

Abstract
Sequencing and microarray samples often are collected or processed in multiple batches or at different times. This often produces technical biases that can lead to incorrect results in the downstream analysis. There are several existing batch adjustment tools for ‘-omics’ data, but they do not indicate a priori whether adjustment needs to be conducted or how correction should be applied. We present a software pipeline, BatchQC, which addresses these issues using interactive visualizations and statistics that evaluate the impact of batch effects in a genomic dataset. BatchQC can also apply existing adjustment tools and allow users to evaluate their benefits interactively. We used the BatchQC pipeline on both simulated and real data to demonstrate the effectiveness of this software toolkit.
Availability and Implementation: BatchQC is available through Bioconductor: http://bioconductor.org/packages/BatchQC and GitHub: https://github.com/mani2012/BatchQC.
Contact: wej@bu.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 Introduction
Non-biological variation, including batch effects, is commonly present across multiple batches of ‘-omics’ data of the same type (e.g. microarray, RNA-seq, DNA methylation, proteomics) and can be caused by technical factors such as differences in profiling platform, lab protocol, experimenter, processing procedures or reagent batch. These data can be affected by technical variation attributable to both observed and unobserved factors (Leek et al., 2010). Batch effects can act as a confounder and produce technical biases that lead to incorrect downstream analyses (Akey et al., 2007; Lambert and Black, 2012). Many effective methods have been developed to filter technical heterogeneity and batch effects from high-throughput biological data (Gagnon-Bartsch et al., 2013; Johnson and Li, 2007; Leek et al., 2012); however, it is often unclear which method should be applied when combining particular sets of experimental data. In some cases, significant batch correction is needed, whereas in other cases no correction is required. Thus, a complete evaluation of each case is necessary before devising an appropriate correction strategy. This process requires preliminary analyses including data visualization, hierarchical clustering, principle components analysis (PCA), and significance testing. These analyses entail considerable effort, and must be repeated for every set of experimental data. BatchQC streamlines batch evaluation by providing interactive diagnostics, visualizations and statistical analyses to explore the extent to which batch variation impacts the data. BatchQC diagnostics guide the user in determining whether batch adjustment needs to be conducted, and how it should be applied before downstream analysis. BatchQC interactively applies multiple batch effect approaches to the data, and the user can see the benefits of each method.
2 Methods
BatchQC is a Shiny App (http://shiny.rstudio.com/) R-package (R ver. 3.3+). The output is organized into multiple tabs, each featuring a part of the batch effect analysis of the data. The package was developed for flexible future development: tabs can be modified, added, or removed as needs change or as new approaches are developed. We highlight many of the interactive features that are provided by BatchQC:
2.1 Summary and sample diagnostics
BatchQC provides diagnostic and measures for the impacts of study design, batch and treatment effects and for individual samples. These include tabular summary of the samples from each condition and batch, and estimates of the level of confounding present in the study design between batch and condition. BatchQC also provides summary statistics and figures for the percentage of the variation explained by the batch and condition variables for each gene, and a P-value analysis that tests statistical significance of both the batch and condition effects across genes (summarizing their distributions using boxplots, histograms and tables). Moreover, BatchQC includes visualizations for between sample correlations to help identify outlying samples and batches.
2.2 Visualization and differential expression
BatchQC provides an interactive box plot of user-inputted genomic values (read count, probe intensity etc.) for each sample, with options to sort and color the samples by condition or batch, which enables the user to visualize differences across conditions and batches (Fig. 1, top). BatchQC also provides heatmap plots for gene-level values and a sample-level circular dendrogram that clusters the samples using the choice of several different agglomeration measures. BatchQC also provides gene-level significance tests.
Fig. 1.
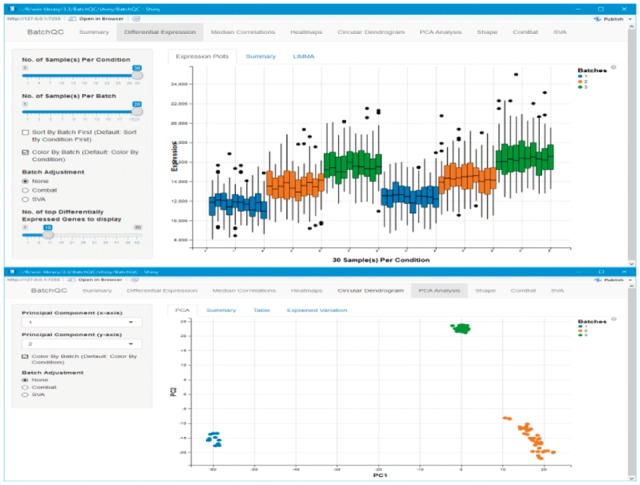
Examples from the BatchQC interface. (top) Boxplots from the simulated dataset showing clear distributional differences between batches. (bottom) The first two PCA components from the signature dataset shows strong batch effects
2.3 PCA and shape analysis
BatchQC conducts PCA on the dataset and produces an interactive plot for displaying the user’s choice of any two components at the same time, with the points colored by the choice of condition or batch (Fig. 1, bottom). BatchQC also provides a summary table that associates the percentage of variation of each PCA component explained by batch and condition, and tests for the statistical significance of these effects. In addition, BatchQC conducts a distributional shape analysis, namely the skewness and kurtosis, to evaluate batch effects in higher moments in the data (Okrah and Bravo, 2015).
2.4 Batch adjustment
BatchQC can interactively adjust the data for batch effects using ComBat or Surrogate Variable Analysis (SVA). BatchQC determines the number of surrogate variables to identify in the given data set, estimates the surrogate variables, and performs the batch adjustment. After batch adjustment, all of the previous diagnostics can be viewed for the raw or adjusted data, and the user can interactively toggle between the two and evaluate the impact of batch adjustment.
3 Data examples and comparisons
The supplementary materials and package vignettes illustrate the functionalities of BatchQC using multiple simulated and real datasets that are included with the package. The need for batch adjustment in the first simulated dataset (details in Supplementary Materials) can be noted in the boxplots (Fig. 1, top), variation analysis, PCA and the clustered heatmap. After adjustment, the boxplots and variation analysis showed no remaining evidence of batch effects, and the samples clustered strongly by condition and not by batch as was previously the case in heatmap and PCA. Interestingly, in the second simulated dataset, PCA did not reveal the batch differences, while the boxplot and heatmaps did show batch differences, indicating the need for multiple diagnostic measures in each data scenario.
As a real-data example, we applied BatchQC to sequencing data captured from human mammary epithelial cells after activating key growth pathway genes (BatchQC examples; GEO accession GSE73628). The data consists of three batches and ten different conditions corresponding to control and activation of nine different pathways (details in Supplement). The PCA plots show a batch effect (Fig. 1, bottom). Similarly, the circular dendrogram and median correlation plots reveal clustering based on batch status. Batch adjustment using either ComBat or SVA diffuses the batch clusters in the PCA and circular dendrogram. These examples demonstrate the utility of all features of the BatchQC package.
4 Conclusions
BatchQC is beneficial for the analyses of ‘-omic’ data and is particularly advantageous in studies where samples are collected in multiple batches or at different times. BatchQC streamlines batch preprocessing and evaluation by providing interactive diagnostics, visualizations and statistical analyses to explore whether batch adjustment needs to be conducted and how correction should be applied (see Supplementary Materials for recommendations on when batch adjustment should be applied). Moreover, BatchQC interactively applies multiple batch effect approaches to the data, and the user can quickly see the benefits of each method interactively. BatchQC is the first software tool to integrate batch diagnostics and correction methods. BatchQC is available as a Shiny user interface that makes the application easy to use and can be easily installed on any computer with standard R installation (R ver. 3.3+) and pandoc (1.12.0+).
Supplementary Material
Funding
This research was supported by funds from the National Institutes of Health (R01 HG005692, R01 ES025002), and the National Science Foundation (DGE 0654108).
Conflict of Interest: none declared.
References
- Akey J.M. et al. (2007) On the design and analysis of gene expression studies in human populations. Nat. Genet., 39, 807–808. [DOI] [PubMed] [Google Scholar]
- Gagnon-Bartsch J.A. et al. (2013) Removing unwanted variation from high dimensional data with negative controls, Tech. Rep. 820, Department of Statistics, University of California, Berkeley.
- Johnson W.E., Li C. (2007) Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics, 8, 118–127. [DOI] [PubMed] [Google Scholar]
- Lambert C.G., Black L.J. (2012) Learning from our GWAS mistakes: from experimental design to scientific method. Biostatistics, 13, 195–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leek J.T. et al. (2012) The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics, 28, 882–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leek J.T. et al. (2010) Tackling the widespread and critical impact of batch effects in high-throughput data. Nat. Rev. Genet., 11, 733–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okrah K., Bravo H.C. (2015) Shape analysis of high-throughput transcriptomics experiment data. Biostatistics, 16, 627–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.