

Abstract
Mutations in the homeobox gene SHOX cause SHOX deficiency, a condition with clinical manifestations ranging from short stature without dysmorphic signs to severe mesomelic skeletal dysplasia. In rare cases, individuals with SHOX deficiency are asymptomatic. To elucidate the factors that modify disease severity/penetrance, we studied a three‐generation family with SHOX deficiency. The variant p.Phe508Cys of the retinoic acid catabolizing enzyme CYP26C1 co‐segregated with the SHOX variant p.Val161Ala in the affected individuals, while the SHOX mutant alone was present in asymptomatic individuals. Two further cases with SHOX deficiency and damaging CYP26C1 variants were identified in a cohort of 68 individuals with LWD. The identified CYP26C1 variants affected its catabolic activity, leading to an increased level of retinoic acid. High levels of retinoic acid significantly decrease SHOX expression in human primary chondrocytes and zebrafish embryos. Individual morpholino knockdown of either gene shortens the pectoral fins, whereas depletion of both genes leads to a more severe phenotype. Together, our findings describe CYP26C1 as the first genetic modifier for SHOX deficiency.
Keywords: clinical variability, genetic modifiers, limb development, retinoic acid, skeletal dysplasia
Subject Categories: Development & Differentiation; Genetics, Gene Therapy & Genetic Disease; Musculoskeletal System
Introduction
In the last decade, many genetic variations responsible for the pathogenesis of disease have been identified. Next‐generation sequencing and high‐throughput genotyping platforms have revolutionized the pursuit of disease‐causing genetic variants. However, disease phenotypes vary—even among individuals carrying the same causative genetic variants within one family—and our understanding of the factors that modify disease severity/penetrance remains incomplete.
Human height is a complex trait with a high heritability. Short stature affects approximately 3% of children worldwide and is diagnosed when height is significantly below the average of the general population for that person's age and sex. More precisely, short stature is statistically defined as two standard deviations (SD) below the mean population height for age, sex, and ethnic group (less than the third percentile) or, when evaluating shortness in relation to family background, more than two SD below the mid‐parental height (Ranke, 1994). To date, many different etiologies of short stature are known and more than two hundred genes underlying growth control have been identified (Marchini et al, 2007; Durand & Rappold, 2013; Baron et al, 2015).
Mutations in the short stature homeobox‐containing (SHOX) gene cause SHOX deficiency, the most frequent monogenic cause of short stature (Marchini et al, 2016). The gene is localized in the pseudoautosomal region 1 (PAR1) shared between the X and Y chromosomes. Its varied clinical manifestations include idiopathic/isolated short stature (ISS), Léri‐Weill dyschondrosteosis (LWD), and Langer mesomelic dysplasia (LD). Heterozygous mutations in its coding or regulatory regions have been identified in up to 10% of patients diagnosed with ISS and 70% of patients with LWD, while homozygous mutations cause LD (Zinn et al, 2002; Rosilio et al, 2012; Marchini et al, 2016). SHOX deficiency also contributes to the short stature and skeletal features in Turner syndrome (Rao et al, 1997; Clement‐Jones et al, 2000). Thus, SHOX deficiency is associated with a broad phenotypic spectrum ranging from short stature without dysmorphic signs to profound dysplasia (Rao et al, 1997; Belin et al, 1998; Shears et al, 1998). Patients with SHOX deficiency often present mesomelic (disproportionate) short stature, a selective shortening of the lower arms/legs. The phenotype may also include Madelung deformity (MD) of the wrist, considered to be the archetypal sign of LWD, which is characterized by shortening and bowing of the radius together with distal hypoplasia of the ulna. Skeletal manifestations are usually more severe in females than in males (Binder, 2011).
The clinical severity of SHOX deficiency varies even in family members carrying the same SHOX mutation (Schiller et al, 2000; Binder, 2011). In rare cases, family members with identical mutation present with stature within the normal range (Huber et al, 2006; Benito‐Sanz et al, 2012; Bunyan et al, 2013). We postulated that SHOX deficiency provides a genetically sensitized background on which genetic modifiers may promote disease progression. To shed light on this phenomenon, we have analyzed a family with five affected individuals displaying LWD. All affected individuals carried a damaging missense mutation in the SHOX gene. Three family members with the same mutation were phenotypically unaffected. To explain this clinical variability, we hypothesized the presence of modifier gene(s). Using whole‐genome linkage analysis and whole‐exome sequencing, we identified a CYP26C1 variant to segregate solely in the affected individuals and carried out subsequent genetic and functional assays to provide evidence for a modifying role of this gene.
Results
SHOX deficiency and clinical variability
We have analyzed a three‐generation German family with five affected individuals displaying LWD (family 1). According to the pattern of transmission of the trait, a dominant inheritance was hypothesized (Fig 1A). All female patients (I:2, I:3, II:7, and III:2, Fig 1A) presented with mesomelic short stature with a SD between −3.14 and −4.51 as well as MD, the archetypal sign of LWD. The father (II:3) of the index patient (III:2) presented with mesomelic short stature with an SD of −2.63 and a borderline MD, consistent with the fact that males are less severely affected than females (Table EV1). The mother (II:4), an aunt (II:8), and the stepsister (III:1) of the index patient (III:2) also presented with short stature, but mesomelia and MD were not diagnosed (Table EV1). Sanger sequencing identified a heterozygous variant, c.482T>C (p.Val161Ala), in the SHOX gene (NG_009385) in all individuals affected with LWD. p.Val161 resides within the DNA binding domain (Fig 2A) and is highly conserved among SHOX vertebrate homologues. To determine the functional relevance of this variant, a luciferase assay in U2OS cells was carried out using the promoter of the fibroblast growth factor receptor 3 (FGFR3) gene, which is a known SHOX target (Decker et al, 2011). We showed a strong influence of the variant on SHOX transcriptional activity (Fig 2A). However, three non‐affected family members also carried the SHOX variant p.Val161Ala (III:4, III:5, and III:6, Fig 1A). Using multiplex ligation‐dependent probe amplification (MLPA), other major genetic lesions in PAR1, including the previously identified enhancer elements in the vicinity of the SHOX locus, were excluded in all individuals.
Figure 1. Damaging variants in SHOX and CYP26C1 in patients with LWD .
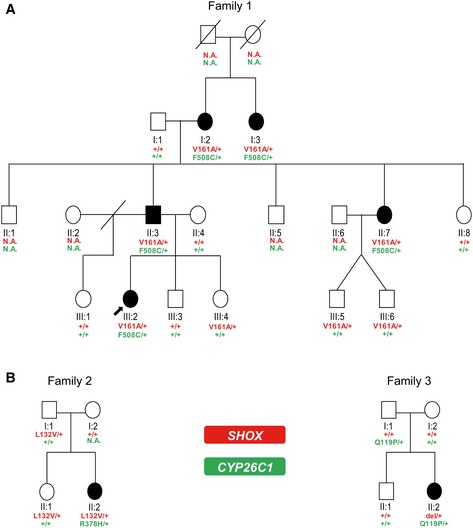
- Pedigree of family 1. Individuals II:3 and III:2 were analyzed by whole‐exome sequencing. Damaging variants in SHOX and CYP26C1 co‐segregate with LWD.
- Pedigrees of families 2 and 3.
Figure 2. Scheme of SHOX and CYP26C1 proteins and luciferase assays.
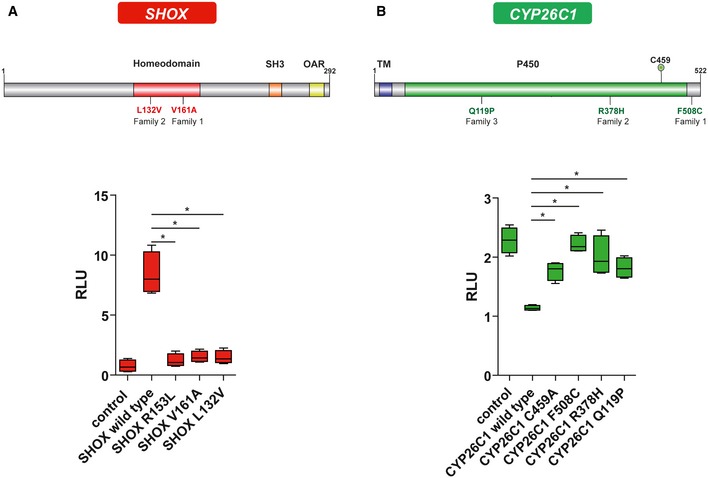
- SHOX protein representation and luciferase assay. The variants found in SHOX are indicated in red. Luciferase assays to test the impact of SHOX mutations on its transcriptional activity were performed on the FGFR3 promoter in U2OS cells (n = 4). Experiments were performed in triplicates. pcDNA4‐TO empty vector was used as control. R153L represents a mutation known to affect SHOX protein activity and was used as a positive control (Schneider et al, 2005). Homeodomain, DNA binding domain; SH3, Src homology 3 domain; OAR, OtpAristalessRax domain; RLU, relative light units.
- CYP26C1 protein representation and luciferase assay. Variants found in CYP26C1 are indicated in green. Cignal RARE system luciferase assays to test the impact of CYP26C1 variants on its RA degradation activity were performed in U2OS cells treated with 250 nM all‐trans retinoic acid (ATRA) for 24 h (n = 4). Experiments were performed in triplicates. pIRES2‐EGFP empty vector was used as control. The residue C459 represents the iron binding residue (Q6VOL0, UniProtKB) and was mutated to Ala and used as a positive control. TM, transmembrane helix; P450, cytochrome p450 domain. RLU, relative light units.
Identification of CYP26C1 as genetic modifier
To address putative genetic causes of this intra‐familiar clinical variability, whole‐genome linkage analysis was performed. This analysis defined a 2.02 Mb interval in the PAR1 of chromosome X (chrX:706800‐2735491; hg19) with a total LOD score of 1.7 (Figs EV1 and EV2). Moreover, a LOD score of 2.4 was obtained in a 19.2 Mb region of chromosome 10 (chr10:85477515–104681710; hg19) (Fig EV3). This region encompasses 263 genes, but could not be further refined. Subsequently, whole‐exome sequencing analysis was performed on the index patient (III:2) and her father (II:3). Variant filtering was performed according to the hypothesized dominant transmission of the phenotype (see Materials and Methods); 98 variants in 97 genes were obtained (Dataset EV1). All variants were tested using the in silico mutation prediction tools PolyPhen‐2, Mutation Taster, SIFT, and PROVEAN, and 36 variants were predicted as disease causing/damaging by at least one of these programs.
Figure EV1. Schematic representation of genome‐wide LOD score calculations.
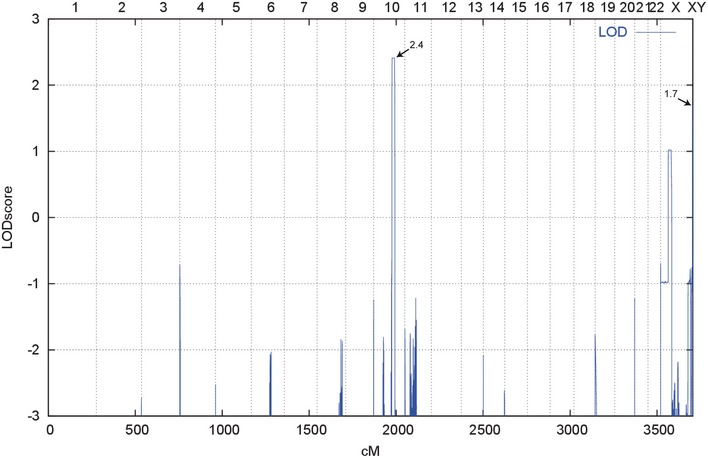
LOD scores calculated with ALLEGRO are given along the y‐axis relative to genomic position cM (centiMorgan) on the x‐axis. Note the highest peak (LOD score 2.4) in the region on chromosome 10 and a second lower peak in the XY PAR1 (LOD score 1.7).
Figure EV2. Haplotype reconstruction for the PAR1 region.
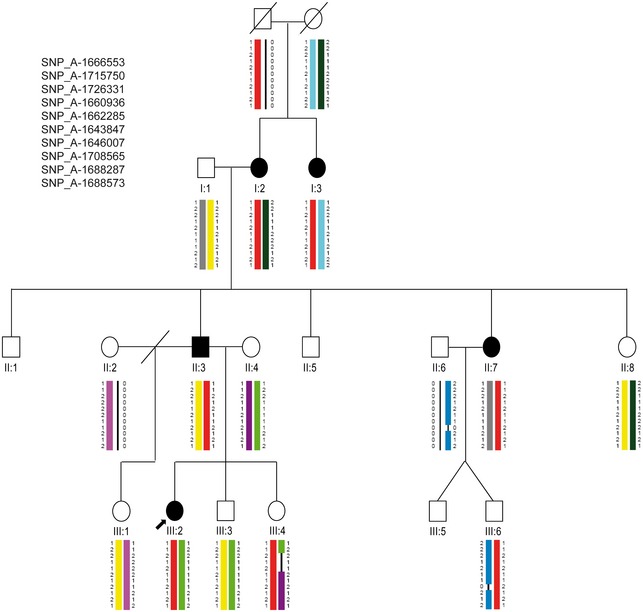
Pedigree of the family with associated SNPs in the pseudoautosomal region 1 (PAR1) of chromosome X is represented. A total LOD score of 1.7 was identified between the flanking markers rs3995646 and rs5939344 and covered a 2.02 Mb sequence (chrX:706800‐2735491; hg19). Filled symbol, LWD‐affected individual; symbol with a slash, deceased individual; slash, divorced; arrow, index patient. Colored chromosomal regions show traceable inheritance: red color regions, common haplotype co‐segregating with LWD; black lines, regions affected by a crossing over of unknown location.
Figure EV3. Haplotype reconstruction for chromosome 10.
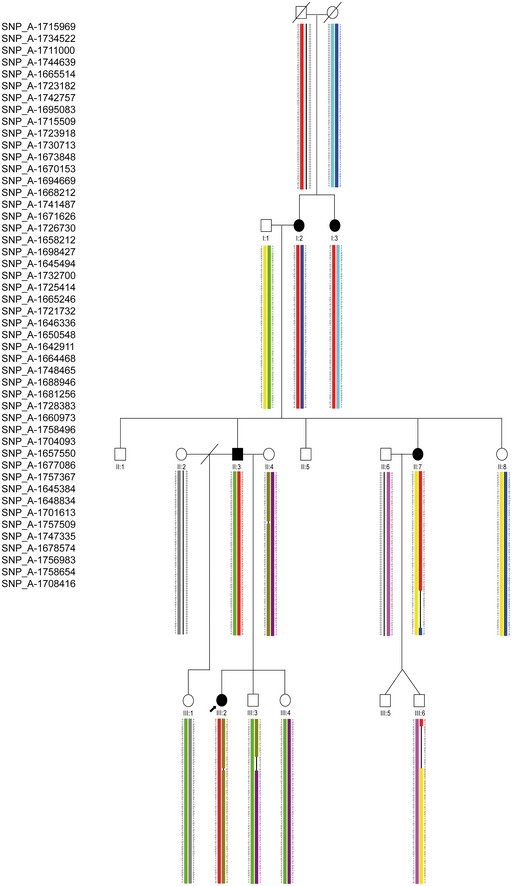
Pedigree of the family with associated SNPs co‐segregating with the disease phenotype on chromosome 10 is represented. A total LOD score of 2.4 was identified between the flanking markers rs10509480 and rs10509758 and covered a 19.2 Mb region (chr10:85477515‐104681710; hg19). Filled symbol, LWD‐affected individual; symbol with a slash, deceased individual; slash, divorced; arrow, index patient. Colored chromosomal regions show traceable inheritance; red color regions, common haplotype co‐segregating with LWD; black lines, regions affected by a crossing over of unknown location.
Sanger sequencing of the 36 identified variants was performed in the five affected and in the eight non‐affected family members where DNA was available. Only variants in two genes, OPN4 (chr10:88419701‐88419701; hg19) and CYP26C1 (chr10:94828408‐94828408; hg19), segregated with the phenotype. Both genes reside within the chromosome 10 interval, which was previously defined by linkage analysis. OPN4 (melanopsin) is a known photopigment of photosensitive retinal ganglion cells and was not considered further (Panda et al, 2002). CYP26C1 (NG_007958.1) encodes for a member of the cytochrome P450 superfamily of enzymes involved in the catabolism of retinoic acid (RA) (Taimi et al, 2004; Uehara et al, 2006; Pennimpede et al, 2010). Loss‐of‐function analyses in mouse and zebrafish have shown that CYP26C1 RA catabolic activity is required for proper hindbrain development (Sirbu et al, 2005; Uehara et al, 2006; Hernandez et al, 2007). RA has been previously shown to play a key role in limb development (Cunningham & Duester, 2015) and CYP26B1, another member of the CYP26 family, is known to be involved in skeleton development (Laue et al, 2011). Moreover, RA has been previously shown to inhibit Shox expression in chicken limbs (Tiecke et al, 2006). Therefore, we decided to further investigate CYP26C1 as a candidate modifier in SHOX deficient LWD patients.
SHOX and CYP26C1 are members of the RA pathway
The heterozygous missense variant c.1523T>G (p.Phe508Cys) in CYP26C1 affected a highly conserved amino acid among vertebrates. p.Phe508Cys was predicted as damaging by all prediction tools that were applied (Dataset EV2). The variant has not yet been described in the three major public variants databases, Exome Aggregation Consortium (ExAC), Exome Variant Server (EVS), and 1000 Genomes Project (TGP). To assess whether this mutation affects the enzymatic activity of CYP26C1, we used the Cignal RARE system, a RA‐responsive luciferase reporter assay, in U2OS cells treated with all‐trans retinoic acid (ATRA) (Laue et al, 2011). Overexpression of wild‐type CYP26C1 reduced the luciferase activity, confirming that it degrades ATRA. In contrast, the CYP26C1 p.Phe508Cys mutant was not able to reduce luciferase activity, suggesting that this mutation impairs CYP26C1 enzymatic activity and consequently RA degradation (Fig 2B).
Next, we tested CYP26C1 expression in human primary chondrocytes, where SHOX is expressed. By performing RT–PCR and Western blot analysis, we could show that CYP26C1 is expressed in these cells (Fig 3A). We then asked whether RA affected SHOX expression in human primary chondrocytes (Appendix Fig S1). Treatment of primary chondrocytes with 100 nM ATRA led to a significant reduction of SHOX levels (Fig 3B). RA exerts its function by regulating the transcriptional activity of nuclear retinoic acid receptors (RARs) that bind as heterodimers with retinoic X receptors (RXRs) to RA response elements. Binding of RA to these receptors triggers activation or repression of their target genes (Weston et al, 2003). To verify whether RA directly or indirectly regulates SHOX expression, we tested a previously described SHOX promoter luciferase reporter assay in U2OS cells (Verdin et al, 2015). Treatment with ATRA led to a significant reduction of luciferase activity compared to mock control (Fig 3C). In silico analysis of the SHOX promoter predicted three putative RXRa binding sites (Fig 3C). However, disruption of these binding sites did not significantly influence the ATRA effect on the SHOX promoter, indicating an indirect effect of RA on SHOX expression (Fig 3C). Altogether, these results provide evidence that CYP26C1 and SHOX are members of the same pathway: CYP26C1 regulates SHOX expression by regulating intracellular levels of RA (Fig 3D).
Figure 3. CYP26C1 is expressed in human primary chondrocytes and retinoic acid affects SHOX expression.
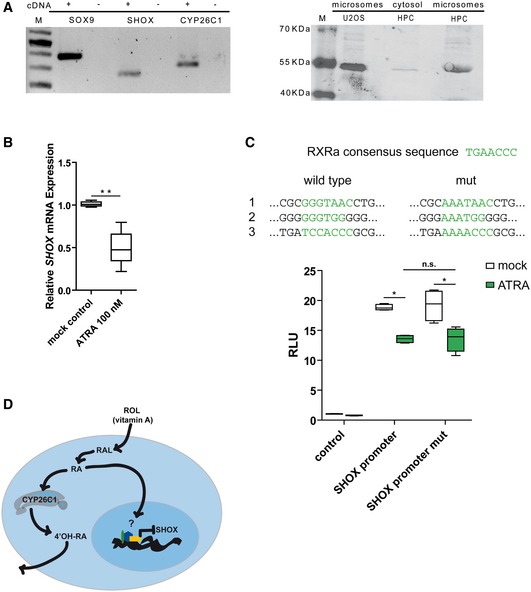
- Left panel, RT–PCR showing the expression of CYP26C1 mRNA in human primary chondrocytes. SOX9 was used as chondrocyte marker. Right panel, Western blot showing the expression of CYP26C1 in human primary chondrocytes on protein level. Overexpression of CYP26C1 in U2OS cells was carried out to compare protein sizes. +, cDNA; −, water; M, marker; HPC, human primary chondrocytes.
- Relative expression of SHOX mRNA normalized to the reference genes SDHA and HPRT in human primary chondrocytes treated with ATRA 100 nM for 6 h (n = 5). One outlier with high relative expression (2.3‐fold) was excluded from ATRA 100 nM treatment (two‐sided Grubbs' test, Z‐value 2.34, P‐value < 0.05).
- SHOX promoter was cloned in pGL3basic for luciferase reporter experiments as previously described (Verdin et al, 2015). In silico analysis of SHOX promoter identified three putative RXRa binding sites which were mutated to test their direct effect on SHOX expression upon treatment with ATRA 250 nM in U2OS cells (n = 4). Experiments were performed in triplicates. RLU, relative light units.
- CYP26C1 and SHOX are members of the retinoic acid pathway. Vitamin A, retinol (ROL), enters the cell and is oxidized to retinaldehyde (RAL). RAL is then oxidized to retinoic acid (RA). RA can enter the nucleus and regulate the expression of its targets. CYP26C1 controls RA intracellular levels by oxidizing this molecule in more hydrosoluble retinoid molecules like 4′‐hydroxy‐retinoic acid (4′‐OH‐RA), which can be readily excreted. High levels of RA downregulate SHOX expression.
Additional genetic evidence in SHOX deficiency patients presenting with LWD
We then investigated whether the co‐occurrence of damaging SHOX and CYP26C1 variants in the severe phenotypes represents a unique finding specific to family 1. We screened CYP26C1 in a cohort of 68 LWD individuals with proven SHOX deficiency and in a cohort of 350 control individuals with normal height. This analysis identified two further cases with co‐occurrence of damaging variants in SHOX and CYP26C1 in the patient cohort (Fig 1B and Dataset EV2). No functional CYP26C1 damaging variants were found in the control individuals with normal height (Dataset EV3; Appendix Fig S2).
In family 2, the affected daughter (II:2) carried a heterozygous missense variant in SHOX, c.349C>G (p.Leu132Val). This variant has been shown to affect SHOX homodimerization and DNA binding (Schneider et al, 2005). The father (I:1) and the sister (II:1) also carried this SHOX variant, but they were reported to be unaffected without dysmorphic signs. Screening of CYP26C1 identified a heterozygous missense variant c.1133G>A (p.Arg378His) only in the daughter with LWD (II:2) (Fig 1B, Table EV1). DNA from the mother (I:2) was not available for the CYP26C1 gene analysis. The third case was an affected girl who carried a de novo heterozygous deletion of SHOX and a missense variant in CYP26C1, c.356A>C (p.Gln119Pro) (Table EV1). Both parents and the brother presented with normal stature and had no dysmorphic signs. The CYP26C1 variant was inherited from the father. Since the SHOX deletion is de novo, this family cannot demonstrate co‐segregation, although it adds to the overall evidence that damaging variants in SHOX and CYP26C1 co‐occur in individuals with severe LWD phenotypes.
We then performed luciferase analysis of all the SHOX and CYP26C1 mutations and could demonstrate their negative impact on protein activity (Fig 2B). We conclude that in addition to family 1, two out of 68 LWD patients with SHOX deficiency presented with functional damaging mutations in CYP26C1, while no damaging mutations with impact on protein activity were identified in 350 control individuals with normal height. In all families where clinical information was available, all individuals with damaging mutations in both SHOX and CYP26C1 presented with short stature and severe skeletal phenotypes.
Modeling SHOX deficiency in zebrafish embryos
Finally, to gain insight into the role of SHOX and CYP26C1 interaction on limb development, we performed antisense morpholino (MO) knockdown experiments in zebrafish embryos. Testing of each MO proofed efficacy (Appendix Figs S4–S6). We first assessed the effect of shox reduction on limb/fin development. Upon shox knockdown, the zebrafish embryos showed an overall delayed growth and a strong impairment of the developing pectoral fins, in concordance with a previous report (Sawada et al, 2015; Fig 4). Using whole‐mount in situ hybridization, we showed that col2a1 expression, an established marker of chondrocytes, was dramatically reduced in the pectoral fin buds upon shox reduction (Fig 4C). Overexpression experiments in zebrafish embryos of human SHOX wild type and the variants identified in families 1 and 2 corroborated the functional significance of SHOX p.Val161Ala (family 1) and p.Leu132Val (family 2) (Appendix Fig S7).
Figure 4. Pattern of defects in zebrafish embryos injected with anti‐shox morpholino.
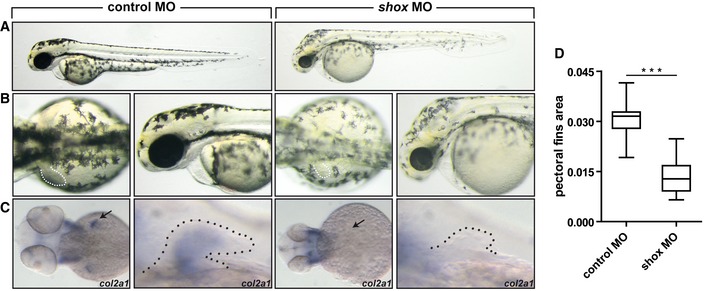
- Lateral views of the embryos at 55 hours post‐fertilization (hpf).
- Dorsal view and magnification on the lateral view of the embryos. Dotted line, pectoral fins. shox morphants show smaller fins compared to controls (n = 30 embryos).
- Expression of col2a1 at 55 hpf was examined by in situ hybridization in wild‐type embryos injected with control MO and with shox MO. Arrow and dotted line indicate the pectoral fin.
- Pectoral fin area was measured by ImageJ (n = 30 embryos).
We then analyzed the effect of cyp26c1 knockdown in zebrafish embryos. Expression of cyp26c1 in the pectoral fins has been previously demonstrated by whole‐mount in situ hybridization (Gu et al, 2005). Knockdown of cyp26c1 resulted in a significant reduction of pectoral fin size, although less strikingly compared to the shox knockdown (Fig 5B–E). The embryos also showed an abnormal development of the otic vesicles and pharyngeal arches (Fig 5B). As reported previously, we also found signs of cardiac dysfunction as evident by the pericardial edema (Rydeen & Waxman, 2014) (Fig 5B). In the pectoral fins, we demonstrated that the expression of col2a1 was strongly reduced or absent (Fig 5C). In addition, the expression of shox was shown to be decreased upon cyp26c1 knockdown (Fig 5D and G). MO knockdown of cyp26c1 led to a significant increase in RA levels compared to control MO, which is in line with its role in RA degradation (Fig 5F). We treated zebrafish embryos with RA and obtained a significant downregulation of shox expression, further corroborating the hypothesis of SHOX as a component of the RA pathway (Fig 5H). Finally, we tested the functional significance of the CYP26C1 variants identified in families 1–3 in zebrafish embryos and could show a striking effect on CYP26C1 activity (Appendix Fig S8). Together, these data indicate that CYP26C1 is involved in limb development and acts by controlling RA levels.
Figure 5. Pattern of defects in zebrafish embryos injected with anti‐cyp26c1 morpholino.
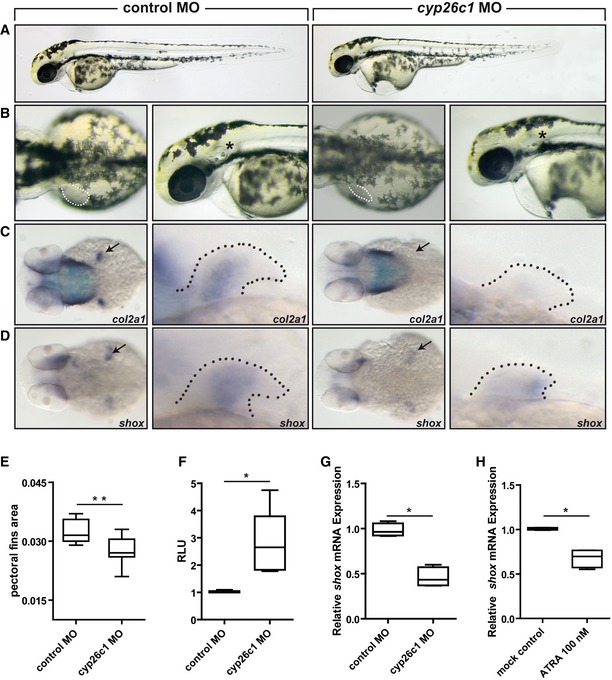
-
A, B(A) Lateral views of the embryos at 55 hours post‐fertilization (hpf). (B) Dorsal view and magnification on the lateral view of the embryos. Dotted line, pectoral fins; *, otic vesicle. cyp26c1 morphants show smaller fins compared to controls (n = 30 embryos).
-
C, DExpression of col2a1 (C) and shox (D) at 55 hpf was examined by in situ hybridization in embryos injected with control MO or with cyp26c1 MO. (C) Dorsal view and magnification on the pectoral fins of col2a1 expression. Arrow and dotted line indicate the pectoral fin. (D) Dorsal view and magnification on the pectoral fins of shox expression. Arrow and dotted line indicate the pectoral fin.
-
EPectoral fin area was measured by ImageJ (n = 30 embryos).
-
FCignal‐RARE system luciferase assay to test cyp26c1 MO knockdown effect on RA acid levels in zebrafish embryos (n = 5 replicates). In each replicate, 20–30 embryos per condition were assayed. RLU, relative light units.
-
GRelative shox mRNA expression normalized to reference genes eef1a and b‐actin in zebrafish embryos injected with control MO or cyp26c1 MO (n = 4). RNA was extracted from 10–15 injected embryos at 55 hpf.
-
HRelative shox mRNA expression normalized to reference genes eef1a and b‐actin in zebrafish embryos treated with 100 nM ATRA (n = 4). Embryos were collected at 24 hpf and treated with mock control or ATRA. RNA was extracted from 10–15 embryos after 6‐h treatment.
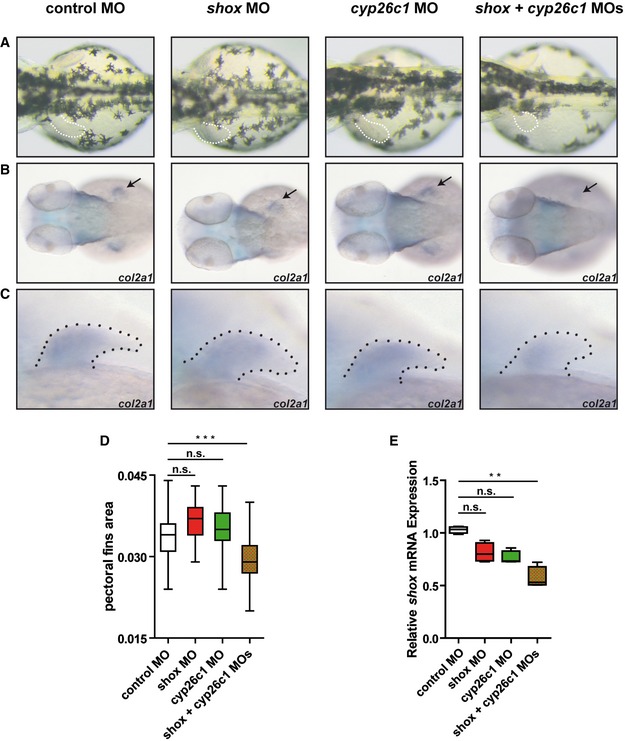
To test the hypothesis that CYP26C1 is a modifier of SHOX deficiency, we tested different concentration of shox and cyp26c1 MOs to determine subphenotypic dosages. While single knockdown of either shox or cyp26c1 using subphenotypic MO doses did not result in any obvious phenotype, double knockdown (simultaneous injection) of shox and cyp26c1 produced significantly smaller pectoral fins (Fig 6A–D). The relative expression of shox mRNA expression was also significantly reduced (Fig 6E). Staining for col2a1 revealed impaired pectoral fin development (Fig 6B). These data further corroborate the hypothesis that CYP26C1 damaging mutations contribute to severe phenotypes in SHOX deficient individuals.
Figure 6. Co‐injection of titrated subphenotypic doses of anti‐shox and anti‐cyp26c1 morpholinos impairs limb development.
-
ADorsal views of the embryos at 55 hours post‐fertilization (hpf). Dotted line, pectoral fins. shox + cyp26c1 double morphants show smaller fins compared to control and single MOs (n = 30 embryos).
-
B, CDorsal view and magnification on the pectoral fins of col2a1 expression at 55 hpf. Arrow and dotted line indicate the pectoral fin.
-
DPectoral fin area was measured by ImageJ (n = 30 embryos). Data are shown as means ± SD. ***P = 0.0001 (one‐way ANOVA with Bonferroni's multiple comparison test).
-
ERelative expression of shox mRNA normalized to reference genes eef1a and b‐actin in zebrafish embryos injected with control MO, shox MO, cyp26c1 MO, or shox + cyp26c1 MOs (n = 4). RNA was extracted from 10–15 injected embryos at 55 hpf. Data are shown as means ± SD. **P = 0.0048 (Kruskal–Wallis with Dunn's multiple comparison test).
Discussion
Variable phenotypes can be attributed to genetic and environmental factors. Our study describes a Mendelian disorder with wide phenotypic variability, which provides a unique opportunity to identify the genetic causes of variability. Individuals with SHOX deficiency but normal stature in a three‐generation family (Family 1) prompted us to postulate genetic modifiers as the possible reasons of this variability. Three individuals with height within the normal range and neither mesomelia nor MD had inherited the same SHOX variant p.Val161Ala as their five affected relatives with LWD. By combining whole‐genome linkage and whole‐exome sequencing analysis in family 1, we identified CYP26C1 as the gene co‐segregating with the clinical phenotype and hypothesized a putative modifier function on SHOX expression. The mother (II:4), an aunt (II:8), and the stepsister (III:1) of the index patient (III:2) presented with short stature, but dysmorphic skeletal signs were not diagnosed. These individuals did not carry damaging variants neither in SHOX nor in CYP26C1, suggesting that different factors contribute to their short stature phenotype. Mesomelia and MD were present only in those individuals carrying damaging variants in both SHOX and CYP26C1.
Defective CYP26C1 and SHOX were found to lead to a more severe phenotype in two further unrelated patients with LWD (Fig 1B). No functionally damaging variants in CYP26C1 were found in 350 control individuals with normal height, suggesting that this co‐occurrence is not coincidental (P‐value = 0.0261, two‐tailed Fisher's exact test). In addition, we browsed TGP for genotypes bearing both SHOX and CYP26C1 damaging variants (predicted as damaging by at least one of the prediction tools used). A limitation of this approach is that not for all variants individual genotypes are available. We could not find any individual from the TGP database carrying damaging variants in both genes (1000 Genomes Project Consortium, 2015). Finally, we browsed the ExAC database and estimated the frequency of SHOX and CYP26C1 damaging variants (Lek et al, 2016). To estimate the frequencies, we summed the allele frequencies of each damaging variant (predicted as damaging by at least one of the prediction tools used) reported in the ExAC database. The estimated allele frequencies were 0.3 and 0.6% for SHOX and CYP26C1, respectively. With these estimated frequencies, the probability for an individual to bear a damaging variant in both genes is 1.8 × 10−5.
CYP26C1 is an enzyme belonging to the cytochrome P450 superfamily and is involved in the oxidation of RA to generate polar retinoid species, which are readily excreted. Thus, CYP26C1 is involved in the catabolism of RA (Taimi et al, 2004; Uehara et al, 2006; Pennimpede et al, 2010). Accordingly, we found that damaging mutations in CYP26C1 reduce its RA catabolizing activity leading to a higher concentration of this retinoid in U2OS cells (Fig 2B).
Retinoic acid plays a key role in development, including formation of the body axis and skeleton (Pennimpede et al, 2010; Cunningham & Duester, 2015). During skeletal development, RA coordinates the development of central body axis, limb axis, and cranium. Moreover, it controls chondroblast differentiation and coordinates maturation and replacement of bone tissue during endochondral ossification (Weston et al, 2003). RA also plays a role in the postnatal maintenance of bone (Green et al, 2015). An excess or deficiency of RA dysregulates the expression of the respective target genes, which has a dramatic effect on development (Weston et al, 2003). Excess or loss of RA has, for example, been shown to impair pectoral fin development in zebrafish and results in reduced forelimb size or complete loss of forelimb in mouse embryos (Uehara et al, 2006; Akimenko & Ekker, 1995; Begemann et al, 2001; Emoto et al, 2005; Cunningham et al, 2013). Hence, a tight regulation of RA metabolism is essential.
We demonstrated for the first time that CYP26C1 is expressed on RNA and protein level in human primary chondrocytes (Fig 3), suggesting a role for CYP26C1 in the fine regulation of RA in these cells. Damaging mutations in CYP26C1 that affect its RA oxidation activity may therefore lead to high levels of this retinoid. In support of this hypothesis, we found that knockdown of cyp26c1 in zebrafish embryos increased RA levels and reduced col2a1 expression (Fig 5F). Previous experiments on chicken limbs have shown that treatment with excess RA strongly reduces Shox expression (Tiecke et al, 2006). Consistent with a role of RA in SHOX regulation, we have shown that treatment of human primary chondrocytes and zebrafish embryos with 100 nM RA significantly reduced SHOX expression, while this was not the case at 10–50 nM RA (Fig 2B and 5H; Appendix Fig S1). Luciferase reporter analyses of the human SHOX promoter suggest an indirect effect of RA on SHOX expression (Fig 3C). RA could lead to SHOX downregulation independently of RAREs, for example, by controlling the activation of other transcription factors, by non‐classical associations of receptors with other proteins, or other mechanisms (Balmer & Blomhoff, 2002). Finally, we showed that reduction of cyp26c1 in zebrafish embryos leads to the downregulation of shox expression (Fig 5D and G). Taken together, our results suggest that SHOX expression is not normally affected by endogenous RA in the nanomolar range (De Leenheer et al, 1995; Cunningham et al, 2013); however, loss of CYP26C1 results in excess RA that downregulates SHOX expression.
Height depends mostly on the longitudinal growth of the limbs. Limbs contain long bones that are formed through endochondral ossification. This process involves the aggregation of mesenchymal progenitors that differentiate into chondrocytes, which form a cartilage template that is then replaced by bone tissue. Endochondral ossification is orchestrated by a complex network of signaling pathways and transcription factors and is highly conserved in vertebrates. SHOX is one of the genes involved in this network (Blaschke & Rappold, 2001). In accordance with a role for SHOX in limb development, we could demonstrate that shox reduction impairs pectoral fin development in zebrafish embryos (Fig 4).
Gene dosage of the SHOX locus has previously been shown to determine height: SHOX deficiency causes short stature, whereas SHOX overdosage has been linked to tall stature in Klinefelter syndrome (Ogata et al, 2001). SHOX has been suggested to repress growth plate fusion and skeletal maturation in the distal limbs. Unlike SHOX deficiency, which is characterized by premature epiphyseal plate fusion and relatively advanced skeletal maturation, SHOX overdosage leads to a delayed growth plate fusion and consequently to longer limbs (Ogata et al, 2001). Thus, different dosages of the SHOX protein play a role in determining the adult height of an individual.
We have found that damaging mutations in CYP26C1 increase RA levels, which affect SHOX dosage and exacerbate SHOX deficiency phenotypes. To corroborate this hypothesis, we have demonstrated that the co‐injection of subphenotypic dosages of shox and cyp26c1 MOs strongly impaired pectoral fins (Fig 6). However, it is possible that damaging mutations in CYP26C1 modify SHOX deficiency not only by directly affecting its expression, but also by misregulating other genes involved in the formation of long bones.
Homozygous/compound heterozygous mutations in CYP26C1 have previously been associated with focal facial dermal dysplasia type IV (Slavotinek et al, 2013), a mild disorder of the skin. However, different CYP26C1 variants were reported and information on height in these individuals was not available. MO knockdown experiments of cyp26c1 in zebrafish embryos revealed abnormal pectoral fin development, but also an impairment of the otic vesicle and pharyngeal arches, structures that encompass the sites of the skin lesions observed in focal facial dermal dysplasia type IV (Slavotinek et al, 2013) (Fig 5B).
In 1963, Grüneberg (1963) defined modifiers as “genes capable of modifying the manifestation of a mutant gene without having an obvious effect on the normal condition”. It would therefore be interesting to find out whether damaging CYP26C1 variants have detectable phenotypes in the absence of SHOX deficiency. In family 3, the father (II:1), for example, carries a CYP26C1 damaging variant but presented height within the normal range and no obvious dysmorphic signs. A systematic analysis on a large cohort of patients with or without dysmorphic signs and short stature is therefore needed to resolve whether CYP26C1 variants alone can contribute to short stature phenotypes.
Taken together, this study represents an effort to elucidate the genetic causes of variability on the clinical manifestation in patients with SHOX deficiency. The RA pathway and one of its components, CYP26C1, were uncovered as biological triggers that we suggest to modify the severity of SHOX deficiency and alter the course of this disease. Only a few genetic modifiers have been reported in the literature so far (Oprea et al, 2008; Ebermann et al, 2010; Bečanović et al, 2015; Corvol et al, 2015; Guo et al, 2015; Lee et al, 2015), but recent technological advances are helping to broaden the understanding of the molecular basis of quantitative or discrete qualitative differences in phenotype. A primary reason to identify disease modifiers is to enable the accurate prediction of disease progression and improve therapeutic development. In the case of SHOX deficiency, manipulating the RA signaling pathway may be therapeutically beneficial.
Materials and Methods
Patients and controls
Family 1
Family 1 comprised 17 German individuals with five affected members diagnosed with LWD. DNA was available from 14 individuals (I:1, I:2, I:3, II:3, II:4, II:6, II:7, II:8, III:1, III:2, III:3, III:4, III:5, III:6). Four females (I:2, I:3, II:7, III:2) presented with mesomelia and MD. In the affected male (II:3), mesomelia and MD were borderline. Individuals (II:4, II:8, III:1) presented with short stature but no dysmorphic signs. The index patient (III:2) was diagnosed with LWD at the age of 8 years and treated with growth hormone from the age of 8.9 years onwards. Age of menarche was of 12 years. Further details on the clinical data are given in Table EV1.
Cohort of patients with SHOX deficiency
The sample comprised 68 unrelated cases with LWD and proven SHOX deficiency; it comprised 60 Europeans (28 Germans) and 9 Japanese. Two out of the 68 cases carried damaging mutations in CYP26C1 and all the available family information was retrieved (see below).
Family 2 comprised four French people with one daughter affected with LWD; parents and sister were unaffected without dysmorphic signs. The affected daughter (II:2) presented with short stature, mesomelia, and MD (available clinical data are given in Table EV1).
Family 3 comprised four Japanese individuals with one daughter presenting with LWD. Parents and brother were reported with normal stature and no dysmorphic signs (available clinical data are given in Table EV1).
Controls
As controls, we screened 350 (240 Germans, 110 Japanese) individuals with normal stature and no dysmorphic signs. We also compared frequencies to publicly available databases (ExAC, TGP, and EVS).
Linkage analysis
We genotyped DNA samples from 12 family members of family 1 using the Affymetrix GeneChip Human Mapping 50K Xba 240 Array (Affymetrix). Genotypes were called by the GeneChip® DNA Analysis Software (GDAS v3.0, Affymetrix). We verified sample genders by counting heterozygous SNPs on the X chromosome. Relationship errors were evaluated with the help of the program Graphical Relationship Representation (Abecasis et al, 2001). The program PedCheck was applied to detect Mendelian errors (O'Connell & Weeks, 1998) and data for SNPs with such errors were removed from the dataset. Non‐Mendelian errors were identified by using the program MERLIN (Abecasis et al, 2002) and unlikely genotypes for related samples were deleted. Non‐parametric linkage analysis using all genotypes of a chromosome simultaneously was carried out with MERLIN.
Parametric linkage analysis was performed by the program ALLEGRO (Gudbjartsson et al, 2000), assuming dominant inheritance with complete penetrance and a disease allele frequency of 0.0001. Haplotypes were reconstructed with ALLEGRO and presented graphically with HaploPainter (Thiele & Nürnberg, 2005). All data handling was performed using the graphical user interface ALOHOMORA (Rüschendorf & Nürnberg, 2005). Using ALLEGRO, we identified one genomic region of 19.2 Mb on chromosome 10 (chr10:85477508‐101462931; hg19) with LOD score of 2.4 (Figs EV1 and EV3). The LOD score obtained was the maximum LOD score expected in this family. A second peak with LOD score of 1.7 localized in a 2.02 Mb region of PAR1 (chrX:706800‐2735491; hg19) (Fig EV2). The reason for the reduced LOD score in PAR1 is individuals III:4 and III:6 who carry the disease haplotype although they are unaffected (Fig EV2).
Whole‐exome sequencing
Whole‐exome sequencing was performed as previously described (Haack et al, 2012). Exomes were enriched in solution provided by the manufacturer and indexed with SureSelect, XT Human All Exon 50 Mb kits (Agilent). Samples were sequenced as 100‐bp paired‐end runs on a HiSeq2000 system (Illumina). Read alignment to the human genome assembly was performed with the Burrows‐Wheeler Aligner (v.0.5.8.). SAMtools (v.0.1.7.) was used for detecting single nucleotide variants and small insertions and deletions. We generated 8.8 and 11.2 gigabases of sequence resulting in 89 and 93% of the target regions being covered at least 20 times for index patient (III:2) and her father (II:3), respectively. To identify putative candidate genes based on dominant inheritance, we filtered all variants that were present in both individuals, but absent in all except two of 1,297 in‐house control exomes.
Sanger sequencing
Sanger sequencing was performed on MegaBACE sequencer using the DYEnamic™ ET Terminator Cycle Sequencing Kit (GE Healthcare Life Sciences) following the manufacturer's protocol. Sequences were analyzed using the MegaBACE Sequence Analyzer (v3.0).
Analysis in human primary chondrocytes
Human primary chondrocytes (mycoplasma‐free) were obtained as previously described (Marchini et al, 2004). Cells were cultured in DMEM (Gibco) containing 10% FBS (Gibco) and penicillin/streptomycin (Gibco) at 37°C, 5% CO2, 95% humidity. To test CYP26C1 mRNA expression, cells were resuspended in Trizol® (Invitrogen) for RNA preparation according to standard protocols. cDNA synthesis was performed with SuperScript™ II (Invitrogen) according to manufacturer's protocol. Specific primers for CYP26C1, SOX9, and SHOX transcripts were designed (Dataset EV4) and PCR performed. PCR products were confirmed by Sanger sequencing.
To test CYP26C1 protein expression, microsomal preparations were obtained as follows: Cells were washed three times with 2 ml cold solution containing 8 mM Na2HPO4, 1.5 mM KH2PO4, and 2.7 mM KCl and subsequently suspended in 1.5 ml of this buffer. Resuspension was centrifuged at 360 g for 2 min at 4°C. The pellet was resuspended in 1 ml of 100 mM potassium phosphate buffer pH 7.4 containing 10 mM EDTA. Resuspension was sonicated for 20 × 1 s. Lysed cells were then centrifuged at 10,700 g for 1 h at 4°C. The pellet was homogenized in 100 μl of 50 mM potassium phosphate buffer pH 7.4, containing 0.1 mM EDTA and 10% glycerol (Biomol). Protein concentration was determined using the BCA method. Western blotting experiments were performed following standard protocol with the LI‐COR Odyssey® system (LI‐COR). hCyp26c1 antibody (PA5‐15059, Thermo Fischer Scientific) was diluted as recommended (dilution 1:100) in LI‐COR Blocking Buffer (LI‐COR), 1× TBS, Tween 0.1%.
SHOX expression analysis was performed as follows: Cells were plated in 6‐well plates (1 × 106 cells per well). After 24 h, cells were treated with ATRA (Sigma‐Aldrich, stock solution 1 mM in ethanol absolute) for 6 h and then resuspended in Trizol for RNA extraction according to standard protocols. cDNA synthesis was performed with SuperScript™ II according to manufacturer's protocol. SHOX expression was analyzed by quantitative PCR using gene‐specific primers and normalized to HPRT and SDHA (Dataset EV4) with SensiFAST™ SYBR® Lo‐ROX Kit (Bioline) in the 7500 Fast Real‐Time PCR System (Applied Biosystems).
Luciferase assays
U2OS cells (human osteosarcoma cells, ATCC; mycoplasma‐free) were cultured in DMEM containing 10% FBS and penicillin/streptomycin at 37°C, 5% CO2, 95% humidity. Luciferase assays to test SHOX variants were performed as follows: Cells were seeded in 24‐well plates at 1 × 105 cells per well. After 24 h, cells were transfected with Lipofectamine 2000 (Invitrogen) according to standard protocols. For each well, cells were transfected with 300 ng of pcDNA4/TO empty vector (Life Technologies), pcDNA4/TO SHOX wild type or mutants; 300 ng of pGL3basic‐FGFR3(−3,430/+464) (Decker et al, 2011) firefly luciferase reporter construct (Promega); 150 ng pRL‐TK Renilla luciferase reporter construct (Promega). Mutations were introduced by site‐directed mutagenesis (QuickChange Lightning Site‐Directed Mutagenesis Kit). After 24 h of transfection, cells were lysed and luciferase activity measured with the Dual Luciferase Assay System (Promega) in a Berthold 96‐microplate luminometer. The experiments were performed each time in triplicate.
Luciferase assays to test CYP26C1 variants were performed as follows: Cells were seeded in 96‐well plates at 1 × 104 cells per well. After 24 h, cells were transfected with Lipofectamine 2000 according to standard protocols. For each well, cells were transfected with 100 ng of pIRES2‐EGFP empty vector (obtained from Dr. Thomas Boettger), pIRES2/EGFP‐CYP26C1 wild type or mutants; 100 ng of Cignal RARE Reporter system plasmids (SABiosciences). Mutations were introduced by site‐directed mutagenesis. After 24‐h transfection, cells were treated with 250 nM ATRA. After 24‐h treatment, cells were lysed and assayed with the Dual Luciferase Assay System in a Berthold 96‐microplate luminometer. The experiments were performed each time in triplicate.
Luciferase assays to test the SHOX promoter were performed as follows: Cells were seeded in 24‐well plates at 1 × 105 cells per well. After 24 h, cells were transfected with Lipofectamine 2000 according to standard protocols. For each well, cells were transfected with 500 ng of pGL3basic‐empty vector (Promega) or pGL3basic‐CNE3‐SHOX promoter as previously described (Verdin et al, 2015); 50 ng of pRL‐TK Renilla luciferase reporter construct. The CNE3 enhancer was chosen because it was reported with the highest activity on the SHOX promoter (Verdin et al, 2015). After 24‐h transfection, cells were treated with 250 nM ATRA. After 24‐h treatment, cells were lysed and assayed with the Dual Luciferase Assay System in a Berthold 96‐microplate luminometer. The experiments were performed each time in triplicate.
Zebrafish experiments
Experiments were carried out in wild‐type Danio rerio strains (AB, TL, Tübingen). Zebrafish were reared according to standard protocols. Morpholino were injected at one‐cell stage embryos as previously described (Renz et al, 2015). The shox and cyp26c1 morpholinos were obtained from Gene Tools (Dataset EV4). Two splicing MOs were designed for shox knockdown: MO1 and MO2. In the main text, figures refer to MO1 (Figs 4 and 6). Specific phenotype was confirmed by MO2 (Appendix Fig S3). For cyp26c1 knockdown, we used a published MO (Liang et al, 2012). The standard control MO from Gene Tools was used as a control. Each embryo was injected with 1–2 ng of control, shox, or cyp26c1 MOs. Embryos for double knockdown experiments were injected with 100 pg of shox MO and/or 800 pg of cyp26c1 MO. RNA was extracted from 20–30 injected embryos at 36 h post‐fertilization (hpf) with Trizol® according to standard protocols. cDNA synthesis was performed with SuperScript™ II according to manufacturer's protocol. Effective splicing blocking was confirmed with test primers (Dataset EV4) and PCR products were cloned in pSTBlue1 vector (Novagen) and sequenced (Appendix Figs S4–S6). Sense‐capped RNA of human SHOX and CYP26C1 wild type and mutants was synthesized using the mMESSAGE mMACHINE system (Ambion) from pCS2 (Hassel et al, 2009).
Expression analyses of shox after MO injection were performed as follows: RNA was extracted from 10–15 injected embryos at 55 h post‐fertilization (hpf) with Trizol® according to standard protocols. cDNA synthesis was performed with SuperScript™ II according to manufacturer's protocol. shox expression was analyzed by quantitative PCR using gene‐specific primers and normalized to eef1a and b‐actin (Dataset EV4) with SensiFAST™ SYBR® Lo‐ROX Kit (Bioline) in the 7500 Fast Real‐Time PCR System (Applied Biosystems).
Luciferase experiments in zebrafish were performed injecting into one‐cell stage embryos doses in the range of 1–2 nl of a 25 nM solution of Cignal RARE Reporter system plasmids and 1–2 ng of control MO or cyp26c1 MO. After 24‐h injection, embryos were separated in groups of 20–30, lysed and assayed with a Dual Luciferase Assay System (Promega) in a Berthold 96‐microplate luminometer.
Treatments of zebrafish embryos with RA were performed as follows: Wild‐type embryos were left developing for 24 h post‐fertilization. At 24 hpf, embryos were separated in groups of 10–15 and treated with mock control or ATRA for 6 h. Finally, RNA was extracted and used for shox expression analysis as described above.
Whole‐mount in situ hybridization
Whole‐mount in situ hybridization was performed as described previously (Jowett & Lettice, 1994). Gene‐specific primers (Dataset EV4) were used to amplify col2a1 and shox cDNA. Amplicons were then cloned using the psTBlue1 vector (Novagen). Digoxigenin‐labeled RNA probes were synthesized using the DIG RNA Labeling Mix (Roche) with the MAXIscript® SP6/T7 Transcription Kit (Ambion). Images were taken with the microscope SZX16, CellDImaging Software (Olympus). Pectoral fin area was measured with Fiji ImageJ (Schindelin et al, 2012).
Statistics
Samples for in vitro and zebrafish embryos experiments were randomly assigned to experimental groups, to processing order, and position in multi‐well devices. No statistical method was used to predetermine sample size. Group sample sizes for experiments were chosen based on previous studies. Zebrafish embryos that died before analysis were excluded. Statistical analyses were performed using GraphPad Prism version 5 for Windows (GraphPad Software). Data were tested for normality using the D'Agostino and Pearson omnibus normality test. When data fitted normal distribution parametric tests were used, otherwise non‐parametric tests were used. Specific tests are described for each group of data in the figure legends. Differences between two groups were analyzed by two‐tailed Student's t‐test. Differences between three or more groups were analyzed by analysis of variance (ANOVA) test. For all experiments, data are expressed as the mean ± SD. P‐values < 0.05 were considered significant.
Bioinformatics resources
Primers for cloning in situ hybridization probes, for sequencing, and for testing MO efficacy were designed using Primer3 (Untergasser et al, 2012). Primers for quantitative PCR were designed using Universal ProbeLibrary Assay Design Center (Roche). Protein and DNA schemes were drawn using Illustrator of Biological Sequences (IBS) (Liu et al, 2015). Mutations were tested with PolyPhen‐2 (Adzhubei et al, 2013), Mutation Taster (Schwarz et al, 2010), SIFT (Vaser et al, 2016), PROVEAN (Choi & Chan, 2015), and CADD (Kircher et al, 2014). Transcription factor binding sites were predicted using PROMO (Messeguer et al, 2002; Farré et al, 2003).
Study approval
Studies involving human material were approved by the Review Board Committee at the University Hospital Heidelberg and at the Hamamatsu University School of Medicine. Written informed consent was received from all participants prior to their inclusion in the study. The study was conducted in accordance with the guidelines of the WMA Declaration of Helsinki and the Department of Health and Human Services Belmont Report. All animal experiments were conducted with approval of local Animal Care Committee and according to institutional guidelines.
Author contributions
GAR designed the project. AM, GAR, and DH designed the experiments and analyzed the data. AM performed the cell culture experiments. LJ and AM performed the experiments in zebrafish embryos. SF‐O, GB, MF, TO, and ED recruited the patients and provided DNA for sequencing. AM, RR, and BW performed the sequencing of patients and controls. GN performed whole‐genome linkage. AM and GAR wrote the manuscript. AM, RR, DH, and GAR contributed to the discussion and all authors commented the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
The paper explained.
Problem
Mutations in the SHOX gene cause SHOX deficiency, the most frequent monogenic cause of short stature. The clinical severity of SHOX deficiency varies widely, ranging from short stature without skeletal signs to pronounced skeletal dysplasia. In rare cases, family members with identical mutations even have normal stature. So far, the underlying factors contributing to the phenotypic variability in individuals with SHOX deficiency are unknown. Genetic factors distinct from the SHOX gene may represent an important source of clinical variability in SHOX deficiency.
Genetic modifiers are genetic factors that influence the phenotypic expression of a disease‐causing gene. Identifying genetic modifiers may result in a better understanding of the clinical variability of SHOX deficiency and enable a more accurate prediction of disease progression.
Results
We describe a large family where some individuals with a damaging SHOX mutation have normal stature, while other family members with the identical mutation have short stature and skeletal anomalies. To identify the genetic factors that could explain such phenotypic differences, a genetic analysis of this family was performed. This led to the identification of the retinoic acid‐degrading enzyme CYP26C1 as a potential modifying genetic factor. Retinoic acid is the most active biological form of vitamin A and has been shown to play a role in skeleton formation. An excess of this molecule impairs limb development. Expression of CYP26C1 in human primary chondrocytes (cells that are involved in bone formation) could be demonstrated. CYP26C1 damaging mutations affect its ability to degrade retinoic acid, leading to higher levels of this vitamin A derivate. High levels of retinoic acid reduced SHOX expression in human primary chondrocytes, suggesting that CYP26C1 regulates SHOX expression within the retinoic acid signaling pathway. Two further families with SHOX and CYP26C1 mutations provided further evidence of the most severe phenotype.
Zebrafish represents an animal model particularly suitable to study bone disorders. Decreasing both SHOX and CYP26C1 levels in zebrafish embryos led to smaller fins, further supporting that SHOX and CYP26C1 interact during skeletal development. Mutations in CYP26C1 result in increased retinoic acid levels which in turn decrease SHOX gene expression leading to more severe SHOX deficiency phenotypes.
Impact
This study represents an effort to understand the genetic causes of clinical variability. Unraveling factors which modify disease toward milder or more severe phenotypes may lead to novel therapeutic approaches. We provide evidence for CYP26C1 as a genetic factor influencing SHOX deficiency phenotypic outcomes through the retinoic acid signaling pathway. Manipulating vitamin A metabolism in SHOX deficiency patients may alleviate the skeletal abnormalities of this condition.
For more information
ExAC
http://exac.broadinstitute.org
TGP
http://browser.1000genomes.org
EVS
http://evs.gs.washington.edu/EVS
PROMO
http://alggen.lsi.upc.es/cgi-bin/promo_v3/promo/promoinit.cgi?dirDB=TF_8.3
SHOX deficiency
http://www.ncbi.nlm.nih.gov/books/NBK1215/
X chromosome gene database
http://grenada.lumc.nl/LOVD2/MR/home.php?select_db=SHOX
OMIM
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Review Process File
Acknowledgements
We thank Tim Strom, Werner Blum, Miriam Rosilio, Dave Bunyan, Christine Fischer, Slavil Peykov, Herbert Steinbeisser, Thomas Holstein, and Christel Herold‐Mende for support. We thank Nagarajan Paramavisam for calculating the CADD scores. Beate Niesler, Simone Berkel, and Antonio Marchini are acknowledged for comments on the manuscript. This study was supported by the DFG (Deutsche Forschungsgemeinschaft), the Medical Faculty of Heidelberg, and the DZHK Heidelberg (German Centre for Cardiovascular Research). AM is a member of the Hartmut Hoffmann‐Berling International Graduate School of Molecular and Cellular Biology (HBIGS); GAR is a member of CellNetworks Cluster for Excellence of the University of Heidelberg, Germany. DH and GAR are members of the DZHK.
EMBO Mol Med (2016) 8: 1455–1469
References
- Abecasis GR, Cherny SS, Cookson WO, Cardon LR (2001) GRR: graphical representation of relationship errors. Bioinformatics 17: 742–743 [DOI] [PubMed] [Google Scholar]
- Abecasis GR, Cherny SS, Cookson WO, Cardon LR (2002) Merlin‐rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet 30: 97–101 [DOI] [PubMed] [Google Scholar]
- Adzhubei I, Jordan DM, Sunyaev SR (2013) Predicting functional effect of human missense mutations using PolyPhen‐2. Curr Protoc Hum Genet 7: Unit7.20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akimenko MA, Ekker M (1995) Anterior duplication of the Sonic hedgehog expression pattern in the pectoral fin buds of zebrafish treated with retinoic acid. Dev Biol 170: 243–247 [DOI] [PubMed] [Google Scholar]
- Balmer JE, Blomhoff R (2002) Gene expression regulation by retinoic acid. J Lipid Res 43: 1773–1808 [DOI] [PubMed] [Google Scholar]
- Baron J, Sävendahl L, De Luca F, Dauber A, Philip M, Wit JM, Nilsson O (2015) Short and tall stature: a new paradigm emerges. Nat Rev Endocrinol 11: 735–746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bečanović K, Nørremølle A, Neal SJ, Kay C, Collins JA, Arenillas D, Lilja T, Gaudenzi G, Manoharan S, Doty CN et al (2015) A SNP in the HTT promoter alters NF‐kB binding and is a bidirectional genetic modifier of Huntington disease. Nat Neurosci 18: 807–816 [DOI] [PubMed] [Google Scholar]
- Begemann G, Schilling TF, Rauch GJ, Geisler R, Ingham PW (2001) The zebrafish neckless mutation reveals a requirement for raldh2 in mesodermal signals that pattern the hindbrain. Development 128: 3081–3094 [DOI] [PubMed] [Google Scholar]
- Belin V, Cusin V, Viot G, Girlich D, Toutain A, Moncla A, Le Merrer M, Munnich A, Cornier‐Daire V (1998) SHOX mutations in dyschondrosteosis (Leri‐Weill syndrome). Nat Genet 19: 67–69 [DOI] [PubMed] [Google Scholar]
- Benito‐Sanz S, Royo JL, Barroso E, Paumard‐Hérnandez B, Barreda‐Bonis AC, Liu P, Gracía R, Lupski JR, Campos‐Barros Á, Gómoez‐Skarmeta JL et al (2012) Identification of the first recurrent PAR1 deletion in Léri‐Weill dyschondrosteosis and idiopathic short stature reveals the presence of a novel SHOX enhancer. J Med Genet 49: 442–450 [DOI] [PubMed] [Google Scholar]
- Binder G (2011) Short stature due to SHOX deficiency: genotype, phenotype, and therapy. Horm Res Paediatr 75: 81–89 [DOI] [PubMed] [Google Scholar]
- Blaschke RJ, Rappold GA (2001) SHOX in short stature syndromes. Horm Res 55: 21–23 [DOI] [PubMed] [Google Scholar]
- Bunyan DJ, Baker KR, Harvey JF, Thomas NS (2013) Diagnostic screening identifies a wild range of mutations involving the SHOX gene, including a common 47.5 kb deletion 160 kb downstream with a variable phenotypic effect. Am J Med Genet 161A: 1329–1338 [DOI] [PubMed] [Google Scholar]
- Choi Y, Chan AP (2015) PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 31: 2745–2747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement‐Jones M, Schiller S, Rao E, Blaschke RJ, Zuniga A, Zeller R, Robson SC, Binder G, Glass I, Strachan T et al (2000) The short stature homeobox gene SHOX is involved in skeletal abnormalities in Turner syndrome. Hum Mol Genet 22: 695–702 [DOI] [PubMed] [Google Scholar]
- Corvol H, Blackman SM, Boëlle PY, Gallins PJ, Pace RG, Stonebraker JR, Accurso FJ, Clement A, Collaco JM, Dang H et al (2015) Genome‐wide association meta‐analysis identifies five modifier loci of lung disease severity in cystic fibrosis. Nat Commun 6: 8382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham TJ, Zhao X, Sandell LL, Evans SM, Trainor PA, Duester G (2013) Antagonism between retinoic acid and fibroblast growth factor signaling during limb development. Cell Rep 3: 1503–1511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham TJ, Duester G (2015) Mechanisms of retinoic acid signaling and its roles in organ and limb development. Nat Rev Mol Cell Biol 16: 110–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Leenheer AP, Lambert WE, Clayes I (1995) All‐trans retinoic acid: measurement of reference values in human serum by high performance liquid chromatography. J Lipid Res 23: 1362–1367 [PubMed] [Google Scholar]
- Decker E, Durand C, Bender S, Rödelsperger C, Glaser A, Hecht J, Schneider KU, Rappold G (2011) FGFR3 is a target of the homeobox transcription factor SHOX in limb development. Hum Mol Genet 20: 1524–1535 [DOI] [PubMed] [Google Scholar]
- Durand C, Rappold GA (2013) Height matters‐from monogenic disorders to normal variation. Nat Rev Endocrinol 9: 171–177 [DOI] [PubMed] [Google Scholar]
- Ebermann I, Phillips JB, Liebau MC, Koenekoop RK, Schermer B, Lopez I, Schäfer E, Roux AF, Dafinger C, Bernd A et al (2010) PDZD7 is a modifier of retinal disease and a contributor to digenic Usher syndrome. J Clin Invest 120: 1812–1823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emoto Y, Wada H, Okamoto H, Kudo A, Imai Y (2005) Retinoic acid‐metabolizing enzyme Cyp26a1 is essential for determining territories of hindbrain and spinal cord in zebrafish. DevBiol 278: 415–427 [DOI] [PubMed] [Google Scholar]
- Farré D, Roset R, Huerta M, Adsuara JE, Roselló L, Albà MM, Messeguer X (2003) Identification of patterns in biological sequences at the ALGGEN server: PROMO and MALGEN. Nucleic Acids Res 31: 3651–3653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1000 Genomes Project Consortium (2015) A global reference for human genetic variation. Nature 526: 68–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green AC, Martin TJ, Purton LE (2015) The role of vitamin A and retinoic acid receptor signaling in post‐natal maintenance of bone. J Steroid Biochem Mol Biol 155: 135–146 [DOI] [PubMed] [Google Scholar]
- Grüneberg H (1963) The pathology of development; a study of inherited skeletal disorders in animals, pp. 309 New York, NY, USA: Wiley J; [Google Scholar]
- Gu X, Xu F, Wang X, Gao X, Zhao Q (2005) Molecular cloning and expression of a novel CYP26 gene (cyp26d1) during zebrafish early development. Gene Expr Patterns 5: 733–739 [DOI] [PubMed] [Google Scholar]
- Gudbjartsson DF, Jonasson K, Frigge ML, Kong A (2000) Allegro, a new computer program for multipoint linkage analysis. Nat Genet 25: 12–13 [DOI] [PubMed] [Google Scholar]
- Guo T, Chung JH, McDonald‐McGinn DM, Kates WR, Hawula W, Coleman K, Zackai E, Emanuel BS, Morrow BE (2015) Histone modifier genes alter conotruncal heart phenotypes in 22q11.2 deletion syndrome. Am J Hum Genet 3: 869–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haack TB, Hogarth F, Kruer MC, Gregory A, Wieland T, Schwarzmayr T, Graf E, Sanford L, Meyer E, Kara E et al (2012) Exome sequencing reveals de novo WDR45 mutations causing a phenotypically distinct, X‐linked dominant form of NBIA. Am J Hum Genet 91: 1144–1149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassel D, Dahme T, Erdmann J, Meder B, Huge A, Stoll M, Just S, Hess A, Ehlermann P, Weichenhan D et al (2009) Nexilin mutations destabilize cardiac Z‐disks and lead to dilated cardiomyopathy. Nat Med 15: 1281–1288 [DOI] [PubMed] [Google Scholar]
- Hernandez RE, Putzke AP, Myers JP, Margeretha L, Moens CB (2007) Cyp26 enzymes generate the retinoic acid response pattern necessary for hindbrain development. Development 134: 177–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber C, Rosilio M, Munnich A, Cormier‐Daire V (2006) French SHOX GeNeSIS Module. High incidence of SHOX anomalies in individuals with short stature. J Med Genet 43: 735–739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jowett T, Lettice L (1994) Whole‐mount in situ hybridizations on zebrafish embryos using a mixture of digoxigenin‐ and fluorescence‐labelled probes. Trends Genet 10: 73–74 [DOI] [PubMed] [Google Scholar]
- Kircher M, Witten DM, Jain P, Cooper GM, Shendure J (2014) A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 46: 310–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laue K, Pogoda HM, Daniel PB, van Haeringen A, Alanay Y, von Ameln S, Rachwalski M, Morgan T, Gray MJ, Breuning MH et al (2011) Craniosynostosis and multiple skeletal anomalies in humans and zebrafish result from a defect in the localized degradation of retinoic acid. Am J Hum Genet 89: 595–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JM, Wheeler VC, Chao MJ, Vonsattel JP, Pinto RM, Lucente D, Abu‐Elneel K, Ramos EM, Mysore JS, Gillis T et al (2015) Identification of genetic factors that modify clinical onset of Huntington's disease. Cell 162: 516–526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O'Donnell‐Luria AH, Ware JS, Hill AJ, Cummings BB et al (2016) Analysis of protein‐coding genetic variation in 60,706 humans. Nature 536: 285–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang D, Jia W, Li J, Li K, Zhao Q (2012) Retinoic acid signaling plays a restrictive role in zebrafish primitive myelopoiesis. PLoS One 7: e30865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Xie Y, Ma J, Luo X, Nie P, Zuo Z, Lahrmann U, Zhao Q, Zheng Y, Zhao Y et al (2015) IBS: an illustrator for the presentation and visualization of biological sequences. Bioinformatics 31: 3359–3361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchini A, Marttila T, Winter A, Caldeira S, Malanchi I, Blaschke RJ, Häcker B, Rao E, Karperien M, Wit JM et al (2004) The short stature homeodomain protein SHOX induces cellular growth arrest and apoptosis and is expressed in human growth plate chondrocytes. J Biol Chem 279: 37103–37114 [DOI] [PubMed] [Google Scholar]
- Marchini A, Ogata T, Rappold GA (2016) A Track Record on SHOX: from Basic Research to Complex Models and Therapy. Endocr Rev 37: 417–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchini A, Rappold G, Schneider KU (2007) SHOX at a glance: from gene to protein. Arch Physiol Biochem 113: 116–123 [DOI] [PubMed] [Google Scholar]
- Messeguer X, Escudero R, Farré D, Nuñez O, Martinez J, Albà MM (2002) PROMO: detection of known transcription regulatory elements using species‐tailored searches. Bioinformatics 18: 333–334 [DOI] [PubMed] [Google Scholar]
- O'Connell JR, Weeks DE (1998) PedCheck: a program for identification of genotype incompatibilities in linkage analysis. Am J Hum Genet 63: 259–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogata T, Matsuo N, Nishimura G (2001) SHOX haploinsufficiency and overdosage: impact of gonadal function status. J Med Genet 38: 1–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oprea GE, Kröber S, McWorther ML, Rossoll W, Müller S, Krawczak M, Bassell GJ, Beattie CE, Wirth B (2008) Plastin 3 is a protective modifier of autosomal recessive spinal muscular atrophy. Science 320: 524–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panda S, Sato TK, Castrucci AM, Rollag MD, DeGrip WJ, Hogenesch JB, Provencio I, Kay SA (2002) Melanopsin (Opn4) requirement for normal light‐induced circadian phase shifting. Science 298: 2213–2216 [DOI] [PubMed] [Google Scholar]
- Pennimpede T, Cameron DA, MacLean GA, Li H, Abu‐Abed S, Petkovich M (2010) The role of CYP26 enzymes in defining appropriate retinoic acid exposure during embryogenesis. Birth Defects Res A Clin Mol Teratol 88: 883–894 [DOI] [PubMed] [Google Scholar]
- Ranke MB (1994) The KIGS aetiology classification system In Progress in growth hormone therapy‐5 years of KIGS, Ranke MB, Gunnarson R. (eds), pp 51–61. Mannheim: J&J Verlag GmBH; [Google Scholar]
- Rao E, Weiss B, Fukami M, Rump A, Niesler B, Mertz A, Muroya K, Binder G, Kirsch S, Winkelmann M et al (1997) Pseudoatusomal deletions encompassing a novel homeobox gene cause growth failure in idiopathic short stature and Turner syndrome. Nat Genet 16: 54–63 [DOI] [PubMed] [Google Scholar]
- Renz M, Otten C, Faurobert E, Rudolph F, Zhu Y, Boulday G, Duchene J, Mickoleit M, Dietrich AC, Ramspacher C et al (2015) Regulation of β1 integrin‐Klf2‐mediated angiogenesis by CCM proteins. Dev Cell 26: 181–190 [DOI] [PubMed] [Google Scholar]
- Rosilio M, Huber‐Lequesne C, Sapin H, Carel JC, Blum WF, Cormier‐Daire V (2012) Genotypes and phenotypes of children with SHOX deficiency in France. J Clin Endocrinol Metab 97: 1257–1265 [DOI] [PubMed] [Google Scholar]
- Rüschendorf F, Nürnberg P (2005) ALOHOMORA: a tool for linkage analysis using 10K SNP array data. Bioinformatics 21: 2123–2125 [DOI] [PubMed] [Google Scholar]
- Rydeen AB, Waxman JS (2014) Cyp26 enzymes are required to balance the cardiac and vascular lineages within the anterior lateral plate mesoderm. Development 141: 1638–1648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawada R, Kamei H, Hakuno F, Takahashi S, Shimizu T (2015) In vivo loss of function study reveals the short stature homeobox‐containing (shox) gene plays indispensable roles in early embryonic growth and bone formation in zebrafish. Dev Dyn 244: 146–156 [DOI] [PubMed] [Google Scholar]
- Schiller S, Spranger S, Schechinger B, Fukami M, Merker S, Drop SL, Tröger J, Knoblauch H, Kunze J, Seidel J et al (2000) Phenotypic variation and genetic heterogeneity in Léri‐Weill syndrome. Eur J Hum Genet 8: 54–62 [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda‐Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B et al (2012) Fiji: an open‐source platform for biological‐image analysis. Nat Methods 9: 676–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider KU, Marchini A, Sabherwal N, Röth R, Niesler B, Marttila T, Blaschke RJ, Lawson M, Dumic M, Rappold G (2005) Alteration of DNA binding, dimerization, and nuclear translocation of SHOX homedodomain mutations identified in idiopathic short stature and Leri‐Weill dyschondrosteosis. Hum Mutat 26: 44–52 [DOI] [PubMed] [Google Scholar]
- Schwarz JM, Roedelsperger C, Schuelke M, Seelow D (2010) MutationTaster evaluates disease‐causing potential of sequence alterations. Nat Methods 7: 575–576 [DOI] [PubMed] [Google Scholar]
- Shears DJ, Vassal HJ, Goodman FR, Palmer RW, Reardon W, Superti‐Furga A, Scambler PJ, Winter RM (1998) Mutation and deletion of the pseudoautosomal gene SHOX cause Leri‐Weill dyschondrosteosis. Nat Genet 19: 70–73 [DOI] [PubMed] [Google Scholar]
- Sirbu IO, Gresh L, Barra J, Duester G (2005) Shifting boundaries of retinoic acid activity control hindbrain segmental gene expression. Development 132: 2611–2622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slavotinek AM, Mehrotra F, Nazarenko I, Tang PL, Cameron D, Li B, Chu C, Chou C, Marqueling AL, Yahyavi M et al (2013) Focal facial dermal dysplasia, type IV, is caused by mutations in CYP26C1. Hum Mol Genet 22: 696–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taimi M, Helvig C, Wisniewski J, Ramshaw H, White J, Amad M, Korcyak B, Petkovich M (2004) A novel human cytochrome P450, CYP26C1, involved in metabolism of 9‐cis and all‐trans isomers of retinoic acid. J Biol Chem 279: 77–85 [DOI] [PubMed] [Google Scholar]
- Thiele H, Nürnberg P (2005) HaploPainter: a tool for drawing pedigrees with complex haplotypes. Bioinformatics 21: 1730–1732 [DOI] [PubMed] [Google Scholar]
- Tiecke E, Bangs F, Blaschke R, Farrell ER, Rappold G, Tickle C (2006) Expression of the short stature homeobox gene Shox is restricted by proximal and distal signals in chick limb buds and affects the length of skeletal elements. Dev Biol 298: 585–596 [DOI] [PubMed] [Google Scholar]
- Uehara M, Yashiro K, Mamiya S, Nishino J, Chambon P, Dolle P, Sakai Y (2006) CYP26A1 and CYP26C1 cooperatively regulate anterior‐posterior patterning of the developing brain and the production of migratory cranial neural crest cells in the mouse. Dev Biol 302: 399–411 [DOI] [PubMed] [Google Scholar]
- Untergasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, Rozen SG (2012) Primer3‐new capabilities and interfaces. Nucleic Acids Res 40: e115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaser R, Adusumalli S, Leng SN, Sikic M, Ng PC (2016) SIFT missense predictions for genomes. Nat Protoc 11: 1–9 [DOI] [PubMed] [Google Scholar]
- Verdin H, Fernández‐Miñán A, Benito‐Sanz S, Janssens S, Callewaert B, De Waele K, De Schepper J, François I, Menten B, Heath KE et al (2015) Profiling of conserved non‐coding elements upstream of SHOX and functional characterization of the SHOX cis‐regulatory landscape. Sci Rep 5: 17667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weston AD, Hoffman LM, Underhill TM (2003) Revisiting the role of retinoid signaling in skeletal development. Birth Defects Res C Embryo Today 69: 156–173 [DOI] [PubMed] [Google Scholar]
- Zinn AR, Wei F, Zhang L, Elder FF, Sctt CI Jr, Marttila P, Ross JL (2002) Complete SHOX deficiency causes Langer mesomelic dysplasia. Am J Med Genet 110: 158–163 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Table EV1
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Review Process File