

Abstract
The death-associated protein (DAP) kinase family includes three protein kinases, DAP kinase, DAP kinase-related protein 1, and ZIP kinase, which display 80% amino acid identity within their catalytic domains and are functionally linked to common subcellular changes occurring during cell death, such as the process of membrane blebbing. Here we show physical and functional cross talk between DAP kinase and ZIP kinase. The two kinases display strong synergistic effects on cell death when coexpressed and physically bind each other via their catalytic domains. Furthermore, DAP kinase phosphorylates ZIP kinase at six specific sites within its extracatalytic C-terminal domain. ZIP kinase localizes to both the nucleus and the cytoplasm and fractionates as monomeric and trimeric forms. Significantly, modification of the DAP kinase phosphorylation sites influences both the localization and oligomerization status of ZIP kinase. A mutant ZIP kinase construct, in which the six serine/threonine residues were mutated to aspartic acid to mimic the phosphorylated state, was found predominantly in the cytoplasm as a trimer and possessed greater cell death-inducing potency. This suggests that DAP kinase and ZIP kinase function in a biochemical pathway in which DAP kinase activates the cellular function of ZIP kinase through phosphorylation, leading to amplification of death-promoting signals.
The death-associated protein kinase (DAPK) family is a recently discovered group of highly related serine/threonine kinases that are involved in cell death. The first family member, DAPK, was originally identified in an unbiased genetic screen which was aimed at cloning genes that are necessary for gamma interferon (IFN-γ)-induced cell death (3). This 160-kDa kinase mediates cell death initiated by various external and internal signals, including tumor necrosis factor alpha (TNF-α), Fas, transforming growth factor β, oncogene expression, C6-ceramide, and detachment from the extracellular matrix (2, 10, 12, 31, 34). Several closely related death-associated kinases were subsequently discovered, based on the high degree of identity which they share with the catalytic domain of DAPK. Altogether, the DAPK family of kinases comprises four additional members (11, 19, 22). DAP kinase-related protein 1 (DRP-1) and ZIP kinase (ZIPK) (also called dual leucine zipper kinase, or DLK) are the most closely related members; their N-terminal catalytic domains share approximately 80% identity with DAPK (11, 18, 19, 21). Two more distantly related proteins are DRAK1 and DRAK2, displaying approximately 50% identity to DAPK in their kinase domains (32). The sequence similarity in the catalytic domains may direct the specificity of these kinases towards common substrates, as shown, for example, in the case of the regulatory light chain of myosin II (MLC) which can be phosphorylated, at least in vitro, by all the family members (2, 3, 11, 18, 19, 32). Common death phenotypes were also detected when activated forms of these kinases were introduced into cells. One common feature of DAPK family induced death is membrane blebbing, which has been attributed to increased phosphorylation of MLC (1, 2, 9, 11, 25). Furthermore, DAPK and DRP-1 have been shown to induce a type II, caspase-indepenent cell death, involving autophagy (9). Sequence alignment of the catalytic domains of DAPK, DRP-1, and ZIPK showed that they share a unique stretch of basic amino acids (amino acids 46 to 56) not found in other kinases that has been referred to as the “fingerprint” of the DAPK family (11, 35). The X-ray crystal structure of the catalytic domain of DAPK showed that this stretch forms a highly ordered, positively charged loop at the surface of the upper lobe of the catalytic domain (35). The extracatalytic regions of the DAPK family members and their cellular localization vary greatly. DAPK is a Ca2+/calmodulin (CaM)-regulated kinase that is associated with actin microfilaments and has a multidomain structure consisting of ankyrin repeats, extracatalytic P-loop motifs, and a C-terminal death domain. DRP-1, the closest member to DAPK, also contains a CaM regulatory domain. Both kinases undergo autophosphorylation at Ser 308 within the CaM regulatory domain, which inhibits catalytic activation (33, 34). This mechanism, which is unique to these two apoptotic kinases, serves to restrain their apoptotic activity under basal conditions and enables their activation only when the right apoptotic signals are imposed on cells (33, 34). Despite these similarities, DRP-1 lacks the other extracatalytic domains of DAPK and possesses instead a short C-terminal tail required for homodimerization (11). In contrast to DAPK, which is resistant to mild detergent extraction, DRP-1 is a soluble protein which displays a diffuse cytoplasmic localization. ZIPK lacks the CaM regulatory domain, suggesting different modes of activation, and also lacks the other motifs of DAPK. This 55-kDa protein kinase contains a leucine zipper domain which is believed to mediate homodimerization and which was shown to be critical for the death-promoting effects of the kinase, at least in the context of the full-length protein (18). Additionally, ZIPK possesses several potential nuclear localization signals (NLS), among which the most C-terminal one was proposed to mediate nuclear localization (20). In fact, both cytoplasmic and nuclear localization profiles have been described for rodent ZIPK depending on the cell type and the presence of other interacting proteins (17, 18, 20, 26, 27). When present in the cytoplasm, human ZIPK, like its family members, possesses death-inducing properties, including phosphorylation of MLC and induction of membrane blebbing (25). The nuclear localization, on the other hand, was correlated with several other functions, including binding to the ATF4 transcription factor (18), binding to Daxx in the PML nuclear bodies (17), and the phosphorylation of centromere-specific histones during mitosis (29). Thus, ZIPK differs from its family members in both its structure and its nuclear functions and thus may require different posttranslational activation mechanisms to activate its cell death-inducing function in response to the right death signals. The molecular mechanisms which restrain the apoptotic activity of ZIPK in healthy cells and activate its function during cell death are yet to be identified. The differences in regulation, structure, and localization of the three family members suggest that each has a unique, nonredundant role in mediating cell death. Each kinase might operate independently to induce cell death or might function in a hierarchy or kinase cascade, whereby one kinase activates another through transphosphorylation. The relationship among the family members is not known. In this paper, we address the functional and physical relationship between two of the family members, DAPK and ZIPK. We report that ZIPK coimmunoprecipitates with DAPK and is phosphorylated by DAPK at multiple sites along its extracatalytic domain, both in vitro and in vivo. Mutations at the phosphorylation sites indicate that they are critical for ZIPK oligomerization, cytoplasmic retention, and the cell death-promoting function. This suggests that DAPK activates the cellular functions of ZIPK by transphosphorylation, a regulatory step that promotes the proper localization and oligomerization status that enables it to acquire proapoptotic activity.
MATERIALS AND METHODS
DNA constructs.
Wild-type (WT), K42A, and deletion mutants of ZIPK were expressed from pcDNA3-FLAG vector as previously described (11). Point mutations were introduced in the WT ZIPK-containing plasmid by the QuikChange site-directed mutagenesis kit (Stratagene) by using a set of primers encompassing the inserted point mutations. ZIPK K42A-FLAG and the hemagglutinin (HA)-tagged DAPK constructs were generated by a PCR-based method as previously described (2, 11).
Cell lines, transfections, and cell death assays.
293T human embryonic kidney cells or HeLa cells were grown in Dulbecco's modified Eagle's medium (Biological Industries) supplemented with 10% fetal calf serum (Bio-Lab). For transient transfections, 2 × 105, 1 × 106, or 4.5 × 106 cells were seeded on 6-, 9-, or 15-cm plates, respectively, 1 day prior to transfection. Transfections were done by the calcium phosphate method. For cell death assays, a mixture containing the cell death-inducing plasmids (pcDNA3 expressing the different ZIPK or DAPK constructs) and pEGFP-NI plasmid (Clontech) or green fluorescent protein (GFP) fused to LC3 (GFP-LC3) was used (see legends of Fig. 1, 9, and 10). When cotransfections of the two kinases were compared to transfection with a single kinase gene, the luciferase (Luc) plasmid was added in the latter case, thus ensuring that the phenotype results from both kinases acting together rather than from an artifact of cotransfections of two randomly expressed plasmids. For each transfection, three fields, each consisting of at least 100 GFP-positive cells, were scored for blebbing or autophagic cells (i.e., cells which display membrane blebbing or contain autophagic vesicles). At the indicated time points, cell lysates were prepared from the transient transfections for protein analysis.
FIG. 1.

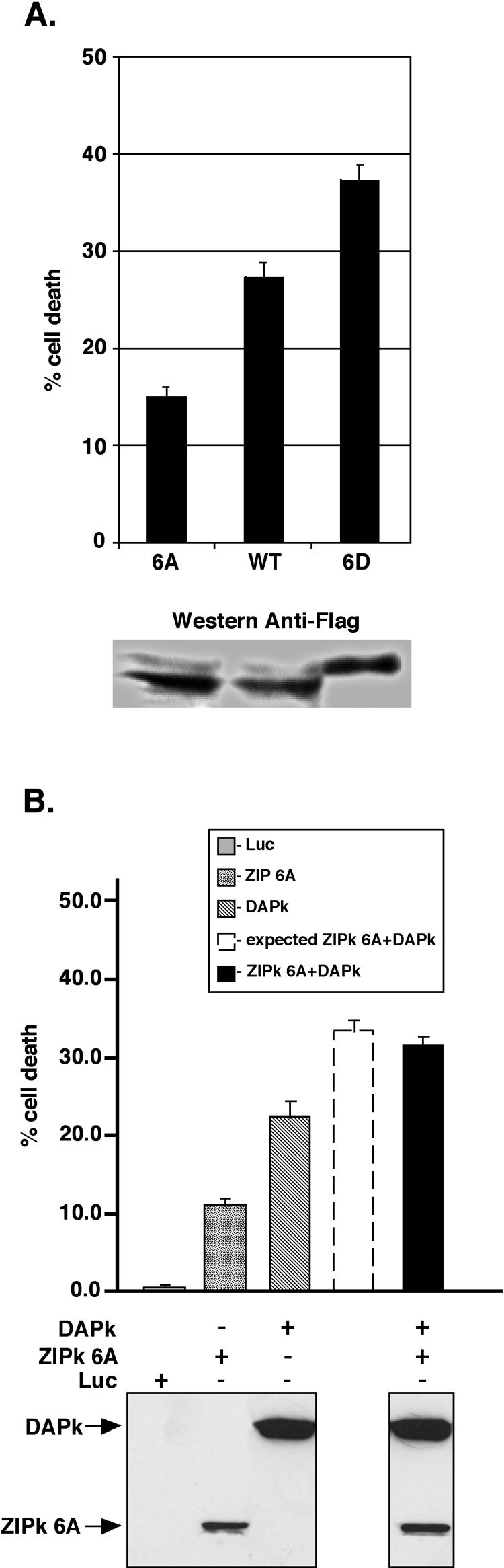
ZIPK synergizes with DAPK to induce cell death. 293T cells grown on 90-mm plates were transiently transfected with 2 μg of FLAG-DAPK or 0.6 μg of FLAG-ZIPK or with both constructs. (A) Percentages of cells showing morphology associated with blebbing among the GFP-positive cells are given as means ± standard deviations and calculated from triplicates of 100 cells each, 12 to 16 h posttransfection. The predicted sum of the first two columns (dashed column) and the actual effect (black column) of the coexpression of ZIPK and DAPK on cell death are shown. (B) Expression of DAPK and ZIPK in cell extracts detected by Western blot analysis by using the anti-Flag antibodies.
FIG. 9.
Negative charge on sites phosphorylated by DAPK elevates the death induction by ZIPK, and the synergy is lost upon mutation of the specific sites. (A) Scores of dying cells (top). Bars showing the percentages of blebbing cells among the GFP-positive cells resulting from cotransfections of 0.7 μg of DNA into 90-mm plates of 293T cells with the specified ZIPK mutants together with GFP. At 17 h posttransfection (means ± standard deviations calculated from triplicates of 100 cells each). Protein extracts from the transfected cells shown were assessed by using anti-Flag antibodies to visualize the protein levels of the different ZIPK mutants (bottom). (B) 293T cells (1.5 ×106) grown in 90-mm plates were transiently transfected with GFP and 1.7 μg of FLAG-DAPK with 0.45 μg of FLAG-ZIPK 6A, with both constructs at the indicated concentrations or with 1.7 μg of Luc as a negative control. Percentages of blebbing cells among the GFP-positive transfectants are given as means ± standard deviations and calculated from triplicates of 100 cells each at 18 h posttransfection (top). The dashed column represents the predicted sum of the first two columns, while the black column shows the actual effect of the coexpression of ZIPK and DAPK on cell death. Expression of DAPK and ZIPK 6A in cell extracts detected by Western blot analysis by using the anti-Flag antibodies (bottom).
FIG. 10.

DAPK and ZIPK induce type II cell death. (A) 293T cells (2 × 105) grown on cover slides in 6-well plates were transiently transfected with 0.2 μg of GFP-LC3 and either 2 μg of FLAG-DAPKΔCaM, 0.6 μg of FLAG-ZIPK 6D, 2 μg of p55 (TNF-α receptor), or 2 μg of Luc as a negative control. The cover slides were fixed by 3.7% paraformaldehyde and analyzed. Examples of typical localization of GFP-LC3 in response to overexpression of the specified genes. The same cells were stained with Hoechst 33342 to visualize the nuclear morphology. (B) Percentages of GFP-positive cells showing a vesicular pattern of GFP-LC3 localization are given as means ± standard deviations and calculated from triplicates of 100 cells each at 72 h posttransfection. (C) Cellular extracts derived from the transfected cells and analyzed by Western blotting by using anti-Flag antibodies, anti-caspase-3 antibodies, and anti-PARP antibodies.
Cell lysates and immunoprecipitations.
Cells were washed with phosphate-buffered saline and lysed in cold B buffer (100 mM KCl, 0.5 mM EDTA, 20 mM HEPES-KOH [pH 7.6], 0.4% NP-40, 20% glycerol, 1 mM dithiothreitol) (3) or PLB (10 mM NaH2PO4 [pH 7.5], 100 mM NaCl, 1% Triton X-100, 0.1% sodium dodecyl sulfate [SDS], 0.5% sodium deoxycholate, 5 mM EDTA) supplemented with protease and phosphatase inhibitors as detailed previously (33). For immunoprecipitation experiments, 1 to 2 mg of protein extract was precleared by Protein G PLUS-Agarose (Santa Cruz Biotechnology) for 2 h at 4°C. The precleared extracts were incubated with 20 μl of agarose-conjugated anti-Flag M2 gel beads (Sigma) for 2 h at 4°C. Immunoprecipitates were washed repeatedly three times with B buffer or PLB and two times with 54K buffer (50 mM Tris [pH 7.9], 150 mM NaCl, 0.5% Triton X-100). When indicated, the proteins were eluted by the addition of 50 μl of Flag peptide (1 μg/μl) (Sigma). For immunoprecipitation of endogenous DAPK, HeLa cells were lysed in PLB, and 7.5 mg of cell lysate was precleared with Protein A PLUS-Agarose beads (Santa Cruz Biotechnology) for 2 h at 4°C. The lysate was then incubated overnight at 4°C with rabbit polyclonal antibodies raised against a synthetic peptide comprising 17 amino acids of DAPK C-terminal tail (3) (SCNSGTSYNSISSVVSR) bound to Protein A PLUS-Agarose beads. Immunoprecipitates were washed repeatedly with PLB and subjected to Western blotting as described below.
Western blotting.
The following antibodies were used for Western blotting: Flag-M2 monoclonal anti-Flag (dilution, 1:500) (Sigma); anti-DAPK monoclonal (clone 55; dilution, 1:2,500) (Sigma); anti-ZIPK monoclonal antibodies (dilution, 1:250) (BD Transduction Laboratories), anti-caspase 3 polyclonal antibodies (dilution, 1:1,000) (Santa Cruz Biotechnology), and anti-poly(ADP-ribose) polymerase (anti-PARP) monoclonal antibodies (dilution, 1:500) (BioMol).
In vitro kinase assay.
293T cells were transfected with various Flag-tagged ZIPK or DAPK constructs. Cells were lysed and proteins were immunoprecipitated and eluted as described above. The immunoprecipitated proteins were quantitated after elution by running samples on gels and comparing the Coomassie blue staining of the bands to a quantified standard protein run in parallel. The specified proteins were then incubated with kinase reaction buffer (50 mM HEPES [pH 7.5], 20 mM MgCl2, 100 mM NaCl, and 0.1 mg of bovine serum albumin per ml). The kinase assay was conducted for 30, 10, and 20 min (see Fig. 5 to 8) at 30°C in 30 μl of reaction buffer containing 10 μCi of [γ-32P]ATP (2 pmol) and 50 μM ATP. When the kinase activity of DAPK and DRP-1 was assayed, 1 μM bovine calmodulin (Sigma) and 0.5 mM CaCl2 were added. In some cases, recombinant human MLC purified from bacteria (kind gift of M. Gautel, European Molecular Biology Laboratory, Heidelberg, Germany) was used for comparison. Protein sample buffer was added to terminate the reaction, and after boiling, the proteins were analyzed on SDS-polyacrylamide gel electrophoresis (PAGE) gels (10 to 15% acrylamide). The gels were blotted onto nitrocellulose membranes, and 32P-labeled proteins were visualized by autoradiography and, in some cases, quantified by PhosphorImager densitometric analysis. Phosphate incorporation was determined by quantifying the intensity of radiolabeled bands in comparison to a dose curve of known [γ-32P]ATP concentrations.
FIG. 5.

DAPK phosphorylates ZIPK in vitro and in vivo. (A) In vitro kinase assay. A 12.5 nM concentration of the indicated kinase and substrate was subjected to in vitro kinase assays. The K42A kinase inactive mutants served as substrates, and the WT forms served as kinases. Arrows on the autoradiogram (top panel) point to the main transphosphorylated bands, and the other bands correspond to autophosphorylation of the WT proteins. The bottom panel is the Western blot of the same membrane reacted with anti-Flag antibodies (B). In vivo kinase assay. 293T cells were cotransfected with the indicated Flag-tagged constructs and metabolically labeled with [P32]orthophosphate. The kinases were immunoprecipitated from cell lysate, resolved by SDS-PAGE, and blotted to nitrocellulose. The top panel corresponds to the autoradiogram and the bottom panel to the Western blot of the same membrane incubated with anti-Flag antibodies. (C) Comparison of the rate of phosphorylation of ZIPK to MLC. A 12.5 nM concentration of DAPK used as kinase and 50 to 400 nM ZIP-K42A or 50 nM to 30 μM MLC serving as substrates were subjected to in vitro kinase assays. The rate of phosphate incorporation into the substrate molecule was plotted against substrate concentration. MW, molecular weight.
FIG. 8.

DAPK phosphorylates ZIPK in vitro on serines 309, 311, 312, 318, and 326. (A) Schematic presentation of ZIPK deletion mutants. The tentative phosphorylation sites, which were each replaced by alanine separately, or in combination, are marked by arrows. (B) Each of the tentative phosphorylation sites was replaced by alanine separately on the background of T299A and K42A substitutions. A 12.5 nM concentration of immunopurified DAPK serving as kinase and, as substrate, 50 nM immunopurified ZIP-K42A-T299A1-337 with various point mutations were assayed in vitro. Proteins were separated on SDS-12% PAGE, transferred and exposed to X-ray film (top panel) and immunoreacted with anti-Flag antibodies (bottom panel). (C) Four out of the five tentative phosphorylation sites were replaced by alanines simultaneously, leaving only one site intact at a time as specified, on the background of T299A and K42A substitutions. A 16.6 nM concentration of DAPK serving as kinase and, as substrates, 100 nM concentrations of ZIPK-K42A-T299A1-337 with multiple point mutations, a ZIPK-K42A-T299A1-337 truncation fragment (no mutations), and a ZIPK-K42A-T299A1-308 truncation fragment were assayed in vitro. The assay was performed as described for panel B. (D) A 12.5 nM concentration of DAPK serving as kinase and 50 nM ZIP-K42A WT or mutated to alanine at the six specific sites (mutant 6A), serving as substrates were assayed in vitro as described for panel B. FL, full length.
In vivo phosphorylation assays.
293T cells grown on 90-mm plates were cotransfected with Flag-tagged DAPK (20 μg) and ZIP-K42A (5 μg) or with Flag-tagged DAPK alone. At 15 h posttransfection, the cells were washed and pulse-labeled with [33P]orthophosphate (5 h in 3.5 ml of PO4−-depleted medium supplemented with 0.33 mCi of 33P-labeled phosphate; 45 min before cell harvesting 150 nM okadaic acid was added). Cells were harvested and lysed in B buffer supplemented with protease and phosphatase inhibitors (Sigma) with 200 nM okadaic acid. The proteins were immunoprecipitated, separated by SDS-PAGE, transferred to nitrocellulose membrane, and exposed overnight to X-ray film. Western blotting of the same membrane was carried out with anti-Flag antibodies.
Immunostaining of cells.
FLAG-ZIPK WT and FLAG-ZIPK mutants were transfected into HeLa cells grown on glass coverslips. After 20 h, the cells were fixed and permeabilized in 3.7% formaldehyde for 12 min or by subsequent incubations in 3.7% formaldehyde, methanol, and acetone for 5, 5, and 2 min, respectively. Cells were then permeabilized and blocked with 0.4% Triton X-100 (Sigma) in 10% normal goat serum (Biological Industries) for 30 min and incubated with anti-Flag antibodies (dilution, 1:250) (M2; Sigma) in 10% normal goat serum for 60 min, followed by Cy3-conjugated goat antimouse secondary antibodies for 30 min (dilution, 1:800) (Jackson Immuno-Research.). All coverslips were finally stained with DAPI (4′,6′-diamidino-2-phenylindole) (0.5 μg/ml; Sigma) and mounted in Fluoromount G (Southern Biotechnology Associates) embedding media. Stained cells were viewed by fluorescent microscopy with a 60× or 100× oil immersion objective (Olympus BX41). Digital imaging was performed with a DP50 charge-coupled device camera and Viewfinder Lite and Studio Lite software (Olympus). Final composites were prepared in Photoshop (Adobe Systems).
Analysis of ZIPK by gel filtration.
293T cells (4.5 × 106) were plated on 15-cm plates and transfected with 8 μg of DNA from the specified constructs. At 20 h posttransfection, cells were collected and proteins were extracted by using PLB supplemented with protease and phosphatase inhibitors (Sigma) at 4°C. The extracts were then centrifuged for 20 min at 20,000 × g to remove insoluble material. Two milligrams of each sample was run on a Superdex 200 gel filtration column (Pharmacia), previously equilibrated with running buffer (50 mM phosphate buffer [pH 7.0], 20 mM MgCl2, 150 mM NaCl, and 5% glycerol) supplemented with protease and phosphatase inhibitors (Sigma) at 4°C. The Superdex 200 column was run at 0.6 ml/min, collecting 0.3-ml fractions. Analysis of the various fractions was done by anti-Flag Western blotting as described above. For determination of molecular weight sizes, the following markers (Sigma) were run in the same conditions on the column: dextran blue, β-amylase, alcohol dehydrogenase, bovine serum albumin, and carbonic anhydrase at 2,000, 200, 150, 66, and 29 kDa, respectively.
RESULTS
DAPK and ZIPK synergize to induce cell death.
In order to assess possible functional relationships within the DAPK-related family of proteins, DAPK and ZIPK were coexpressed in 293T human embryonic kidney cells, and cell death was scored by quantitating cells exhibiting membrane blebbing. For each kinase, suboptimal concentrations of DNA were transfected, achieving relatively low expression levels which individually induced moderate levels of cell death (19 and 28% blebbing cells for ZIPK and DAPK, respectively) (Fig. 1A). In contrast, simultaneous expression of both kinases resulted in strong synergy; 84% of the transfected cell population showed the apoptotic blebbing morphology. A 99% confidence interval for the percentage of apoptotic cells in the cotransfection experiment is 77.96 and 90.04%. The confidence interval is far above the sum of the single transfections, thus further implying that the value of 84% is significantly higher than the predicted percentage of cell death, assuming an additive effect of the two kinases (47.5%). The Western blot in Fig. 1B confirms that equal protein levels were achieved in single and double transfections, suggesting that the two kinases influence each other in a manner independent of protein expression.
DAPK and ZIPK physically interact through their kinase domains.
To further investigate the basis for the strong functional interaction between the two protein kinases, we tested whether they physically interact with each other by using coimmunoprecipitation assays. Flag-tagged ZIPK (FLAG-ZIPK) and HA-tagged DAPK (HA-DAPK) were transfected into 293T cells, and ZIPK was immunoprecipitated via its Flag epitope. The immunoprecipitated complexes were subjected to Western blotting and reacted with anti-Flag and anti-HA antibodies (Fig. 2A, top). HA-DAPK, but not an irrelevant HA-tagged protein (RFX), was specifically coimmunoprecipitated with FLAG-ZIPK (Fig. 2A, compare lanes 1 and 2 in top right panel). The levels of DAPK brought down were significantly higher than the background levels seen upon coimmunoprecipitation with an irrelevant protein (FLAG-RFX) (Fig. 2A, compare lanes 1 and 3 in top right panel). Assessment of the levels of the four proteins in total cell lysates confirmed that they were all expressed to a similar extent in the different combinations (Fig. 2A, bottom). Importantly, the interaction between DAPK and ZIPK was not limited to the overexpressed proteins but also was observed between the endogenous proteins. Immunoprecipitation of endogenous DAPK from HeLa cells by using an antibody raised to the C-terminal tail of DAPK (anti-DAPK-tail) resulted in specific coimmunoprecipitation of the endogenous ZIPK protein (Fig. 2B). Thus, DAPK and ZIPK physically interact, either directly or indirectly.
FIG. 2.

DAPK physically interacts with ZIPK. (A) 293T cells grown on 90-mm plates were transiently cotransfected with the following constructs: lane 1,2.5 μg of FLAG-ZIPK plus 25 μg of HA-DAPK; lane 2, 2.5 μg of FLAG-ZIPK plus 25 μg of HA-RFX (used as a control); lane 3, 25 μg of FLAG-RFX plus 25 μg of HA-DAPK. Protein samples (2 mg) were extracted in PLB and immunoprecipitated with anti-Flag antibodies. The immunoprecipitated proteins and the total cell lysates were assayed by Western blotting by using the anti-Flag and anti-HA antibodies. Asterisks correspond to nonspecific bands. Note that immunoprecipitated ZIPK runs beneath the immunoglobulin heavy chain. (B) Endogenous DAPK interacts with endogenous ZIPK. HeLa cell lysate (7.5 mg) was immunoprecipitated in the presence or absence of rabbit polyclonal antibodies to DAPK-tail. The immunoprecipitated proteins and the total cell lysates were assayed by Western blotting by using monoclonal antibodies to DAPK and ZIPK. IP, immunoprecipitation; α, anti; TCL, total cell lysate; IgG, immunoglobulin G.
In order to map the domain of ZIPK which is responsible for its interaction with DAPK, a set of Flag-tagged C-terminal ZIPK deletion mutants was constructed (Fig. 3A) and tested for their ability to pull down wild-type DAPK, as described above. Full-length ZIPK (ZIPKfl) and all the deletion mutants were capable of pulling down the full-length DAPK (DAPKfl) to a similar extent (Fig. 3B, lanes 1 to 5 of top right panels). All of these interactions were significantly higher than the background of nonspecific binding of HA-DAPK to the empty anti-Flag beads (Fig. 3B, lane 6 of top right panel). The expression of each protein in total cell lysates was verified by Western blot analysis with anti-HA and anti-Flag antibodies (Fig. 3B). Of note, the smallest ZIPK deletants, which consisted of only the kinase domain from residues 1 to 276 (FLAG-ZIPK1-276) or the kinase domain plus NLS2 (FLAG-ZIPK1-308; residues 1 to 308), were expressed at relatively low levels (Fig. 3B, lanes 1 and 2 of left panels) but were still capable of proportionally immunoprecipitating FLAG-DAPK (Fig, 3B, lanes 1 and 2 of top right panel). Thus, the kinase domain of ZIPK is sufficient by itself to mediate interactions with DAPK.
FIG. 3.

The kinase domain of ZIPK is sufficient to bind DAPK. (A) Schematic presentation of ZIPK deletion mutants. The different structural domains and tentative NLS are shown (20, 21). (B) 293T cells were transiently transfected with the following constructs: lane 1, FLAG-ZIPK1-276 plus HA-DAPK; lane 2, FLAG-ZIPK1-308 plus HA-DAPK; lane 3, FLAG-ZIPK1-337 plus HA-DAPK; lane 4, FLAG-ZIPK1-400 plus HA-DAPK; lane 5, FLAG-ZIPKfl plus HA-DAPK; lane 6, HA-DAPK. The anti-Flag immunoprecipitated proteins and the total cell lysates were assayed by Western blotting by using the anti-Flag and anti-HA antibodies on separate membranes. Asterisks indicate nonspecific bands. IP, immunoprecipitation; IgG, immunoglobulin G; α, anti.
Parallel deletion analysis was performed with DAPK in order to identify the domains of DAPK responsible for its interaction with ZIPK (Fig. 4A). HA-tagged DAPKΔDD1-1273, which lacks the C-terminal death domain, and HA-tagged DAPK1-305, which contains only the kinase domain, both coimmunoprecipitated with ZIPKfl to a similar extent as DAPKfl (Fig. 4B, lanes 1 to 3 of top right panel). Although low levels of nonspecific binding of the DAPK truncation mutants to the empty beads was observed, the interaction of these mutants with ZIPK was significantly higher in each case (Fig. 4B, lanes 4 to 6 or top right panel). The expression levels of all the constructs in total cell lysates (Fig. 4B, bottom) and the amounts of the immunoprecipitated ZIPK which pulled down DAPK (Fig. 4B, top left panel) were similar in the different combinations. This set of experiments indicated that the kinase domain of DAPK is sufficient to bind ZIPK. Moreover, deletion of the kinase domain (HA-DAPKΔKD) completely abolished the ability of DAPK to specifically interact with ZIPK (Fig. 4C, compare lanes 1 and 2 of top right panel). Levels of coimmunoprecipitated HA-DAPKΔKD did not significantly exceed the background levels of nonspecific binding (Fig. 4C, compare lanes 2 to 4 of top right panel). Thus, the kinase domain of DAPK is both necessary and sufficient to mediate interactions with ZIPKfl.
FIG. 4.

The kinase domain of DAPK is necessary and sufficient to bind ZIPK and the interaction is mediated via the basic loop. (A) Schematic presentation of DAPK deletion mutants. The different structural domains are shown (2). (B) 293T cells were transiently transfected with the following constructs: lane 1, FLAG-ZIPK plus HA-DAPKfl; lane 2, FLAG-ZIPK plus HA-DAPKΔDD; lane 3, FLAG-ZIPK plus HA-DAPK1-305;lane 4, HA-DAPKfl; lane 5, HA-DAPKΔDD; lane 6, HA-DAPK1-305. Proteins were immunoprecipitated with anti-Flag antibodies. The immunoprecipitated proteins and the total cell lysates were assayed by Western blotting by using the anti-Flag and anti-HA antibodies on separate membranes. (C) 293T cells were transiently transfected with the following constructs: lane 1, FLAG-ZIPK plus HA-DAPKfl; lane 2, FLAG-ZIPK plus HA-DAPKΔKD; lane 3, HA-DAPKfl; lane 4, HA-DAPKΔKD. Proteins were immunoprecipitated with anti-Flag antibodies. The immunoprecipitated proteins and the total cell lysates were assayed by Western blotting by using the anti-Flag and anti-HA antibodies on separate membranes. (D) 293T cells grown in two 90-mm plates were transiently transfected with the following constructs: lane 1, 15 μg of FLAG-ZIPK1-276 plus 20 μg of HA-DAPK1-305; lane 2, 15 μg of FLAG-ZIPKBM1-276 (in which part of the basic loop has been mutated) plus 20 μg of HA-DAPK1-305; lane 3, 16 μg of HA-DAPK1-305. Proteins were immunoprecipitated with anti-Flag antibodies, and the immunoprecipitated proteins and the total cell lysates were assayed by Western blotting by using the anti-Flag and anti-HA antibodies on separate membranes. IP, immunoprecipitation; α, anti.
In order to demonstrate that both kinase domains are sufficient for the interaction and to investigate the importance of the unique basic loop of the kinase domain in this regard, four arginin residues (47, 48, 53, 54) out of the seven amino acids comprising the basic loop were mutated to alanine in the kinase domain of ZIPK (FLAG-ZIPKBM1-276). While HA-tagged DAPK1-305 successfully coimmunoprecipitated with FLAG-ZIPK1-276, this interaction was markedly reduced upon mutation of the basic loop of ZIPK (FLAG-ZIPKBM1-276) (Fig. 4D, lanes 1 and 2 of top right panel). The expression levels of the HA-tagged constructs in total cell lysates (Fig. 4D, bottom) and the amounts of the Flag-tagged immunoprecipitated ZIPK which pulled down DAPK (Fig. 4D, top left panel) were similar in the different combinations.
These experiments demonstrate that ZIPK and DAPK interact via their respective catalytic domains, thus highlighting, quite unexpectedly, the importance of the two catalytic domains in the process of protein-to-protein interaction.
ZIPK serves as substrate for DAPK in vitro and in vivo.
Since DAPK and ZIPK form a kinase complex, it was possible that one, or both, might phosphorylate its partner. This was first tested by in vitro kinase assays with DAPK and ZIPK. The third closely related family member, DRP-1, was also included in these assays to determine whether transphosphorylation processes within this family of death kinases are robust in their directionality. Catalytically inactive mutants (K42A) of DAPK, DRP-1, and ZIPK were used as substrates in order to avoid autophosphorylation, which might mask possible transphosphorylation. All six possible combinations were tested by using the three different inactive forms as substrates with the other two active WT kinases and were compared to the extent of phosphorylation in the absence of functional kinase. In each case, the kinase and substrate were separately prepared by immunopurification from 293T cell lysate and mixed together in a kinase assay in the presence of [32P]ATP. As previously reported, each kinase underwent autophosphorylation, indicating that the kinases were fully active (Fig. 5A). Among the six combinations, only one pairing showed a strong transphosphorylation; that is, DAPK phosphorylated the catalytically inactive ZIPK substrate (Fig. 6A, arrow). DAPK was also capable, to a much smaller extent, of phosphorylating DRP-1 (Fig. 5A, arrow). Neither ZIPK nor DRP-1 showed any significant ability to phosphorylate the other family members, suggesting the specificity and unidirectionality of these transphosphorylations.
FIG. 6.

Gross mapping of DAPK-dependent phosphorylation sites on ZIPK. (A) DAPK and ZIPK-K42A mutants were immunoprecipitated and eluted via anti-Flag antibodies and Flag peptide, respectively. A 12.5 nM concentration of DAPK used as kinase and 50 nM ZIP-K42A truncation mutants serving as substrates were assayed in vitro in standard conditions, and the proteins were separated by SDS-12% PAGE, transferred, and exposed to X-ray film. (B) Western blot of the same membrane reacted with anti-Flag antibodies. MW, molecular weight.
In order verify that ZIPK is a legitimate substrate for DAPK, we compared its efficiency as a DAPK substrate to that of DAPK's known in vivo substrate, MLC (1, 23). The data indicate that within physiological protein concentrations (nanomolar range), ZIPK proved to be a more efficient substrate for DAPK than MLC, exhibiting a 10-fold higher reaction rate (picomoles of ATP per minute per milligram of DAPK) (Fig. 5C).
Importantly, ZIPK also served as a substrate for DAPK in vivo. Both kinase and substrate (DAPK and ZIPK-K42A kinase inactive form) were cotransfected into 293T cells, followed by in vivo labeling of the cells with orthophosphate, immunoprecipitation of the proteins, and detection of phosphate incorporation into the substrate molecule. ZIP-K42A was clearly phosphorylated in 293T cells in which DAPK was present but not in its absence (Fig. 5B). No phosphorylation of DAPK by ZIPK was observed in the reciprocal cotransfection assay (data not shown). Thus, DAPK phosphorylates ZIPK both in vitro and in vivo upon coexpression.
Mapping the DAPK-dependent ZIPK phosphorylation site(s).
In order to grossly map the phosphorylation site(s) on ZIPK, which are targets for DAPK, four successive C-terminal truncation mutants of ZIPK-K42A were generated and assayed as substrates for DAPK in an in vitro kinase assay (Fig. 6). Deletion of the entire extracatalytic domain, leaving only the kinase domain intact (residues 1 to 276) (Fig. 3A), completely abolished the phosphorylation by DAPK. In contrast, phosphorylation was observed in the slightly longer deletion mutant (residues 1 to 308) (Fig. 6A). Therefore, a DAPK phosphorylation site(s) is likely to reside within the stretch of amino acids spanning amino acids 276 to 308, while the catalytic domain is devoid of phosphorylation sites. Moreover, the phosphorylation of the fragment comprising residues 1 to 308 was lower than that of the fragment comprising residues 1 to 337 (by a factor of six after normalization to protein levels), suggesting that either an additional site(s) resides within the region spanning amino acids 308 to 337 or that this region facilitates or promotes phosphorylation on the previous sites. Interestingly, there was a substantial increase in the DAPK phosphorylation of the two longest deletion mutants (fragments comprising residues 1 to 400 and 1 to 337) over the full-length protein (Fig. 6A), suggesting that the region from amino acid 400 to the end may act as an inhibitory module, for example, by interfering with the proper exposure of the sites to DAPK.
Four potential phosphorylation sites are located in the stretch spanning residues 276 to 308 (Fig. 7A). In order to identify which of these are phosphorylated by DAPK, each Ser or Thr was individually mutated to Ala in the context of the ZIPK-K42A1-308 construct. All the mutants were properly expressed (Fig. 7C). The substitution of Ala for Thr 299 completely eliminated the phosphorylation of ZIPK1-308 by DAPK, while all the other mutations had no effect (Fig. 7B). Thus, Thr 299 is the exclusive phosphorylation site within the first 308 residues of ZIPK. Substitution of Thr 299 by Ala in the context of the full-length protein or the truncation mutant spanning residues 1 to 337, however, neither abolished nor attenuated the phosphorylation by DAPK (data not shown), which is consistent with our previous suggestion that an additional site(s) may be present, specifically in the stretch spanning amino acids 308 to 337. To identify these additional sites, five putative phosphorylation sites in this stretch were individually replaced by Ala, in addition to the Thr299Ala mutation (Fig. 8A). Interestingly, none of these single-site substitutions had any effect on the phosphorylation by DAPK (Fig. 8B), suggesting the existence of multiple sites. Moreover, stoichiometric analysis of the ATP utilized upon phosphorylation of the deletion mutant at residues 1 to 337 indicated that 1.4 mol of phosphate was incorporated into 1 mole of substrate, reflecting the existence of more than one site. In order to confirm the significance of the multiple phosphorylation sites, additional mutants were generated in which four of the Ser residues and Thr 299 were mutated to Ala, leaving only one site intact at a time. These were compared to the constructs spanning residues 1 to 308 and 1 to 337 and harboring the Thr 299 mutation, with the former construct serving as a negative control since it lacked all five serines and the latter acting as a positive control, with all five sites intact. Each of the single intact Ser constructs underwent phosphorylation by DAPK (Fig. 8C), indicating that each of the five tentative sites can accept phosphate from DAPK in an independent manner.
FIG. 7.

DAPK phosphorylates ZIPK in vitro on threonine 299. (A) Schematic presentation of ZIPK deletion mutants. The tentative phosphorylation sites, which were each replaced by alanine separately, are marked by arrows. (B) DAPK WT, ZIPK-K42A1-308 fragments (comprising residues 1 to 308) carrying the different point mutations, and a ZIPK-K42A1-276 truncation fragment were immunoprecipitated and eluted with anti-Flag antibodies and Flag peptide, respectively. A 12.5 nM concentration of DAPK serving as kinase and 50 nM concentrations of ZIP-K42A1-308 point mutation and ZIP-K42A1-276 truncation fragments serving as substrates were assayed in vitro. The proteins were separated by SDS-12% PAGE, transferred, and exposed to X-ray film. (C) Western blot of the same membrane reacted with anti-Flag antibodies. MW, molecular weight.
In order to verify that DAPK phosphorylates ZIPK on the six specific sites (T299 and the five serines) in the context of the full-length protein, these six sites were mutated to alanine within the full-length K42A ZIPK protein (named 6A), and the construct generated was used as a substrate for DAPK in vitro. Replacement of these six sites by Ala completely abolished the DAPK-dependent phosphorylation of ZIPK (Fig. 8D), suggesting that these are the specific sites recognized by DAPK.
Functional implications of ZIPK phosphorylation by DAPK.
In order to determine the functional significance of the DAPK-mediated phosphorylation of ZIPK, we assessed whether the phosphorylation status of ZIPK affected its ability to induce cell death. To this end, two additional constructs mimicking the phosphorylated and dephosphorylated forms of full-length, catalytically active ZIPK were generated. In the first construct, all six phosphorylation sites mapped above were converted to Asp (mutant 6D), while in the second construct, the six sites were mutated to nonphosphorylatable Ala (mutant 6A). These two constructs were transfected into 293T cells, and their death-promoting effects were monitored and compared to those of the wild-type ZIPK by scoring the blebbing phenotype among the transfected cells. All three proteins were expressed in the cells to the same extent (Fig. 9A, bottom). The nonphosphorylatable ZIPK 6A mutant induced significantly lower levels of blebbed cells compared to the WT ZIPK, which unlike the mutant, has the potential to be phosphorylated by endogenous DAPK. In contrast, the 6D mutant, which mimics the constitutively phosphorylated form of ZIPK, showed an enhanced ability to induce membrane blebbing (Fig. 9A). Likewise, the death-promoting synergy observed with DAPK and WT ZIPK (Fig. 1) is abolished upon mutation of these six sites to alanines (Fig. 9B). Thus, these DAPK phosphorylation sites on ZIPK are critical for the ability of the two kinases to promote each other's death-inducing capabilities. This suggests that phosphorylation of ZIPK by DAPK elevates its cell death-promoting capacity and that the synergy depends on the transphosphorylation of ZIPK rather than being an artifact of cotransfection.
To investigate the exact mode of cell death induced by the activation of ZIPK by DAPK, various hallmarks of cell death were monitored, and the formation of autophagic vesicles was assessed. Unlike expression of the p55 TNF receptor, expression of activated DAPK deleted of the CaM regulatory domain (ΔCaM) or ZIPK 6D did not lead to activation of caspase-3 or PARP cleavage (Fig. 10C). While no signs of caspase-dependent apoptosis were evident, autophagosome formation was observed. This was assessed by monitoring the localization of LC3, an early marker of autophagy that is involved in autophagic vesicle formation and is recruited to newly formed autophagosome membranes (16). To this end, a GFP-LC3 fusion was coexpressed with DAPK ΔCaM or ZIPK 6D. In control luciferase-expressing cells, GFP-LC3 was localized in a diffuse manner to the cytoplasm and nucleus (Fig. 10A). Expression of the active forms of DAPK or ZIPK 6D, however, led to a redistribution of GFP-LC3 to punctate cytoplasmic structures corresponding to autophagic vesicles. These structures were not apparent in p55-expressing cells. The quantitation of transfected cells displaying autophagy is shown in Fig. 10B. Thus, as with DAPK or DRP-1 (9), overexpression of an activated form of ZIPK induced caspase-independent, autophagic type II cell death in 293T cells.
In order to elucidate the mechanism by which these specific phosphorylations influence the cell death functions of ZIPK, the effects of phosphorylation on ZIPK's intracellular localization was examined. The two phosphorylation mutants and the WT protein were transfected into HeLa cells and stained with anti-Flag antibodies. The following four localization patterns were observed: group I, exclusive cytoplasmic localization with ZIPK completely excluded from the nucleus; group II, cytoplasmic localization with minor nuclear staining (partial nuclear exclusion); group III, even distribution between the nucleus and cytoplasm; and group IV, a pattern with more prominent staining in the nucleus than in the cytoplasm. Examples of these different localization patterns are illustrated in Fig. 11A. Examination of the three ZIPK proteins indicated that they clearly differ in their localization patterns as quantified in Fig. 11B. The wild-type ZIPK was mainly cytoplasmic, with some of the protein decorating the nucleus (group II). The 6A mutant, on the other hand, displayed a stronger tendency towards nuclear staining; the majority of cells showed an even distribution of ZIPK between the cytoplasm and the nucleus (group III), and a substantial fraction of cells showed a more prominent nuclear staining (group IV). Of note, confocal microscopic imaging confirmed that the signal in the nucleus reflects intranuclear staining (data not shown). In contrast, cells expressing the 6D mutant exhibited a greater tendency towards the cytoplasmic pattern of staining, with a significant proportion of cells in which ZIPK was completely excluded from the nucleus (group I). Thus, the negatively charged or phosphorylated form of ZIPK is anchored in the cytoplasm and/or prevented from being shuttled to the nucleus, whereas nuclear translocation or retention is maximal when ZIPK is not phosphorylated.
FIG. 11.

The cellular localization of ZIPK is regulated by phosphorylation on six sites. (A) Immunostaining of ZIPK mutants ectopically expressed in HeLa cells with anti-Flag antibodies to visualize the cellular localization of the different mutants. The same cells were stained with DAPI to visualize the nucleus. (B) Graph showing the distribution of cellular localizations, displayed by different ZIPK mutants. Quantitation was calculated (means ± standard deviations) from triplicates of 200 cells each. N/C, nuclear/cytoplasm.
The oligomerization state of ZIPK is affected by its phosphorylation status.
It has been previously suggested that ZIPK may homodimerize through its leucine zipper domain, based on interactions in the yeast two-hybrid system (18). The oligomerization status of ZIPK was directly tested here for the first time by using size fractionation by gel filtration. To this end, a Superdex 200 column was used to separate WT ZIPK according to its native size, following its overexpression in 293T cells. The ZIPK protein eluted in two distinct peaks at fractions 26 and 32, which correspond to a molecular weight of approximately 154 and 64 kDa, respectively. Thus, ZIPK seems to fluctuate between monomeric and trimeric states. No distinct peak that corresponds to the position of the dimer could be detected, although the presence of low levels of dimers cannot be excluded (Fig. 12A). This equilibrium between trimer and monomer was affected by the phosphorylation status of ZIPK. The elution profiles of the two ZIPK mutants that mimicked the phosphorylated and the dephosphorylated forms, (mutants 6D and 6A, respectively) were compared. While the major peak of the 6D mutant eluted in fraction 28, which corresponds approximately to 148 kDa or the trimeric form (Fig. 12B), the major peak of the 6A mutant eluted in fraction 33, which corresponds approximately to 66 kDa or the monomer. Therefore, while the WT ZIPK molecules exist in equilibrium between trimeric and monomeric forms, phosphorylation on DAPK target sites increases the trimeric form, and its dephosphorylation shifts the equilibrium towards the monomeric form. Thus, the phosphorylation status of ZIPK is critical for both its intracellular localization and its ability to oligomerize.
FIG. 12.

The oligomerization state of ZIPK is subjected to regulation by DAPK. (A) Cellular extract prepared of 293T cells transfected with WT ZIPK was run on a Superdex 200 column, and fractions were analyzed by Western blotting by using anti-Flag antibodies. Arrowheads indicate the estimated molecular weights of the various fractions. (B) Graph showing the distribution of ZIPK mutants 6A and 6D upon fractionation. The quantity of protein was determined by densitometric analysis of the relevant Western blots.
DISCUSSION
We report in this work that two kinases from the DAPK family, DAPK and ZIPK, not only share sequence and functional properties but also physically and functionally cooperate to activate cell death. The physical interaction results in the phosphorylation of ZIPK by DAPK but not the reverse. This transphosphorylation was directly assessed by in vitro kinase assays and was confirmed in vivo upon coexpression of both kinases in cells. DAPK's activity towards ZIPK, at least at physiologic protein concentrations, was greater than that displayed towards MLC. While it has been argued that DAPK is much less efficient at phosphorylating MLC compared to other MLC kinases, such as MLCK (15), MLC has been shown to be an in vivo substrate of DAPK whose phosphorylation is critical for DAPK's induction of membrane blebbing (1, 23). Thus, ZIPK is a better substrate for DAPK than a known, functionally relevant substrate. The phosphorylation on ZIPK was mapped to multiple sites within the extracatalytic region of ZIPK. While the mapping was performed by in vitro kinase assays, the finding that the replacement of the Ser/Thr sites by Asp amplified the death-promoting capacity of ZIPK proved the physiological relevance of this phosphorylation. This suggests that the physical association between the two closely related proteins, shown at the level of the endogenous proteins, serves as a means to enhance the death signal through DAPK-mediated phosphorylation of ZIPK, which ultimately leads to activation of the cellular function of ZIPK to promote cell death.
Direct interactions between kinases resulting in transphosphorylation frequently serve as a means of turning on the catalytic activity of kinases and further provide a mechanism of signal amplification in response to a stimulus. This classical kinase cascade has been demonstrated in much detail with regard to the mitogen-activated protein (MAP) kinase pathway (4). Here we propose the utilization of transphosphorylation for promoting the cellular functions of the target kinase in controlling cell death. Both DAPK and ZIPK are continuously expressed in viable cells (2, 3, 18, 21), and, therefore, their death-promoting functions need to be tightly controlled so that their cell-destructive effects are activated only when necessary. For DAPK and its relative DRP-1, both Ca2+-activated CaM and a relief from a unique inhibitory autophosphorylation, occurring on Ser308 within the CaM regulatory domain, have been shown to be required for activation (33, 34). ZIPK, in contrast, lacks the CaM regulatory domain and, therefore, lacks the autoinhibitory mechanism common to its close family members. Based on the evidence presented here, we propose that it is, instead, activated by an alternative mechanism, which depends on a DAPK-initiated kinase pathway. Interestingly, the DAPK-ZIPK kinase-substrate relationship was unique among the family members. Neither ZIPK nor DRP-1 appears to be capable of phosphorylating the other family members, and DAPK phosphorylation of DRP-1 is relatively weak when compared to ZIPK. Thus, the uniqueness of the DAPK-ZIPK phosphorylation suggests that a very definite hierarchy exists in the DAPK family, with DAPK positioned upstream of ZIPK. Unlike the MAP kinase cascade, where the catalytic activity of downstream kinases is directly turned on by phosphorylation of the activation loop within the catalytic domain, here the phosphorylation targets on ZIPK do not reside within the catalytic domain but, rather, in the C-terminal region that has other important cellular functions. It should be noted that the 6D mutant, which mimics the phosphorylated form of ZIPK, did not show increased catalytic activity towards MLC in an in vitro kinase assay but, rather, was even slightly less active when compared to the WT protein (data not shown). This suggests that the catalytic activity per se is not significantly influenced by transphosphorylation, at least towards MLC. It is therefore more likely that the phosphorylation of ZIPK influences its accessibility to specific substrates in vivo, either due to the localization change and/or to novel leucine zipper-mediated interactions that may occur due to conformational changes at the C terminus of ZIPK.
The physical interaction between DAPK and ZIPK was assessed in this work by coimmunoprecipitation experiments. Quite unexpectedly, the minimal region within the two proteins sufficient for this protein-protein interaction was mapped to the catalytic domains of each kinase. In the case of DAPK, we found that the catalytic domain is not only sufficient but also necessary to form this complex. Interestingly, other domains of DAPK, including those previously implicated in mediating protein-protein interactions, such as the ankyrin repeats or the death domain, were not capable of binding ZIPK. Classical enzyme-substrate interactions that occur via the catalytic domain are usually transient in nature and terminate after the phospho-transfer reaction. In this case, however, a unique high-affinity docking site is present in the catalytic domain of both kinases, which maintains the complex in a more permanent state, so that it can be recovered following immunoprecipitation. Moreover, the unique basic loop of the catalytic domain of ZIPK, known as the fingerprint of the DAPK family, takes an active part in this physical binding, since mutation of this stretch significantly interferes with the physical association between ZIPK and DAPK. The exposed nature of this basic loop at the surface of the catalytic domain (35) is consistent with the finding that it mediates these important interactions between the family members.
It is not possible at this stage to determine whether the ZIPK-DAPK interaction is direct or whether there is a third scaffold protein that joins the two kinases together through their catalytic domains. In the MAP kinase cascade, for example, MEK and extracellular signal-regulated kinase are held in close proximity by a scaffold protein. Elegant work in the yeast system showed that tethering the appropriate kinases to a scaffold is sufficient to elicit the signaling and, in fact, the specificity of MAP kinases entails recruitment of the enzymes to the proper substrate via the scaffold protein (4, 28, 30). Future work with highly purified ZIPK and DAPK should determine whether a scaffold protein is necessary to tether the two kinases together and how the basic loop is involved in such a process.
DAPK phosphorylates ZIPK on several sites, including one Thr and five specific Ser residues. These phosphate-accepting sites are located next to a unique and positively charged region of the ZIPK protein molecule (26). It is plausible that the introduction of multiple negative charges to this region, either by phosphorylation or by forced mutation to Asp, severely alters the nature of this domain and, by this means, its function. In fact, two dramatic changes are observed with the negatively charged form of ZIPK. First, the equilibrium between the monomeric and oligomeric forms of ZIPK shifts towards the oligomeric state, probably due to conformational changes of the protein associated with exposure of certain domains that promote the homotypic interactions. Second, ZIPK is shuttled away from the nucleus to the cytoplasm. This may be due to increased retention in the cytoplasm, as a result of increased affinity for specific cytoplasmic anchoring proteins, or, alternatively, it may be due to decreased nuclear transport or retention. In fact, there are numerous examples in the literature of proteins whose nuclear translocations are regulated by phosphorylation, such as T antigen, NF-κB, c-rel, dorsal, yeast SW15, cofilin, xnf7, lamin B2, v-Jun, MEK1, MAP kinase, and others (14). In some cases, phosphorylation affects interactions with the transport machinery itself, while in others it influences association with cytoplasmic partners (7, 8, 13, 24, 36). The region of ZIPK incorporating the DAPK phosphorylation sites contains the second of four potential NLS dispersed throughout the C terminus. Although the most C-terminal NLS was shown to be critical for nuclear transport (20), modification of the second NLS may negatively affect overall nuclear transport.
Most significantly, the negatively charged pseudophosphorylated form of ZIPK was a more potent inducer of type II cell death-associated morphological changes. This may result from its increased propensity to trimerize or from its increased cytoplasmic localization or both. Its presence in the cytoplasm may expose it to cell death-relevant substrates, such as MLC, whose phosphorylation is proposed to mediate membrane blebbing and/or other potential autophagy-related, still unidentified, substrates (25). The relationship between subcellular localization and ZIPK function has been a subject of debate in the literature. Overexpressed rodent ZIPK was shown to be localized to the nucleus, in particular, to nuclear speckles characterized as nuclear PML oncogenic domains (17, 18, 21). While some of these studies associated the nuclear localization of ZIPK with its cell death capabilities (17, 18), others related its nuclear localization to its involvement in other nuclear activities, such as transcription, mRNA splicing, and mitosis (21, 27, 29). In fact, ZIPK binds to several transcription and splicing factors, including ATF4 (18), AATF (27), and CDC5 (5) and can phosphorylate, at least in vitro, the splicing factor ASF/SF2 (6) and histone H3 (21, 29). Based on this latter phosphorylation and the fact that ZIPK localizes to centromeres in a mitotic-specific manner that correlates with the timing and position of the phosphorylation of histone H3, it has been suggested that ZIPK may play a role in preparing chromatin for mitosis (29). However, when forced to the cytoplasm, either as a result of the truncation of the C-terminal NLS or upon coexpression of its interacting partner Par-4, rodent ZIPK has been shown to acquire cell-killing properties (20, 26, 27). Furthermore, it has been reported that human ZIPK localizes to the cytoplasm, where it induces phosphorylation of MLC, reorganization of the actin cytoskeleton, and membrane blebbing (25). While the differences in localization and functional activity may reflect species or cell type divergence, the data presented in this work offer an alternate explanation for the discrepant results. The cellular localization of ZIPK appears to be under tight regulation by its upstream family member DAPK, which, through transphosphorylation, controls ZIPK's shuttling between the cytoplasm and the nucleus. ZIPK may possess functional roles in both the nucleus and the cytoplasm, but its death-promoting effects would require redistribution to the cytoplasm.
In summary, DAPK and ZIPK form a kinase complex that potentially serves to transduce the signals that lead to the activation of DAPK to additionally activate ZIPK, thus forming a unique kinase cascade employed to amplify autophagic type II death signals. It will be interesting to determine whether this concept can be extended to include additional family members, such as DRP-1, as well. In addition, it is also not known if the kinases act in a way whereby DAPK activates ZIPK, and activated ZIPK serves as the effector kinase to phosphorylate specific cellular substrates such as MLC. Alternatively, all the family members may directly phosphorylate cellular substrates, and the transphosphorylation acts as a means of regulating and/or amplifying cellular functions of the target kinase. Future work involving all three family members will be critical to elucidating this novel death-regulating kinase network.
Acknowledgments
We thank Hanna Berissi and Ofer Cohen for the preparation of the anti-DAPK-tail antibodies. We thank Tamotsu Yoshimori for providing the GFP-LC3 construct.
This work was supported by the Israel Science Foundation administered by the Israel Academy of Sciences and Humanities. A.K. is an incumbent of the Helena Rubinstein Chair of Cancer Research.
REFERENCES
- 1.Bialik, S., A. R. Bresnick, and A. Kimchi. DAP-kinase-mediated morphological changes are localization dependent and involve myosin-II phosphorylation. Cell Death Differ. 11:631-644. [DOI] [PubMed]
- 2.Cohen, O., E. Feinstein, and A. Kimchi. 1997. DAP-kinase is a Ca2+/calmodulin-dependent, cytoskeletal-associated protein kinase, with cell death-inducing functions that depend on its catalytic activity. EMBO J. 16:998-1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Deiss, L. P., E. Feinstein, H. Berissi, O. Cohen, and A. Kimchi. 1995. Identification of a novel serine/threonine kinase and a novel 15-kD protein as potential mediators of the gamma interferon-induced cell death. Genes Dev. 9:15-30. [DOI] [PubMed] [Google Scholar]
- 4.Elion, E. A. 1998. Routing MAP kinase cascades. Science 281:1625-1626. [DOI] [PubMed] [Google Scholar]
- 5.Engemann, H., V. Heinzel, G. Page, U. Preuss, and K. H. Scheidtmann. 2002. DAP-like kinase interacts with the rat homolog of Schizosaccharomyces pombe CDC5 protein, a factor involved in pre-mRNA splicing and required for G2/M phase transition. Nucleic Acids Res. 30:1408-1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Engemann, H., D. Kogel, S. Manderscheid, U. Preuss, and K. H. Scheidtmann. 2002. Elucidation of transcriptional elements and the genomic structure of DAP-like kinase. Ann. N. Y. Acad. Sci. 973:363-367. [DOI] [PubMed] [Google Scholar]
- 7.Hodel, M. R., A. H. Corbett, and A. E. Hodel. 2001. Dissection of a nuclear localization signal. J. Biol. Chem. 276:1317-1325. [DOI] [PubMed] [Google Scholar]
- 8.Hubner, S., C. Y. Xiao, and D. A. Jans. 1997. The protein kinase CK2 site (Ser111/112) enhances recognition of the simian virus 40 large T-antigen nuclear localization sequence by importin. J. Biol. Chem. 272:17191-17195. [DOI] [PubMed] [Google Scholar]
- 9.Inbal, B., S. Bialik, I. Sabanay, G. Shani, and A. Kimchi. 2002. DAP kinase and DRP-1 mediate membrane blebbing and the formation of autophagic vesicles during programmed cell death. J. Cell Biol. 157:455-468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Inbal, B., O. Cohen, S. Polak-Charcon, J. Kopolovic, E. Vadai, L. Eisenbach, and A. Kimchi. 1997. DAP kinase links the control of apoptosis to metastasis. Nature 390:180-184. [DOI] [PubMed] [Google Scholar]
- 11.Inbal, B., G. Shani, O. Cohen, J. L. Kissil, and A. Kimchi. 2000. Death-associated protein kinase-related protein 1, a novel serine/threonine kinase involved in apoptosis. Mol. Cell. Biol. 20:1044-1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jang, C. W., C. H. Chen, C. C. Chen, J. Y. Chen, Y. H. Su, and R. H. Chen. 2002. TGF-beta induces apoptosis through Smad-mediated expression of DAP-kinase. Nat. Cell Biol. 4:51-58. [DOI] [PubMed] [Google Scholar]
- 13.Jans, D. A. 1995. The regulation of protein transport to the nucleus by phosphorylation. Biochem. J. 311:705-716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jans, D. A., and S. Hubner. 1996. Regulation of protein transport to the nucleus: central role of phosphorylation. Physiol. Rev. 76:651-685. [DOI] [PubMed] [Google Scholar]
- 15.Jin, Y., E. K. Blue, S. Dixon, L. Hou, R. B. Wysolmerski, and P. J. Gallagher. 2001. Identification of a new form of death-associated protein kinase that promotes cell survival. J. Biol. Chem. 276:39667-39678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kabeya, Y., N. Mizushima, T. Ueno, A. Yamamoto, T. Kirisako, T. Noda, E. Kominami, Y. Ohsumi, and T. Yoshimori. 2000. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 19:5720-5728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kawai, T., S. Akira, and J. C. Reed. 2003. ZIP kinase triggers apoptosis from nuclear PML oncogenic domains. Mol. Cell. Biol. 23:6174-6186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kawai, T., M. Matsumoto, K. Takeda, H. Sanjo, and S. Akira. 1998. ZIP kinase, a novel serine/threonine kinase which mediates apoptosis. Mol. Cell. Biol. 18:1642-1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kawai, T., F. Nomura, K. Hoshino, N. G. Copeland, D. J. Gilbert, N. A. Jenkins, and S. Akira. 1999. Death-associated protein kinase 2 is a new calcium/calmodulin-dependent protein kinase that signals apoptosis through its catalytic activity. Oncogene 18:3471-3480. [DOI] [PubMed] [Google Scholar]
- 20.Kogel, D., H. Bierbaum, U. Preuss, and K. H. Scheidtmann. 1999. C-terminal truncation of Dlk/ZIP kinase leads to abrogation of nuclear transport and high apoptotic activity. Oncogene 18:7212-7218. [DOI] [PubMed] [Google Scholar]
- 21.Kogel, D., O. Plottner, G. Landsberg, S. Christian, and K. H. Scheidtmann. 1998. Cloning and characterization of Dlk, a novel serine/threonine kinase that is tightly associated with chromatin and phosphorylates core histones. Oncogene 17:2645-2654. [DOI] [PubMed] [Google Scholar]
- 22.Kogel, D., J. H. Prehn, and K. H. Scheidtmann. 2001. The DAP kinase family of pro-apoptotic proteins: novel players in the apoptotic game. Bioessays 23:352-358. [DOI] [PubMed] [Google Scholar]
- 23.Kuo, J. C., J. R. Lin, J. M. Staddon, H. Hosoya, and R. H. Chen. 2003. Uncoordinated regulation of stress fibers and focal adhesions by DAP kinase. J. Cell Sci. 116:4777-4790. [DOI] [PubMed] [Google Scholar]
- 24.Li, X., W. Shou, M. Kloc, B. A. Reddy, and L. D. Etkin. 1994. Cytoplasmic retention of Xenopus nuclear factor 7 before the mid blastula transition uses a unique anchoring mechanism involving a retention domain and several phosphorylation sites. J. Cell Biol. 124:7-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Murata-Hori, M., Y. Fukuta, K. Ueda, T. Iwasaki, and H. Hosoya. 2001. HeLa ZIP kinase induces diphosphorylation of myosin II regulatory light chain and reorganization of actin filaments in nonmuscle cells. Oncogene 20:8175-8183. [DOI] [PubMed] [Google Scholar]
- 26.Page, G., D. Kogel, V. Rangnekar, and K. H. Scheidtmann. 1999. Interaction partners of Dlk/ZIP kinase: co-expression of Dlk/ZIP kinase and Par-4 results in cytoplasmic retention and apoptosis. Oncogene 18:7265-7273. [DOI] [PubMed] [Google Scholar]
- 27.Page, G., I. Lodige, D. Kogel, and K. H. Scheidtmann. 1999. AATF, a novel transcription factor that interacts with Dlk/ZIP kinase and interferes with apoptosis. FEBS Lett. 462:187-191. [DOI] [PubMed] [Google Scholar]
- 28.Park, S. H., A. Zarrinpar, and W. A. Lim. 2003. Rewiring MAP kinase pathways using alternative scaffold assembly mechanisms. Science 299:1061-1064. [DOI] [PubMed] [Google Scholar]
- 29.Preuss, U., G. Landsberg, and K. H. Scheidtmann. 2003. Novel mitosis-specific phosphorylation of histone H3 at Thr11 mediated by Dlk/ZIP kinase. Nucleic Acids Res. 31:878-885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ptashne, M., and A. Gann. 2003. Signal transduction. Imposing specificity on kinases. Science 299:1025-1027. [DOI] [PubMed] [Google Scholar]
- 31.Raveh, T., G. Droguett, M. S. Horwitz, R. A. DePinho, and A. Kimchi. 2001. DAP kinase activates a p19ARF/p53-mediated apoptotic checkpoint to suppress oncogenic transformation. Nat. Cell Biol. 3:1-7. [DOI] [PubMed] [Google Scholar]
- 32.Sanjo, H., T. Kawai, and S. Akira. 1998. DRAKs, novel serine/threonine kinases related to death-associated protein kinase that trigger apoptosis. J. Biol. Chem. 273:29066-29071. [DOI] [PubMed] [Google Scholar]
- 33.Shani, G., S. Henis-Korenblit, G. Jona, O. Gileadi, M. Eisenstein, T. Ziv, A. Admon, and A. Kimchi. 2001. Autophosphorylation restrains the apoptotic activity of DRP-1 kinase by controlling dimerization and calmodulin binding. EMBO J. 20:1099-1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shohat, G., T. Spivak-Kroizman, O. Cohen, S. Bialik, G. Shani, H. Berrisi, M. Eisenstein, and A. Kimchi. 2001. The pro-apoptotic function of death-associated protein kinase is controlled by a unique inhibitory autophosphorylation-based mechanism. J. Biol. Chem. 276:47460-47467. [DOI] [PubMed] [Google Scholar]
- 35.Tereshko, V., M. Teplova, J. Brunzelle, D. M. Watterson, and M. Egli. 2001. Crystal structures of the catalytic domain of human protein kinase associated with apoptosis and tumor suppression. Nat. Struct. Biol. 8:899-907. [DOI] [PubMed] [Google Scholar]
- 36.Xiao, C. Y., S. Hubner, and D. A. Jans. 1997. SV40 large tumor antigen nuclear import is regulated by the double-stranded DNA-dependent protein kinase site (serine 120) flanking the nuclear localization sequence. J. Biol. Chem. 272:22191-22198. [DOI] [PubMed] [Google Scholar]