

Significance
NOV1 is a stilbene cleavage oxygenase (SCO). SCOs cleave the central double bond of stilbenes, forming two phenolic aldehydes. Many stilbenes, such as resveratrol, are produced by plants as secondary metabolites. They are also formed from lignin during kraft pulping. SCOs are related to carotenoid cleavage oxygenases (CCOs), which cleave β-carotene or apocarotenoids. Carotenoids play important roles in photosynthesis and light perception in the eye. We present the structure of an SCO and the structure of a CCO-related enzyme in ternary complex with oxygen and substrate. This structure allows us to propose a mechanism relevant to both SCOs and CCOs, where the substrate is activated for reaction with a ferric-superoxo electrophile by active site base-catalyzed deprotonation of a phenol group.
Keywords: stilbene, dioxygenase, structure, carotenoid
Abstract
Stilbenes are diphenyl ethene compounds produced naturally in a wide variety of plant species and some bacteria. Stilbenes are also derived from lignin during kraft pulping. Stilbene cleavage oxygenases (SCOs) cleave the central double bond of stilbenes, forming two phenolic aldehydes. Here, we report the structure of an SCO. The X-ray structure of NOV1 from Novosphingobium aromaticivorans was determined in complex with its substrate resveratrol (1.89 Å), its product vanillin (1.75 Å), and without any bound ligand (1.61 Å). The enzyme is a seven-bladed β-propeller with an iron cofactor coordinated by four histidines. In all three structures, dioxygen is observed bound to the iron in a side-on fashion. These structures, along with EPR analysis, allow us to propose a mechanism in which a ferric-superoxide reacts with substrate activated by deprotonation of a phenol group at position 4 of the substrate, which allows movement of electron density toward the central double bond and thus facilitates reaction with the ferric superoxide electrophile. Correspondingly, NOV1 cleaves a wide range of other stilbene-like compounds with a 4′-OH group, offering potential in processing some solubilized fragments of lignin into monomer aromatic compounds.
Stilbenes are diphenyl ethene compounds that are produced naturally in a wide variety of plant species and some bacteria. One stilbene derivative of note is resveratrol, which is a plant phytoalexin abundant in grapes and peanuts. Studies have demonstrated numerous health benefits related to the consumption of resveratrol, which is correlated with reduced cardiovascular disease and cancer (1). Lignostilbene α,β-dioxygenase (LSD, EC 1.13.11.43), originally observed in Sphingomonas paucimobilis, was the first enzyme shown to cleave the central double bond of stilbenes, forming two phenolic aldehydes (2, 3). Subsequently, NOV1 and NOV2 (4) from Novosphingobium aromaticivorans, Rco1 (5) from Ustilago maydis, and CAO-1 (6) from Neurospora crassa were also shown to be stilbene cleaving oxygenases (SCOs). SCOs are related to carotenoid cleavage oxygenases (CCOs), which are enzymes that oxidatively cleave β-carotene or apocarotenoids. Carotenoids are a diverse class of molecules that play important roles in photosynthesis, immune function, and light perception in the eye. CCOs have been studied in great detail, including several crystal structures (7–9).
Here, we present the X-ray structure of an SCO, NOV1 from N. aromaticivorans (NOV1). The structure was determined in complex with a representative substrate (resveratrol), a representative product (vanillin), and without ligand bound. We have also observed the ternary complex with oxygen and substrate or product bound, which has not been previously detected in a crystal structure of any CCO-related enzyme. Despite being related to CCOs, this structure of NOV1 shows several key differences that are indicative of their disparate substrate specificities. Moreover, the observed placement of Fe, O2, and the phenolic substrate resveratrol in the ternary complex suggests a mechanism initiated by enzyme-catalyzed deprotonation of the phenol group, which allows electron delocalization toward an Fe3+-superoxo electrophile.
Results and Discussion
Overall Structure of NOV1.
The crystal structures of NOV1 were determined to 1.61 Å without ligand, 1.89 Å with resveratrol (a substrate), and 1.75 Å with vanillin (a product from reaction with isoeugenol) (Table 1). The electron density map is generally well ordered throughout the entire polypeptide chain, with the exception of a short loop (amino acids 383–390) and the last five residues at the C terminus. The overall fold of NOV1 is a seven-bladed β-propeller (Fig. 1A). A mononuclear iron coordinated by four histidines (Fig. 1) is located down the central axis of the propeller. Long loops and short helical stretches connecting the strands of the β-propeller form a cap over one face of the enzyme. This cap houses the substrate-binding cavity.
Table 1.
X-ray data collection and refinement statistics
| Apo | Resveratrol | Vanillin | |
| PDB ID code | 5J53 | 5J54 | 5J55 |
| Resolution, Å | 50–1.61 (1.64–1.61) | 50–1.89 (1.94–1.89) | 50–1.75 (1.78–1.75) |
| Space group | I222 | I222 | I222 |
| Unit cell, Å | a = 92.4 | a = 93.6 | a = 92.2 |
| b = 101.3 | b = 100.1 | b = 101.1 | |
| c = 144.5 | c = 145.6 | c = 144.6 | |
| α = γ = β = 90° | α = γ = β = 90° | α = γ = β = 90° | |
| Total reflections | 612,990 | 306,969 | 429,868 |
| Unique reflections | 87,570 | 54,816 | 68,233 |
| Average multiplicity | 7.0 (6.8) | 5.6 (2.5) | 6.3 (2.8) |
| Completeness, % | 93.2 (94.5) | 95.0 (71.1) | 93.6 (53.2) |
| 〈I/σ(I)〉 | 33.8 (1.35) | 10.4 (1.34) | 22.3 (1.44) |
| CC1/2 | 0.928 (0.666) | 0.991 (0.522) | 0.996 (0.820) |
| Rmerge* | 0.05 (0.988) | 0.11 (0.52) | 0.10 (0.51) |
| Rfactor | 0.15 | 0.15 | 0.14 |
| Rfree | 0.18 | 0.18 | 0.17 |
| Wilson B | 22.3 | 24.1 | 19.5 |
| Geometry statistics | |||
| rms deviations from ideal geometry† | |||
| Bonds, Å | 0.010 | 0.013 | 0.010 |
| Angles, ° | 0.995 | 0.997 | 0.975 |
| Molprobity analysis‡ | |||
| Ramachandran plot | |||
| Favored, % | 98 | 97 | 98 |
| Outliers, % | 0 | 0 | 0 |
| Rotamer outliers, % | 1.2 | 0.8 | 1.5 |
| Clash score | 1.2 | 1.7 | 1.1 |
Fig. 1.

(A) Side view of NOV1 (lignostilbene α,β-dioxygenase). (B) A top view down the center of the β-propeller axis. Resveratrol is rendered as orange sticks. The iron cofactor is represented as a sphere, and the four coordinating histidines are shown as sticks.
The overall fold and active site arrangement has been previously described for the CCOs apocarotenoid-15,15′-oxygenase (ACO) from cyanobacteria Synechocystis (7), RPE65 from Bos taurus (9), and viviparous14 (VP14) from Zea mays (8). NOV1 and ACO superimpose with an rmsd of 1.9 Å over 399 matched Cα positions. The structures of NOV1 and the CCOs match very closely in the β-sheet region but differ considerably in the cap region. It is these variations in the cap region that define several key structural differences between NOV1 and CCOs. First, the active site cavity is much smaller in NOV1, reflecting the smaller size of the substrate (Fig. 2). Also, substrates for CCOs are highly lipophilic and tend to reside in hydrophobic environments, such as plant thylakoid membranes (10) or liposomes in mammals (11–13). Accordingly, CCOs show varying levels of membrane association, from soluble to integral (14–16). ACO, VP14, and RPE65 all have surface-exposed hydrophobic patches in analogous regions that may serve as membrane interaction domains. In ACO, two small helical segments from residues 121–128 and residues 259–265 contain surface-facing hydrophobic residues (Fig. 2B, white surface) (7). The backbone of region 259–265 in ACO is structurally conserved in NOV1 (residues 238–244). However, NOV1 has no outward-facing hydrophobic residues. The region composed of residues 121–128 in ACO contains an insertion compared with NOV1 and, therefore, is not structurally analogous. No prominent hydrophobic patches are observed on the surface of NOV1, which is consistent with its expression and behavior as a stable soluble protein.
Fig. 2.

Surface slice outlining the active site cavities of (A) NOV1 (cyan) and (B) ACO yellow. ACO has a discrete entrance proximal to the membrane binding region (white) and exit near the β-propeller axis. NOV1 has no membrane binding region and retains the cavity opening only near the β-propeller axis.
The membrane interaction regions of all three CCOs are proximal to large hydrophobic cavities that extend from the surface to the active site center (Fig. 2B). These tunnels may channel substrate from the membrane to the active site. In ACO and VP14, the product may then exit through a second tunnel on the cap side of the central axis of the β-propeller. In NOV1, this substrate entrance tunnel is closed off by an extended loop (residues 183–191) and W238 (Fig. 2A). There is only a single tunnel with access to the active site. Therefore, substrate must enter through the same tunnel that product exits. RPE65 also has only a single tunnel. However, the axial tunnel is closed off, and the membrane tunnel must serve dual duty, which is consistent with the isomerase activity of RPE65, which would not produce a soluble product, and, therefore, the product would return to the membrane (9). Thus, the enzymes with insoluble substrates and soluble products (ACO and VP14) have both the membrane tunnel and the axial tunnel. The enzyme with insoluble substrate and insoluble product (RPE65) has only the membrane tunnel, and the enzyme with soluble substrate and soluble product (NOV1) has only the axial tunnel. It is clear that each of these tunnels is specialized to interact with ligands of a particular hydrophobicity.
Active Site.
The iron, which is required for dioxygenase activity, is located down the central axis of the β-propeller and has coordination provided by four histidine residues: 167, 218, 284, and 476. An elongated density consistent with a dioxygen molecule is also present in the substrate-free and ligand-bound structures (Fig. S1). The dioxygen clearly coordinates the iron in a side-on manner. This mode of oxygen binding was previously shown for naphthalene dioxygenase (17) and homoprotocatechuate 2,3-dioxygenase (18). The refined iron–oxygen distances are 2.4 Å and 2.5 Å, which are the same as was described for homoprotocatechuate 2,3-dioxygenase and similar to naphthalene dioxygenase (2.2 Å and 2.3 Å). Even at a resolution of 1.61 Å, it is difficult to determine unambiguously whether the oxygen species present in NOV1 is dioxygen, superoxide, or peroxide. Dioxygen gave a slightly better fit to the electron density and was, therefore, modeled into all three NOV1 structures. However, the better fit of dioxygen does not conclusively rule out the presence of either superoxide or peroxide. There were no bound oxygen atoms observed in any of the structures of ACO (7, 19) or RPE65 (9). However, the structure of VP14 does contain dioxygen bound to the active site iron in an angular, end-on geometry (8). However, because the resolution of the VP14 is only 3.2 Å, it is difficult to confirm whether the dioxygen binds in an end-on manner or whether it actually binds in a side-on manner as we have observed.
Fig. S1.

An omit electron density map of the dioxygen species is shown with the 2Fo − Fc (blue) contoured to 1s and the Fo − Fc (green) contoured to 6σ. To determine the identity of the observed density, first, water was modeled using Coot (36) and refined with Phenix.refine (37). However, residual Fo − Fc density was observed. Two water molecules were spaced too far apart to fit the observed density. No molecule other than O2 that can bind iron was present in the crystallization buffer. Therefore, dioxygen, peroxide, and superoxide were each separately placed by Coot and refined by Phenix.refine. The results were all very similar, with slightly better B-factors for dioxygen. However, at 1.61-Å resolution, it is difficult to tell the difference between these three states, and there may be a mixture. Early rounds of refinement with dioxygen were done unrestrained by metal coordination to the iron. After a stable position was converged upon, the dioxygen was restrained at that position.
NOV1 was crystallized in the presence of the substrate analog resveratrol. Oxyresveratrol and isoeugenol were also tried, but no useful crystals resulted. Resveratrol is composed of a 4-hydroxyphenyl group and a 3,5-hydroxyphenyl group joined together by an ethylene bridge. Well-resolved electron density was observed for the entire substrate molecule. Resveratrol binds with the central ethylene bridge oriented roughly perpendicular to and directly facing the iron-coordinated dioxygen moiety at a distance of 2.7–2.9 Å (Fig. 3A). Hydrogen bonds to the hydroxyl groups helped to stabilize the position of the substrate for catalysis. Y101 and K134 hydrogen bond to the 4-hydroxyphenyl moiety (Fig. 3A), which is conjugated to the central double bond. On the other side of the molecule, both S283 and E353 hydrogen bond to the same hydroxyl group of the 3,5-hydroxyphenyl. Neither of these phenol groups is conjugated to the central double bond. Additionally, aromatic stacking of F59 with the 4-hydroxyphenyl and F307 with the 3,5-hydroxyphenyl help position the substrate for catalysis. A secondary shell of aromatic residues lines the entrance to the active site cavity. Ternary complexes of Fe, O2 and substrate have been trapped in crystal structures of related iron-containing enzymes (17, 18), perhaps arising from changes in rate of reaction associated with crystal packing effects that dampen motions required for catalysis.
Fig. 3.

Detailed view of the active site. Stick representations of resveratrol (orange) bound to NOV1 (A) and vanillin bound to NOV1 (B). Dioxygen is represented as a red stick, and iron is shown as an orange sphere. Dotted lines denote hydrogen bonds, whereas solid lines show metal coordination.
NOV1 was also crystallized in the presence of vanillin, a product from the oxidation of isoeugenol. Attempts were also made to cocrystallize NOV1 with 4-hydroxybenzaldehyde and 3,5-dihydroxybanzaldehyde, but no crystals were observed. Because it is composed of a single ring, vanillin has the potential to bind to either or both sites occupied by the phenyl groups of resveratrol. However, density was observed only at the position occupied by the 4-hydroxyphenyl moiety in the resveratrol-bound structure (Fig. 3B), and Y101 and K134 were hydrogen bonded to the 4-hydroxy group. In addition, K134 was also able to hydrogen bond with the 3-methoxy group. As with the resveratrol-bound structure, residue F59 provided aromatic stacking interactions. The aldehyde group of vanillin was in nearly the same position as the central double bond of resveratrol. Almost all of the active site residues were positioned the same as the resveratrol-bound structure, with the exception of L475, which has a 160° rotation of the Cβ–Cγ bond to accommodate the methoxy group of vanillin. The dioxygen was also present and was in the same position as the apo and resveratrol-bound structures.
Substrate Specificity.
The substrate specificity of NOV1 has previously been explored (4). NOV1 was shown to react with resveratrol and piceatannol (having a 4-OH group) and was reported to react much more slowly with rhapontigenin and rhaponticin (4-OMe group).
We tested additional stilbene and stilbene-like compounds for activity (Table S1). Products of the reactions were identified based on the mass fragmentation pattern of the external standards; all peaks were well separated (Fig. S2). As previously demonstrated, the enzyme was active with resveratrol and piceatannol. NOV1 was also active with oxyresveratrol, and, because of its relatively higher solubility, this compound was chosen as a model substrate for catalysis studies. The specific activity of oxyresveratrol was 0.554 (± 0.019) μmol⋅min−1⋅mg−1 at the optimal condition of pH 8.0 and temperature 25 °C (Fig. S3). All of the above-mentioned stilbenoid compounds that are substrates for NOV1 (resveratrol, piceatannol, and oxyresveratrol) contain a 4′-OH group that can hydrogen bond with Y101 and K134 in the manner observed for resveratrol in the crystal structure. Deprotonation of the 4′-OH group should increase electron density at the central double bond, and so help to facilitate reaction with the Fe3+-superoxo electrophile (Fig. 3A). No activity was observed for trans-stilbene, cis-stilbene, or 3,4′,5-trimethoxy-stilbene, which, despite their overall similarity to the reactive compounds, lack a 4′-OH group. Moreover, the presence of 4′-OMe in 3,4′,5-trimethoxy-stilbene eliminates the potential that base catalysis from Y101 and K134 might facilitate electronic redistribution and may also introduce unfavorable steric clashes.
Table S1.
Reactivity and specific activity of various substrates were analyzed at pH 8.0 and 25 °C for 5 min
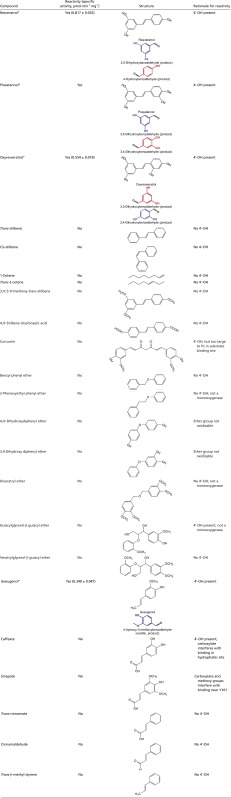 |
Substrates with detectable enzymatic activity and the products (blue and red) of the reaction are shown.
Fig. S2.

The chromatograms of the product distribution from the enzymatic cleavage of resveratrol and oxyresveratrol by HPLC/DAD/MS. Resveratrol was cleaved into 4-hydroxybenzaldehyde and 3,5-dihydroxybenzaldehyde whereas oxyresveratrol was cleaved into 2,4-dihydroxybenzaldehyde and 3,5-dihydroxybanzaldehyde. Products of the reaction were identified by their mass fingerprints. To further verify the products, ethyl acetate was used to extract the reaction products before analysis by gas chromatography/mass spectrometry (GC/MS). The mass spectrum of the products is shown in the Inset of the chromatogram.
Fig. S3.

The pH and temperature activity profiles for NOV1 were conducted using oxyresveratrol as a substrate. The pH activity profiles were investigated at 30 °C for 5 min, and pH 8.0 was found to be optimal (A). The optimal pH was then applied for the temperature activity profile for 5 min, and the optimal temperature was found to be ∼25 °C (B).
Dioxygenase activity was also observed for isoeugenol, which is not a stilbene but does have an electron-donating 4′-OH group. Instead of a second phenyl ring, the presence of the methyl group suggests that acetaldehyde would be a product of the dioxygenation reaction. Two isoeugenol oxygenases have previously been discovered, IE27 from Pseudomonas putida (20) and IEM from Pseudomonas nitroreducens (21). NOV1 has ∼40% identity with these two. All four of the iron-coordinating histidines are conserved, and three of the four residues (Tyr, Lys, and Glu) that hydrogen bond with resveratrol in NOV1 are conserved in IEM. However, residue S283 in NOV1 is a phenylalanine (F281) in IEM. This change reflects the smaller size of isoeugenol compared with resveratrol and the lack of polar groups on that end of the molecule.
No activity was detected for diphenyl ether compounds, which would seemingly require that the enzyme function as a monooxygenase to cleave the ether bond via hydroxylation at an adjacent position (22).
Reaction Mechanism.
EPR spectroscopy showed that purified NOV1 contains Fe(II) that can readily form a stable EPR-active S = 3/2 {Fe-NO}7 complex when exposed to NO (EPR Analysis). S = 3/2 {Fe-NO}7 EPR spectra were also sensitive to the presence of either resveratrol or vanillin, consistent with the close proximity of Fe, O2, and aromatic compounds observed in the crystal structures. The NOV1 reaction has similarities to that of carotenoid-cleaving oxygenases (23), which also use a 4-His Fe(II) center and O2 to cleave an alkene double bond into aldehyde products. Fig. 4 places a plausible reaction mechanism in the context of the active site determined in this work. Starting with the resting Fe(II) enzyme (Fig. 4, step A), both dioxygen and substrate bind to form a ternary complex, where O2 interacts with Fe(II) to form a side-on bound initial complex described as either Fe3+-superoxo or Fe(II)-O2 (5); the case for the reaction starting with Fe3+-superoxo is shown. The observed formation of an Fe-O2 moiety in the absence of substrate suggests that NOV1 has no obligate binding of substrate before O2 leading to the ternary complex, which is consistent with the lack of binding interactions between substrate and Fe shown in the NOV1 structures, and distinct from the ordered binding observed in aromatic ring-cleaving dioxygenases (18), where the catecholic substrate binds directly to the Fe center. The observation of a NOV1–NO complex in the absence or in the presence of either substrate (resveratrol) or a product (vanillin) is consistent with these findings.
Fig. 4.

Proposed reaction mechanism for NOV1. (A) Formation of ternary complex of Fe(III)-superoxo Lig13, O2 and alkene substrate, and contributions of deprotonation of 4′-OH by Y101 and K135 in activation of the substrate. (B) Intermediate formed prior to formation of a C–O bond. (C) Intermediate formed after formation of the first C–O bond. (D) Cleavage of the O–O bond and formation of the second C–O bond. (E) Cleavage of the C–C bond and restoration of the Fe(II) enzyme. (F) Reaction products.
Fig. 4, step A also shows how activation of the 4′-OH group can help to delocalize electron density toward the central double bond and the Fe3+-superoxo electrophile. The NOV1-resveratrol structure shows that the activated Fe complex and reactive π orbitals of substrate approach closer than 2.8 Å. For resveratrol and other typical substrates of NOV1 possessing a 4′-OH, extensive resonance with the appended aromatic rings would stabilize an intermediate radical (Fig. 4, step B), and would be enhanced by interactions of the 4′-OH with Tyr101 and Lys135. The close approach, seemingly enforced by steric interactions with the active site cavity, should promote formation of the first C–O bond (Fig. 4, step C), and also provide an interesting constraint to subsequent O-atom transfer steps of the reaction, where breakage of the O–O bond (Fig. 4, step D), and displacement of Fe3+-bound O atoms toward the alkenyl radical are needed to form the two C–O bonds to substrate (bond length of ∼1.4 Å) (Fig. 4, steps C–E). Subsequent electron rearrangements within the Fe3+-bound dioxygenated intermediate would lead to C–C bond cleavage and collapse to the aldehyde products (Fig. 4, step F). There are several other mechanisms by which the ternary complex of Fig. 4, step A might proceed to catalysis, including dioxetane, epoxide, or Criegee rearrangements to yield aldehyde products (5).
The results reported here provide crystallographic and spectroscopic lines of evidence supporting formation of the initiating ternary complex (Fig. 4, step A) and also provide distance constraints relevant to different mechanisms contemplated for this enzyme family. Answering the question of whether CCOs and SCOs are monooxygenases or dioxygenases has not been straightforward. An initial study using labeled oxygen and water with the reaction of the CCO BCO1 from chicken showed labeling patterns consistent with monooxygenase activity (i.e., one atom from O2 incorporated into products) (14). Likewise, earlier labeling studies with the SCO NOV2 (4), which is similar to NOV1, and the isoeugenol-cleaving enzyme IEM (21) gave monooxygenase-labeling patterns. However, subsequent studies with human BCO1 (24), NOV2, and CCO ACO (25) that accounted for solvent back exchange into the aldehyde products showed dioxigenase-labeling patterns (i.e., both O atoms from O2 incorporated into products, albeit transiently). Due to the sequence similarity to NOV2, it is highly likely that NOV1 has dioxygenase activity. The assignment of dioxygenase activity is also consistent with the structure of NOV1, where both iron-bound oxygen atoms are in close proximity to the substrate double bond and no waters are observed in coordination with the iron or anywhere proximal to resveratrol.
EPR Analysis
Purified NOV1 has an unremarkable g = 4.3 EPR resonance from high-spin Fe(III) (<5% total iron) (Fig. S4, trace A). This result is expected from a 4-His oxygenase containing a high-spin Fe(II) metal center (5). EPR silent S = 2 Fe(II) centers can be effectively studied using nitric oxide (NO) (an S = 1/2 radical) and EPR spectroscopy. Upon NO binding, the EPR silent Fe(II) site converts to an EPR active S = 3/2 Fe-NO complex that is sensitive to changes in the active site structure caused by ligands, substrates, and products.
Fig. S4.

The 9.39-GHz EPR spectra of 190-μM NOV1 and 190-μM NOV1-NO along with simulations of the S = 3/2 species observed with NOV1-NO. Spectra were collected at either 4 or 10 K with 10 G modulation amplitude and 0.05 mW power. (Trace A) Purified NOV1 at 10 K. (Trace B) EPR spectrum of NOV1 plus NO at 4 K. (Trace C) Simulation of an S = 3/2 iron center with g = 4.01, 3.98, and 2.00, and E/D = 0.002. The simulation was scaled to match the intensity of the experimental data shown in trace C. The temperature dependence and g values obtained from the simulations indicate that the signals arise from the ground state mS = 1/2 doublet of the S = 3/2 spin system.
Fig. S4, trace B shows that NOV1-NO has a prominent axial EPR signal near g = 4. As the temperature was increased to 10 K, the intensity of g = 4 signal decreased. Thus, this resonance arises from a ground state. Fig. S4, trace C shows a simulation assuming an S = 3/2 spin system with E/D = 0.002 and g = 4.012, 3.988, and 2.00 (species 1). Although the g = 2 region of the experimental data are dominated by a broad EPR resonance from unbound NO (Fig. S4, trace B), the simulation of the resonance near g = 4 matched the experimental data (Fig. S4, trace C, blue line). Spin quantification revealed that species 1 accounted for ∼80% of the total enzyme concentration, with about 15% likely remaining in an NO-free Fe(II) state. Table S2 summarizes the spin quantification results.
Table S2.
Summary of spin quantification results
| Enzyme | E/D | % Yield | ||
| NOV1-NO | NOV1-NO-resveratrol | NOV1-NO-vanillin | ||
| Species 1 | 0.002 | 80 | 7 | — |
| Species 2 | 0.128 | — | 80 | — |
| Species 3 | 0.039 | — | 8 | — |
| Species 4 | 0.01 | — | — | 40 |
| Species 5 | 0.03 | — | — | 15 |
| Low spin | — | <5 | <5 | 18 |
| Unbound* | — | 15 | — | 28 |
Species not detected are represented by dashes.
Remaining material balance assumed to be Fe(II) not bound by NO.
Fig. S5, trace A shows the EPR spectrum of an anaerobic sample of NOV1 mixed with NO and resveratrol, a substrate. The NOV1-NO-resveratrol spectrum was simulated as three different S = 3/2 species (sum shown as the red line, Fig. S5, trace B, with individual simulations shown as blue lines in Fig. S5, traces C–E). Species 2 (Fig. S5, trace C), with spin quantitation corresponding to ∼80% of the total enzyme concentration, has the greatest rhombic distortion, with g = 4.70, 3.20, and 1.90 and E/D = 0.128. Species 3 (Fig. S5, trace D), accounting for ∼8% of total enzyme concentration, had g = 4.22, 3.76, and 1.99 with E/D = 0.039. The marked rhombic distortions of the EPR spectra of species 2 and 3 must arise from active site perturbations induced by the binding of resveratrol in the proximity of the Fe–NO complex, particularly as the crystal structure indicates that direct coordination of resveratrol to Fe(II) is not likely. In light of the limited solubility of resveratrol, the remaining ∼6% of the total enzyme concentration corresponded to species 1, NOV1-NO (compare Fig. S4, trace B with Fig. S5, trace E).
Fig. S5.

The 9.39-GHz EPR spectra of 190-μM NOV1-NO-resveratrol and corresponding simulations of the S = 3/2 species observed. All spectra were collected at 10 K with 10 G modulation amplitude and 0.25 mW power. (Trace A) EPR spectrum of NOV1-NO-resveratrol. (Trace B) Sum of simulations shown in traces C, D, and E. (Trace C) Simulation assuming S = 3/2, g = 4.70, 3.20, and 1.90, and E/D = 0.128. (Trace D) Simulation assuming S = 3/2, g = 4.22, 3.76, and 1.99, and E/D = 0.039. (Trace C) Simulation assuming S = 3/2, g = 4.01, 3.98, and 2.00, and E/D = 0.002 (same parameters as used for species 1) (Fig. S4, trace C). The simulations are scaled to match the intensities of the experimental spectrum shown in trace A.
Fig. S6, trace A shows the EPR spectrum of the NOV1 mixed with NO and vanillin, a product analog. The NOV1-NO-vanillin spectrum was simulated as two different S = 3/2 species (sum shown as the red line, Fig. S6, trace B, with individual contributions to the simulation shown as blue lines in Fig. S6, traces C and D). Species 4 (Fig. S6, trace C), ∼40% of the total enzyme concentration, was simulated with g = 4.00, 3.94, and 1.99 and E/D = 0.010. Species 5 (Fig. S6, trace D), ∼15% of the total concentration, was simulated with g = 4.17, 3.81, and 1.99 and E/D = 0.030. All NO-treated samples with substrate or product also showed an S = 1/2 EPR signal at g ≈ 2 that accounted for <5% to ∼18% of total enzyme concentration depending on sample preparation conditions (e.g., Fig. S6, trace A). Further analysis of this variable species was not considered here.
Fig. S6.

The 9.39-GHz EPR spectra of NOV1-NO-vanillin and corresponding simulations of S = 3/2 species observed. All spectra were collected at 10 K with 10 G modulation amplitude and 0.25 mW power. (Trace A) EPR spectrum of NOV1-NO-vanillin. All NO-treated samples show an S = 1/2 EPR signal at g ≈ 2.1 that accounted for <5% to ∼30% of total enzyme concentration, depending on sample preparation conditions (i.e., substrate, product, or incubation time). Further analysis of this variable species was not considered here. (Trace B) Sum of simulations shown in traces C and D. (Trace C) Simulation assuming S = 3/2, g = 4.00, 3.94, and 1.99, and E/D = 0.01. (Trace D) Simulation assuming S = 3/2, g = 4.177, 3.81, and 1.99, and E/D = 0.030. The simulations are scaled to match the intensities of the experimental spectrum shown in trace A.
Conclusions
We have presented the crystal structure of the SCO NOV1, a class of CCO-related enzymes that has previously been uncharacterized structurally. Because of the hydrophobic nature of their substrates, it has been difficult to obtain high-resolution crystal structures of CCOs with substrate bound. The ongoing debate over whether CCOs and SCOs are monooxygenases or dioxygenases has recently turned in favor of dioxygenases upon consideration of exchange of isotopic labels in the aldehyde products. The structure of NOV1, including the close contacts observed in the ternary complex of iron, O2, and substrate presented here, is consistent with dioxygenase activity.
Enzymatic analysis of NOV1 shows that it cleaves a range of stilbene compounds with a 4′-OH group that can be deprotonated by Tyr101 and Lys135. Molecules that lacked the 4′-OH or that had a 4′-OCH3, which could not be deprotonated, did not react. This pattern suggests that deprotonation and rearrangement of electrons toward the central double bond and the Fe3+-superoxo electrophile are important early steps in the reaction mechanism. Stilbene derivatives such as resveratrol are antimicrobials, and it is possible NOV1 plays a role in pathogen resistance to plant defenses. This role has been suggested for the SCO CAO-1 (6) from N. crassa. Little is known about this antimicrobial resistance pathway, and further research may have implications for the production of food crops. Stilbenes are also produced from lignin during the kraft pulping process (26) used to make paper and other products. The conversion of lignin, which accounts for ∼30% of plant cell wall carbon, into chemicals or fuels could have a significant impact on the economics of processing lignocellulosic biomass (27, 28). Ultimately, enzymes like NOV1 could assist in the biological valorization of dimeric fragments derived from lignin and so contribute to the sustainable operation of a biorefinery for the production of biofuels and other bioproducts.
Methods
Protein Expression and Purification.
Escherichia coli BL21 derivative (NEB Express Competent E. coli) transformed with the NOV1 expression plasmid was grown in 4 mL of Overnight Express Instant TB medium (EMD Millipore) containing 50 μg/mL kanamycin for 16 h at 37 °C with 300 rpm shaking. The overnight culture was transferred to 400 mL of fresh media in a 2-L flask and incubated at 37 °C. The optical density (OD) was monitored, and isopropyl β-d-1-thiogalactopyranoside (IPTG) was added (final concentration 0.5 mM) to induce protein expression when the OD600 reached 0.3–0.5. After induction, the culture was incubated at 18 °C for 48 h. Cells were pelleted and lysed at room temperature with HN buffer (50 mM Hepes, 100 mM NaCl, pH 7.4) using a homogenizer. Soluble proteins were collected after centrifugation of lysed cells at 20,000 × g for 15 min at 4 °C. His-tagged NOV1 was purified from the soluble fraction using Ni-NTA spin columns (Qiagen) according to the manufacturer’s protocol. NOV1 was cleaved from columns using 2 mM phytic acid. The eluted pure proteins were dialyzed in 20 mM Tris buffer, pH 8.0, using 96 D-tube dialyzers (EMD Millipore). Glycerol (10% vol/vol) was added to the dialyzed pure protein and stored at −80 °C. Purified protein (7.5 μL) was electrophoresed on 10−20% polyacrylamide gel (Bio-Rad) under denaturing condition. The gel was stained with Bio-Safe Coomassie Stain (Bio-Rad) and destained with water. The protein concentration was determined by SDS/PAGE densitometry using a number of protein standards of known concentration using Image Lab software (Bio-Rad).
Crystallization.
Crystallization screening was carried out on a Phoenix robot (Art Robbins Instruments), using a sparse matrix screening method (29). NOV1 concentrated to 10 mg/mL was crystallized by sitting drop vapor diffusion in drops containing a 2:1 ratio of protein solution and 0.16 M MgCl2, 0.08 M Tris, pH 8.5, 18% (wt/vol) PEG 4000, and 20% glycerol. An additional 5 mM vanillin was added to the crystallization buffer to obtain the vanillin-bound structure. Cube-like crystals were observed within 2 d. To obtain resveratrol-bound crystals, an excess of resveratrol powder was added to drops containing previously formed crystals and allowed to soak for 1–2 h. For data collection, crystals were flash frozen in liquid nitrogen directly from the crystallization drop.
X-Ray Data Collection and Structure Determination.
The X-ray datasets for NOV1 were collected at the Berkeley Center for Structural Biology on beamlines 8.2.1 and 8.2.2 of the Advanced Light Source at Lawrence Berkeley National Laboratory. Diffraction data were recorded using ADSC Q315R detectors (Area Detector Systems Corporation). Processing of image data was performed using the HKL2000 suite of programs (30). The high resolution cutoff was set according to the methods described by Karplus and Diederichs (31). Phases were calculated by molecular replacement with the program Phaser (32), using the structure of ACO (PDB ID code 2BIW) (7) as a search model. Automated model building was conducted using AutoBuild (33, 34) from the Phenix (35) suite of programs and resulted in a model that was 85% complete. Manual building using Coot (36) was alternated with reciprocal space refinement using Phenix (37). Waters were automatically placed using Phenix and manually added or deleted with Coot according to peak height (3.0σ in the Fo − Fc map) and distance from a potential hydrogen bonding partner (<3.5 Å). Translation–libration–screw-rotation (TLS) refinement (38) using 10 groups, chosen using the TLSMD web server (39), was used in later rounds of refinement. An initial rigid body refinement using Phenix (35) was performed with the final WT model (all waters removed) for NOV1. This model was then refined and built in the same manner as the WT model. All data collection, phasing, and refinement statistics are summarized in Table 1.
Biochemical Reaction Assays.
To determine specific activities toward substrates, we first determined the optimal pH and temperature of the reaction condition. Oxyresveratrol was used as a substrate, unless noted otherwise. Solutions of 100 mM sodium acetate, 50 mM Hepes, and 50 mM Mes, pH 3.9, were mixed at various ratios to make buffers, with pH ranging from 4.0 to 8.0. The purified enzymes were diluted in buffers of different pH to a final concentration of 200 nM stock enzymes. All substrate solutions were prepared in dimethyl sulfoxide to a final concentration of 5 mM. Substrate solutions were diluted in buffers of different pH to a final concentration of 100 µM stock substrates. To determine the optimal pH, we mixed the stock enzyme and stock substrate at 50:50 (vol/vol). The reaction mixture was incubated at 30 °C for 5 min, unless noted otherwise. The reaction was stopped by adding an equal volume of 2% (vol/vol) formic acid in water with 200 µM phenol as an internal standard. To determine the optimal temperature, the stock substrate and stock enzyme at the optimal pH were used. The reaction mixture was incubated at temperatures ranging from 10 to 35 °C for 5 min. The reaction was stopped as above. All resulting reaction mixtures were analyzed by HPLC/diode array detector (DAD)/MS. Products of reactions were identified by Agilent Technologies 7890 Gas Chromatography (GC) with a 5975 Mass Spectrometry Detector (MSD). Then, 1 µL of samples was injected through a 7683B Series Injector using a split mode of 50:1. Product separation was carried out using an Agilent J&W DB1701 column at a flow rate of 1 mL/min He (UHP). The oven temperature was programmed using an initial temperature of 50 °C and heating rate of 25 °C/min to 280 °C (hold for 5 min).
Product Profiling Analysis.
All samples were introduced into the HPLC/DAD/MS via an autosampler as a full-loop injection volume (25 mL) for high reproducibility. Buffer A (0.1% formic acid in water) and acetonitrile (buffer B) were used as mobile phase solvents. A Kinetix C18 reversed-phase HPLC column was used to separate the reaction mixtures. A nonlinear gradient elution was used for this column. The column was equilibrated with buffer A for 10 min before each run and placed in a column compartment that maintained a temperature of 30 °C to increase the reproducibility of the retention times and peak widths. The analysis was carried out using an Agilent 1100 HPLC (Agilent Technologies Inc.) equipped with a diode array detector (DAD) and API 2000 liquid chromatography-mass spectrometry/mass spectrometry (LC/MS/MS) system (AB SCIEX) to identify products. The amounts of substrates consumed and products formed were quantified by peak area of the chromatogram at 280 nm. Our electrospray ionization conditions were 4.5-kV spray voltage with an auxiliary gas (N2) flow of 20 and 8 (arbitrary units) and a heated ion transfer capillary/mass spectrometer inlet temperature of 350 °C.
EPR Sample Preparation and Analysis.
Resveratrol (≥99% pure) and vanillin (99% pure) were purchased from Sigma-Aldrich. The nitrosyl complexes of NOV1 were prepared as described below. All experiments were done in 50 mM Hepes, pH 7.4, 25% glycerol, and 75 mM NaCl. Resveratrol, vanillin, and 1-mL aliquots of NOV1 (280 µM) contained in 5-mL crimp-top glass vials were made anaerobic using an Ar Schlenk line fitted with an Agilent BOT O2 trap and an Agilent GC-4 POP O2 indicating cartridge. Anaerobic, hydrated Ar was continuously passed through the head space of the sample vial with mild stirring for 30 min before the sample was transferred into a Coy anaerobic glove box without exposure to air. In the glove box, an anaerobic NOV1 plus resveratrol complex was prepared by mixing 2 mg of resveratrol with 1 mL of NOV1 (170 or 190 μM enzyme). The NOV1 plus vanillin complex was prepared with either 170 or 190 µM enzyme and a final vanillin concentration of 10 mM. Anaerobic vanillin or resveratrol complexes were allowed to equilibrate with stirring in the glove box for 45 min. Aliquots of the anaerobic enzyme or enzyme–substrate complex (300 µL) were transferred into an EPR tube. Additional aliquots of the anaerobic enzyme or enzyme–substrate complex were transferred into septum vials, sealed with gas-tight septa, and transferred out of the glove box for exposure to NO delivered by use of a Schlenk apparatus connected to an NO tank. The tubing used to deliver NO gas ended in a needle. The tubing was purged with anaerobic Ar for 5 min, and then the needle was used to puncture the septum of the sample vial so that NO gas could be delivered into the sample vial. After ∼5 min, the samples exposed to NO gas were transferred back into the glove box and transferred into anaerobic EPR tubes. All EPR samples were capped with a rubber stopper, transferred out of the glove box, flash frozen in a methanol/dry bath, and stored in liquid N2. X-band (9.25 GHz) EPR spectra were recorded at 4 K and 10 K under nonsaturating conditions. Spin quantitation was calibrated using a Cu(II)EDTA standard, and EPR simulations were done using Biomolecular EPR spectroscopy software (40).
Acknowledgments
We thank the staff of the Berkeley Center for Structural Biology at the Advanced Light Source of Lawrence Berkeley National Laboratory. This work was performed as a collaboration between the Joint BioEnergy Institute (https://www.jbei.org/) and the Great Lakes Bioenergy Research Center (https://www.glbrc.org/). The Joint BioEnergy Institute is supported by the US Department of Energy, Office of Science, Office of Biological and Environmental Research, through contract DE-AC02-05CH11231 between Lawrence Berkeley National Laboratory and the US Department of Energy. The Great Lakes Bioenergy Research Center is supported by the US Department of Energy, Office of Science, Office of Biological and Environmental Research, through Grant DE-FG02-07ER64495. The Berkeley Center for Structural Biology is supported in part by the National Institutes of Health, National Institute of General Medical Sciences. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the US Department of Energy under Contract DE-AC02-05CH11231. A part of this work was supported by the National Science Foundation under Cooperative Agreement 1355438.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 5J53, 5J54, and 5J55).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1608917113/-/DCSupplemental.
References
- 1.Yu W, Fu YC, Wang W. Cellular and molecular effects of resveratrol in health and disease. J Cell Biochem. 2012;113(3):752–759. doi: 10.1002/jcb.23431. [DOI] [PubMed] [Google Scholar]
- 2.Kamoda S, Saburi Y. Structural and enzymatical comparison of lignostilbene-alpha,beta-dioxygenase isozymes, I, II, and III, from Pseudomonas paucimobilis TMY1009. Biosci Biotechnol Biochem. 1993;57(6):931–934. doi: 10.1271/bbb.57.931. [DOI] [PubMed] [Google Scholar]
- 3.Kamoda S, Saburi Y. Cloning, expression, and sequence analysis of a lignostilbene-alpha,beta-dioxygenase gene from Pseudomonas paucimobilis TMY1009. Biosci Biotechnol Biochem. 1993;57(6):926–930. doi: 10.1271/bbb.57.926. [DOI] [PubMed] [Google Scholar]
- 4.Marasco EK, Schmidt-Dannert C. Identification of bacterial carotenoid cleavage dioxygenase homologues that cleave the interphenyl alpha,beta double bond of stilbene derivatives via a monooxygenase reaction. ChemBioChem. 2008;9(9):1450–1461. doi: 10.1002/cbic.200700724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brefort T, et al. Cleavage of resveratrol in fungi: Characterization of the enzyme Rco1 from Ustilago maydis. Fungal Genet Biol. 2011;48(2):132–143. doi: 10.1016/j.fgb.2010.10.009. [DOI] [PubMed] [Google Scholar]
- 6.Díaz-Sánchez V, Estrada AF, Limón MC, Al-Babili S, Avalos J. The oxygenase CAO-1 of Neurospora crassa is a resveratrol cleavage enzyme. Eukaryot Cell. 2013;12(9):1305–1314. doi: 10.1128/EC.00084-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kloer DP, Ruch S, Al-Babili S, Beyer P, Schulz GE. The structure of a retinal-forming carotenoid oxygenase. Science. 2005;308(5719):267–269. doi: 10.1126/science.1108965. [DOI] [PubMed] [Google Scholar]
- 8.Messing SA, et al. Structural insights into maize viviparous14, a key enzyme in the biosynthesis of the phytohormone abscisic acid. Plant Cell. 2010;22(9):2970–2980. doi: 10.1105/tpc.110.074815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kiser PD, Golczak M, Lodowski DT, Chance MR, Palczewski K. Crystal structure of native RPE65, the retinoid isomerase of the visual cycle. Proc Natl Acad Sci USA. 2009;106(41):17325–17330. doi: 10.1073/pnas.0906600106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Markwell J, Bruce BD, Keegstra K. Isolation of a carotenoid-containing sub-membrane particle from the chloroplastic envelope outer membrane of pea (Pisum sativum) J Biol Chem. 1992;267(20):13933–13937. [PubMed] [Google Scholar]
- 11.Britton G. Structure and properties of carotenoids in relation to function. FASEB J. 1995;9(15):1551–1558. [PubMed] [Google Scholar]
- 12.Socaciu C, Jessel R, Diehl HA. Carotenoid incorporation into microsomes: Yields, stability and membrane dynamics. Spectrochim Acta A Mol Biomol Spectrosc. 2000;56(14):2799–2809. doi: 10.1016/s1386-1425(00)00375-9. [DOI] [PubMed] [Google Scholar]
- 13.Socaciu C, Jessel R, Diehl HA. Competitive carotenoid and cholesterol incorporation into liposomes: Effects on membrane phase transition, fluidity, polarity and anisotropy. Chem Phys Lipids. 2000;106(1):79–88. doi: 10.1016/s0009-3084(00)00135-3. [DOI] [PubMed] [Google Scholar]
- 14.Leuenberger MG, Engeloch-Jarret C, Woggon WD. The reaction mechanism of the enzyme-catalyzed central cleavage of beta-carotene to retinal. Angew Chem Int Ed Engl. 2001;40(14):2613–2617. doi: 10.1002/1521-3773(20010716)40:14<2613::AID-ANIE2613>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 15.Scherzinger D, Ruch S, Kloer DP, Wilde A, Al-Babili S. Retinal is formed from apo-carotenoids in Nostoc sp. PCC7120: In vitro characterization of an apo-carotenoid oxygenase. Biochem J. 2006;398(3):361–369. doi: 10.1042/BJ20060592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Golczak M, Kiser PD, Lodowski DT, Maeda A, Palczewski K. Importance of membrane structural integrity for RPE65 retinoid isomerization activity. J Biol Chem. 2010;285(13):9667–9682. doi: 10.1074/jbc.M109.063941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Karlsson A, et al. Crystal structure of naphthalene dioxygenase: Side-on binding of dioxygen to iron. Science. 2003;299(5609):1039–1042. doi: 10.1126/science.1078020. [DOI] [PubMed] [Google Scholar]
- 18.Kovaleva EG, Lipscomb JD. Crystal structures of Fe2+ dioxygenase superoxo, alkylperoxo, and bound product intermediates. Science. 2007;316(5823):453–457. doi: 10.1126/science.1134697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sui X, et al. Analysis of carotenoid isomerase activity in a prototypical carotenoid cleavage enzyme, apocarotenoid oxygenase (ACO) J Biol Chem. 2014;289(18):12286–12299. doi: 10.1074/jbc.M114.552836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yamada M, Okada Y, Yoshida T, Nagasawa T. Purification, characterization and gene cloning of isoeugenol-degrading enzyme from Pseudomonas putida IE27. Arch Microbiol. 2007;187(6):511–517. doi: 10.1007/s00203-007-0218-9. [DOI] [PubMed] [Google Scholar]
- 21.Ryu JY, et al. Characterization of an isoeugenol monooxygenase (Iem) from Pseudomonas nitroreducens Jin1 that transforms isoeugenol to vanillin. Biosci Biotechnol Biochem. 2013;77(2):289–294. doi: 10.1271/bbb.120715. [DOI] [PubMed] [Google Scholar]
- 22.Rogge CE, Fox BG. Desaturation, chain scission, and register-shift of oxygen-substituted fatty acids during reaction with stearoyl-ACP desaturase. Biochemistry. 2002;41(31):10141–10148. doi: 10.1021/bi020306d. [DOI] [PubMed] [Google Scholar]
- 23.Sui X, Kiser PD, Lintig Jv, Palczewski K. Structural basis of carotenoid cleavage: From bacteria to mammals. Arch Biochem Biophys. 2013;539(2):203–213. doi: 10.1016/j.abb.2013.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.dela Seña C, et al. The human enzyme that converts dietary provitamin A carotenoids to vitamin A is a dioxygenase. J Biol Chem. 2014;289(19):13661–13666. doi: 10.1074/jbc.M114.557710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sui X, et al. Utilization of dioxygen by carotenoid cleavage oxygenases. J Biol Chem. 2015;290(51):30212–30223. doi: 10.1074/jbc.M115.696799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gierer J, Pettersson I. Studies on the condensation of lignins in alkaline media. Part II. The formation of stilbene and arylcoumaran structures through neighbouring group participation reactions. Can J Chem. 1977;55(4):593–599. [Google Scholar]
- 27.Bugg TD, Ahmad M, Hardiman EM, Rahmanpour R. Pathways for degradation of lignin in bacteria and fungi. Nat Prod Rep. 2011;28(12):1883–1896. doi: 10.1039/c1np00042j. [DOI] [PubMed] [Google Scholar]
- 28.Bugg TD, Ahmad M, Hardiman EM, Singh R. The emerging role for bacteria in lignin degradation and bio-product formation. Curr Opin Biotechnol. 2011;22(3):394–400. doi: 10.1016/j.copbio.2010.10.009. [DOI] [PubMed] [Google Scholar]
- 29.Jancarik J, Kim SH. Sparse matrix sampling: A screening method for crystallization of proteins. J Appl Cryst. 1991;24:409–411. [Google Scholar]
- 30.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 31.Karplus PA, Diederichs K. Linking crystallographic model and data quality. Science. 2012;336(6084):1030–1033. doi: 10.1126/science.1218231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McCoy AJ, et al. Phaser crystallographic software. J Appl Cryst. 2007;40(Pt 4):658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Terwilliger TC, et al. Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Crystallogr D Biol Crystallogr. 2008;64(Pt 1):61–69. doi: 10.1107/S090744490705024X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Terwilliger TC. Automated main-chain model building by template matching and iterative fragment extension. Acta Crystallogr D Biol Crystallogr. 2003;59(Pt 1):38–44. doi: 10.1107/S0907444902018036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Adams PD, et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 2):213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Emsley P, Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 12 Pt 1):2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 37.Afonine PV, et al. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr D Biol Crystallogr. 2012;68(Pt 4):352–367. doi: 10.1107/S0907444912001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Painter J, Merritt EA. Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr D Biol Crystallogr. 2006;62(Pt 4):439–450. doi: 10.1107/S0907444906005270. [DOI] [PubMed] [Google Scholar]
- 39.Painter J, Merritt EA. TLSMD web server for the generation of multi-group TLS models. J Appl Cryst. 2006;39(1):109–111. [Google Scholar]
- 40.Hagen WR. Biomolecular EPR spectroscopy. CRC; Boca Raton, FL: 2008. [Google Scholar]
- 41.Engh RA, Huber R. Accurate bond and angle parameters for X-ray protein-structure refinement. Acta Crystallogr A. 1991;47:392–400. [Google Scholar]
- 42.Chen VB, et al. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 1):12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]