐SB) were restimulated with matDC and interferon (IFN)‐γ (a) and IL‐10 (B) production, measured using enzyme‐linked immunosorbent assay (ELISA), was assessed on day 3. Horizontal lines represent median values, n (IFN‐γ production) = 7; n (IL‐10 production) = 6. *P < 0·05 calculated with Wilcoxon signed‐rank test. (e) These results depicted as the percentage cytokine production of Tmat cell lines to Fig. 2e shows these results depicted as the percentage cytokine production of Tmat cell lines.
Fig. S3. Rheumatoid arthritis (RA) CD4+ T cells have a reduced response to transforming growth factor (TGF)‐β1 stimulation. Peripheral blood mononuclear cells (PBMC) from healthy controls (HC) (n = 6) and PBMC and synovial fluid mononuclear cells (SFMC) from RA patients (n = 5) were stimulated with αCD3αCD28 beads in the absence or presence of 10 ng/ml TGF‐β1 for 3 days. Interferon (IFN)‐γ was measured by enzyme‐linked immunosorbent assay (ELISA). Horizontal lines represent median values. *P < 0·05 calculated using Wilcoxon signed‐rank test. (b) These results depicted as the percentage suppression of cultures containing TGF‐β1 compared to those without to Fig. 3b shows these results depicted as the percentage suppression of cultures containing TGF‐β1 compared to those without.
Fig. S4. Rheumatoid arthritis (RA) CD4+ T cells express human mothers against decapentaplegic homologue (Smad)7. CD4+ T cells from healthy controls (HC) (n = 4) and RA patients (n = 6) were isolated and lysed in radioimmunoprecipitation assay (RIPA) buffer before analysis using Western blot. Blots were probed with anti‐Smad7 [clone 293039; R&D systems – molecular weight (MW) 50 KDa] and anti‐human glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) (clone D16H11; Cell Signalling – MW 37 KDa). HeLa cell lysate was used as a positive control.
Fig. S5. Tolerogenic dendritic cells (tolDC) induce transforming growth factor (TGF)‐βRII expression on T cells. CD4+ T cells (1 × 106 cells/well) were co‐cultured with allogeneic tolDC (1 × 105 cells/well) for 1, 3 or 6 days. Cells were stained with anti‐human CD3, anti‐human CD4, and anti‐human TGF‐βRII antibodies and were assessed by flow cytometry. Debris and dead cells were excluded on the basis of forward‐ and side‐scatter. A CD3+CD4+ gate was used to identify CD4+ T cells. (a) Representative plots of TGF‐βRII expression on CD4+ T cells. The gate shows the percentage of CD4+ T cells expressing TGF‐βRII. (b) The percentage of CD4+ T cells expressing TGF‐βRII at the different time‐points. Bars represent median values for n = 3.
Acknowledgements
We thank Christine Routledge, Julie Norris and Sarah Grant for their help with patient recruitment. We thank the Flow Cytometry Core Facility (FCCF) of Newcastle University. The authors thank colleagues within the network A FACTT (Action to Focus and accelerate Cellular Tolerance‐inducing Therapies; COST action BM1305) for helpful discussions. This work was funded by the JGWP foundation. Research within the Musculoskeletal Research Group is supported by the National Institute for Health Research Newcastle Biomedical Research Centre based at Newcastle Hospitals NHS Foundation Trust and Newcastle University.
References
- 1. Boissier MC, Semerano L, Challal S, Saidenberg‐Kermanac'h N, Falgarone G. Rheumatoid arthritis: from autoimmunity to synovitis and joint destruction. J Autoimmun 2012; 39:222–8. [DOI] [PubMed] [Google Scholar]
- 2. Anderson AE, Sayers BL, Haniffa MA et al Differential regulation of naive and memory CD4+ T cells by alternatively activated dendritic cells. J Leukoc Biol 2008; 84:124–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Anderson AE, Swan DJ, Sayers BL et al LPS activation is required for migratory activity and antigen presentation by tolerogenic dendritic cells. J Leukoc Biol 2009; 85:243–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Harry RA, Anderson AE, Isaacs JD, Hilkens CM. Generation and characterisation of therapeutic tolerogenic dendritic cells for rheumatoid arthritis. Ann Rheum Dis 2010; 69:2042–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Stoop JN, Harry RA, von Delwig A, Isaacs JD, Robinson JH, Hilkens CM. Therapeutic effect of tolerogenic dendritic cells in established collagen‐induced arthritis is associated with a reduction in Th17 responses. Arthritis Rheum 2010; 62:3656–65. [DOI] [PubMed] [Google Scholar]
- 6. Hilkens CM, Isaacs JD. Tolerogenic dendritic cell therapy for rheumatoid arthritis: where are we now? Clin Exp Immunol 2013; 172:148–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bell GM, Anderson AE, Diboll J et al Autologous tolerogenic dendritic cells for rheumatoid and inflammatory arthritis. Ann Rheum Dis 2016; doi: 10.1136/annrheumdis‐2015‐208456. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Holter W, Kalthoff FS, Pickl WF et al Transforming growth factor‐beta inhibits IL‐4 and IFN‐gamma production by stimulated human T cells. Int Immunol 1994; 6:469–75. [DOI] [PubMed] [Google Scholar]
- 9. Shevach EM, Davidson TS, Huter EN, Dipaolo RA, Andersson J. Role of TGF‐beta in the induction of Foxp3 expression and T regulatory cell function. J Clin Immunol 2008; 28:640–6. [DOI] [PubMed] [Google Scholar]
- 10. Taylor AW. Review of the activation of TGF‐beta in immunity. J Leukoc Biol 2009; 85:29–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schmierer B, Hill CS. TGFbeta‐SMAD signal transduction: molecular specificity and functional flexibility. Nat Rev Mol Cell Biol 2007; 8:970–82. [DOI] [PubMed] [Google Scholar]
- 12. Yan X, Liu Z, Chen Y. Regulation of TGF‐beta signaling by Smad7. Acta Biochim Biophys Sin (Shanghai) 2009; 41:263–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fava R, Olsen N, Keski‐Oja J, Moses H, Pincus T. Active and latent forms of transforming growth factor beta activity in synovial effusions. J Exp Med 1989; 169:291–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lafyatis R, Thompson NL, Remmers EF et al Transforming growth factor‐beta production by synovial tissues from rheumatoid patients and streptococcal cell wall arthritic rats. Studies on secretion by synovial fibroblast‐like cells and immunohistologic localization. J Immunol 1989; 143:1142–8. [PubMed] [Google Scholar]
- 15. Chu CQ, Field M, Abney E et al Transforming growth factor‐beta 1 in rheumatoid synovial membrane and cartilage/pannus junction. Clin Exp Immunol 1991; 86:380–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Taketazu F, Kato M, Gobl A et al Enhanced expression of transforming growth factor‐beta s and transforming growth factor‐beta type II receptor in the synovial tissues of patients with rheumatoid arthritis. Lab Invest 1994; 70:620–30. [PubMed] [Google Scholar]
- 17. Szekanecz Z, Haines GK, Harlow LA et al Increased synovial expression of transforming growth factor (TGF)‐beta receptor endoglin and TGF‐beta 1 in rheumatoid arthritis: possible interactions in the pathogenesis of the disease. Clin Immunol Immunopathol 1995; 76:187–94. [DOI] [PubMed] [Google Scholar]
- 18. Lettesjo H, Nordstrom E, Strom H et al Synovial fluid cytokines in patients with rheumatoid arthritis or other arthritic lesions. Scand J Immunol 1998; 48:286–92. [DOI] [PubMed] [Google Scholar]
- 19. Pohlers D, Beyer A, Koczan D, Wilhelm T, Thiesen HJ, Kinne RW. Constitutive upregulation of the transforming growth factor‐beta pathway in rheumatoid arthritis synovial fibroblasts. Arthritis Res Ther 2007; 9:R59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bucht A, Larsson P, Weisbrot L et al Expression of interferon‐gamma (IFN‐gamma), IL‐10, IL‐12 and transforming growth factor‐beta (TGF‐beta) mRNA in synovial fluid cells from patients in the early and late phases of rheumatoid arthritis (RA). Clin Exp Immunol 1996; 103:357–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yamane K, Ihn H, Asano Y, Jinnin M, Tamaki K. Antagonistic effects of TNF‐alpha on TGF‐beta signaling through down‐regulation of TGF‐beta receptor type II in human dermal fibroblasts. J Immunol 2003; 171:3855–62. [DOI] [PubMed] [Google Scholar]
- 22. Cottrez F, Groux H. Regulation of TGF‐beta response during T cell activation is modulated by IL‐10. J Immunol 2001; 167:773–8. [DOI] [PubMed] [Google Scholar]
- 23. Bitzer M, von Gersdorff G, Liang D et al A mechanism of suppression of TGF‐beta/SMAD signaling by NF‐kappa B/RelA. Gene Dev 2000; 14:187–97. [PMC free article] [PubMed] [Google Scholar]
- 24. Lord P, Spiering R, Aguillon JC et al Minimum information about tolerogenic antigen‐presenting cells (MITAP): a first step towards reproducibility and standardisation of cellular therapies. PeerJ 2016; 4:e2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sung JL, Lin JT, Gorham JD. CD28 co‐stimulation regulates the effect of transforming growth factor‐beta 1 on the proliferation of naive CD4(+) T cells. Int Immunopharmacol 2003; 3:233–45. [DOI] [PubMed] [Google Scholar]
- 26. Schramm C, Kriegsmann J, Protschka M et al Susceptibility to collagen‐induced arthritis is modulated by TGFbeta responsiveness of T cells. Arthritis Res Ther 2004; 6:R114–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Brennan FM, Chantry D, Turner M, Foxwell B, Maini R, Feldmann M. Detection of transforming growth factor‐beta in rheumatoid arthritis synovial tissue: lack of effect on spontaneous cytokine production in joint cell cultures. Clin Exp Immunol 1990; 81:278–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yin Z, Siegert S, Neure L et al The elevated ratio of interferon gamma‐/interleukin‐4‐positive T cells found in synovial fluid and synovial membrane of rheumatoid arthritis patients can be changed by interleukin‐4 but not by interleukin‐10 or transforming growth factor beta. Rheumatology (Oxf) 1999; 38:1058–67. [DOI] [PubMed] [Google Scholar]
- 29. Xibillé Friedmann DX, Mejía Cristobal LM, Garay Sánchez SA et al Defective TGF‐β signaling pathway of mononuclear cells in patients with Rheumatoid Arthritis (Conference Abstract). American College of Rheumatology, Annual Scientific Meeting, San Francisco, CA, USA, 2008.
- 30. Verrecchia F, Pessah M, Atfi A, Mauviel A. Tumor necrosis factor‐alpha inhibits transforming growth factor‐beta/Smad signaling in human dermal fibroblasts via AP‐1 activation. J Biol Chem 2000; 275:30226–31. [DOI] [PubMed] [Google Scholar]
- 31. Sobel ES, Brusko TM, Butfiloski EJ et al Defective response of CD4(+) T cells to retinoic acid and TGF beta in systemic lupus erythematosus. Arthritis Res Ther 2011; 13:R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Monteleone G, Kumberova A, Croft NM, McKenzie C, Steer HW, MacDonald TT. Blocking Smad7 restores TGF‐beta 1 signaling in chronic inflammatory bowel disease. J Clin Invest 2001; 108:601–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fantini MC, Rio A, Fina D et al Smad7 controls resistance of colitogenic T cells to regulatory T cell‐mediated suppression. Gastroenterology 2009; 136:1308–16. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting information may be found in the online version of this article at the publisher's web‐site:
Fig. S1. Transforming growth factor (TGF)‐β1 diminishes the ability of tolerogenic dendritic cells (tolDC) to stimulate healthy control (HC) T cells at different dendritic cell (DC) : T cell ratios. Mature lipopolysaccharide (LPS)‐activated dendritic cells (matDC) or tolDC were co‐cultured with allogeneic HC CD4+ T cells (1 × 105 cells/well) at different DC : T cell ratios in the absence or presence of 1 μM SB‐505124, a small molecule inhibitor of transforming growth factor (TGF)‐βRI. Proliferation (a), measured using [3H]‐thymidine uptake, and interferon (IFN)‐γ production (b), measured using a Meso Scale Discovery (MSD) immunoassay, were assessed at day 6. Columns represent median values for n = 3.
Fig. S2. Cytokine concentrations in restimulation assays. Allogeneic healthy control (HC) CD4+ T cells were primed with dendritic cells (DC) (10 : 1) for 6 days and rested for 4 days with 10 IU/ml interleukin (IL)−2. T cell lines primed by mature lipopolysaccharide (LPS)‐activated dendritic cells (matDC) (Tmat), tolerogenic dendritic cells (tolDC) (Ttol) and tolDC + SB‐505124 (Ttol‐SB) were restimulated with matDC and interferon (IFN)‐γ (a) and IL‐10 (B) production, measured using enzyme‐linked immunosorbent assay (ELISA), was assessed on day 3. Horizontal lines represent median values, n (IFN‐γ production) = 7; n (IL‐10 production) = 6. *P < 0·05 calculated with Wilcoxon signed‐rank test. (e) These results depicted as the percentage cytokine production of Tmat cell lines to Fig. 2e shows these results depicted as the percentage cytokine production of Tmat cell lines.
Fig. S3. Rheumatoid arthritis (RA) CD4+ T cells have a reduced response to transforming growth factor (TGF)‐β1 stimulation. Peripheral blood mononuclear cells (PBMC) from healthy controls (HC) (n = 6) and PBMC and synovial fluid mononuclear cells (SFMC) from RA patients (n = 5) were stimulated with αCD3αCD28 beads in the absence or presence of 10 ng/ml TGF‐β1 for 3 days. Interferon (IFN)‐γ was measured by enzyme‐linked immunosorbent assay (ELISA). Horizontal lines represent median values. *P < 0·05 calculated using Wilcoxon signed‐rank test. (b) These results depicted as the percentage suppression of cultures containing TGF‐β1 compared to those without to Fig. 3b shows these results depicted as the percentage suppression of cultures containing TGF‐β1 compared to those without.
Fig. S4. Rheumatoid arthritis (RA) CD4+ T cells express human mothers against decapentaplegic homologue (Smad)7. CD4+ T cells from healthy controls (HC) (n = 4) and RA patients (n = 6) were isolated and lysed in radioimmunoprecipitation assay (RIPA) buffer before analysis using Western blot. Blots were probed with anti‐Smad7 [clone 293039; R&D systems – molecular weight (MW) 50 KDa] and anti‐human glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) (clone D16H11; Cell Signalling – MW 37 KDa). HeLa cell lysate was used as a positive control.
Fig. S5. Tolerogenic dendritic cells (tolDC) induce transforming growth factor (TGF)‐βRII expression on T cells. CD4+ T cells (1 × 106 cells/well) were co‐cultured with allogeneic tolDC (1 × 105 cells/well) for 1, 3 or 6 days. Cells were stained with anti‐human CD3, anti‐human CD4, and anti‐human TGF‐βRII antibodies and were assessed by flow cytometry. Debris and dead cells were excluded on the basis of forward‐ and side‐scatter. A CD3+CD4+ gate was used to identify CD4+ T cells. (a) Representative plots of TGF‐βRII expression on CD4+ T cells. The gate shows the percentage of CD4+ T cells expressing TGF‐βRII. (b) The percentage of CD4+ T cells expressing TGF‐βRII at the different time‐points. Bars represent median values for n = 3.