

Abstract
An integrated multi‐target small molecule capable of altering dynamic epigenetic and transcription programs associated with the brain and nervous system has versatile applications in the regulation of therapeutic and cell‐fate genes. Recently, we have been constructing targeted epigenetic ON switches by integrating sequence‐specific DNA binding pyrrole‐imidazole polyamides with a potent histone deacetylase inhibitor SAHA. Here, we identified a DNA‐based epigenetic ON switch termed SAHA‐L as the first‐ever multi‐target small molecule capable of inducing transcription programs associated with the human neural system and brain synapses networks in BJ human foreskin fibroblasts and 201B7‐iPS cells. Ingenuity pathway analysis showed that SAHA‐L activates the signaling of synaptic receptors like glutamate and γ‐aminobutyric acid, which are key components of autism spectrum disorders. The long‐term incubation of SAHA‐L in 201B7‐iPS cells induced morphology changes and promoted a neural progenitor state. Our finding suggests that the tunable SAHA‐L could be advanced as a cell‐type‐independent multi‐target small molecule for therapeutic and/or cell‐fate gene modulation.
Keywords: brain and nervous system, DNA recognition, multi-target small molecules, polymerase chain reaction, synthetic biology
The brain and nervous system functions involve precise orchestration of diverse bioactive factors, intricate receptor signaling, and alteration in epigenetic programs.1 Therefore, defective modifications in the intertwined brain synapses and neurotransmitter networks are difficult to repair.2 Epigenetic cell‐fate regulation often supersedes the local microenvironmental cues.3 Mainly, histone deacetylase (HDAC)‐governed acetylation and deacetylation of histone proteins play a critical role in neural fate decisions by changing the accessibility of DNA towards transcription factors (TFs).4 Several HDAC inhibitors, including valproic acid and sodium butyrate, have been shown to promote transcription associated with neuronal differentiate and brain development.5 Besides, reversal of histone deacetylation could also promote differentiation of human pluripotent stem cells (hPSCs) towards neural progenitor cells (NPCs).6 SAHA is known to enhance neuronal differentiation/regeneration and is particularly attractive, as it is the most common HDAC inhibitor already approved by the FDA for therapeutic applications.7
However, lack of gene specificity and tunable targeting efficacy is a major shortcoming of SAHA as a potential drug to treat the pathological nervous system. Also, there is a need to employ multiple small molecules specific for individual signaling pathways and factors. However, for clinical translation, fewer factors need to be used.8
Considering the exponential increase in the number of annotated brain synapses and neurotransmitter genes, there is an increasing need to develop an epigenetically active multi‐target synthetic strategy that mimics the structural simplicity and functional efficacy of natural TFs.[9] Hairpin pyrrole‐imidazole polyamides (PIPs) represent a class of DNA minor‐groove‐binding small molecules that specifically recognize each of the four Watson–Crick base pairs.10 Distinct PIPs were conjugated with alkylating agents to attain dual‐functional OFF switches for different cancer‐associated genes that closely mimicked the natural TFs in terms of structure and repressive function.11 As a novel chemical approach to activate the specific gene of interest, recently, we integrated HDAC inhibitor SAHA with PIPs to create a new type of synthetic TFs called SAHA‐PIPs. We first created a library of 16 SAHA‐PIPs with unique chemical architectures and named them alphabetically from “A” to “P”.12 Likewise, we constructed a different set of 16 SAHA‐PIPs termed “Q” to “Φ” and identified hit SAHA‐PIPs with bioactivity towards pluripotency genes.13 Through independent lines of evidence, we have previously shown that distinct SAHA‐PIPs could modulate epigenetic programs at distinct DNA sequences to induce selective genes and noncoding RNAs.14 Stem cell scientists prefer human dermal fibroblasts (HDFs) as the cell source for cellular reprogramming, as fibroblasts are readily available and can be acquired in a non‐invasive manner. Hence, we evaluated the effect of constructed SAHA‐PIPs on genome‐wide gene expression in HDF cells and identified certain SAHA‐PIPs as ON switches for germ‐cell, stem‐cell, and retinal‐cell genes,15 However, the effect of SAHA‐PIP was confined to gene expression analysis. Also, it was unclear whether SAHA‐PIP could retain bioactivity on pluripotent cell types and direct them to specific cell differentiation.
Herein, through functional gene expression analysis in BJ human foreskin fibroblasts, we identified the first multi‐target ON switch SAHA‐L (Figure 1) for multiple genes associated with the brain synapses and neurotransmitter networks. Furthermore, we evaluated the effect of SAHA‐L on 201B7 iPS cells to validate whether SAHA‐L could retain the bioactivity and induce neural progenitor markers like NESTIN, PAX 6, and NGN2.
Figure 1.
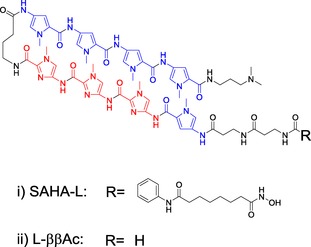
Chemical structure of the i) hit SAHA‐PIP L or SAHA‐L encompassing the PIP targeting 5′‐WGGGW‐3’ (W=A/T) and epigenetically active SAHA, and ii) L‐ββAc, the control PIP lacking SAHA moiety.
Initially, we carried out gene ontology studies of preliminary microarray data in 54 year‐old breast HDFs, which indicated that SAHA‐L could activate few Wnt/β‐catenin signaling‐regulated neural genes (see Figure S1 in the Supporting Information). However, the effect was not remarkable. As BJ foreskin fibroblasts are more amenable to reprogramming, we carried out further studies in this cell line by using purified SAHA‐L and l‐ ββAc lacking the SAHA moiety as the control (Figure S2). Initial standardization studies suggested that 1 μm treatment for 48 h as the optimal condition for attaining regular gene induction activity. Subsequently, the whole transcriptional gene profile of BJ fibroblasts was performed by a human gene 2.1 ST array strip after treatment with DMSO, l‐ββAc (1 μm), and SAHA‐L (1 μm), respectively, over 48 h. The initial microarray data were annotated by using the expression console and the transcriptome analysis console V2.0. The result showed that SAHA‐L exhibited increased bioactivity in BJ fibroblasts compared to HDFs and about 1470 genes were significantly upregulated compared with that in DMSO (Fold Change>2 and ANOVA p‐value<0.05). QIAGEN's ingenuity pathway analysis (IPA) of the up‐regulated genes showed that, in SAHA‐L‐treated BJ fibroblasts, the top five networks are remarkably related to 1) cell‐to‐cell signaling and interaction and 2) nervous system development and function (Table S1). A gene ontology study clearly substantiated that SAHA‐L‐induced genes (10/15, green bars) are associated with neurotransmitter signaling and neuronal development (Figure 2 A).
Figure 2.
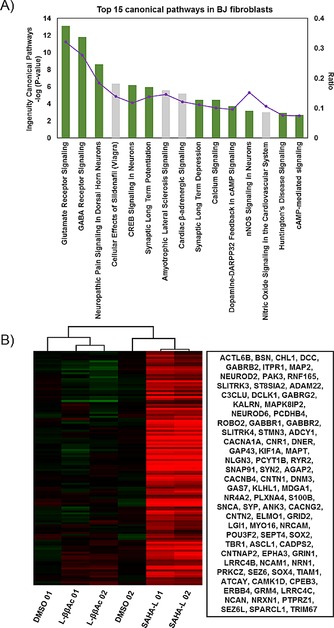
A) Top 15 canonical pathways of SAHA‐L‐treated BJ fibroblasts gene change (Fold Change>2 and ANOVA p‐value <0.05) compared with DMSO. Green bars show the neural system‐related pathways. B) Heat map of selected neural related gene expression of DMSO, l‐ββAc, and SAHA‐L‐treated HDFs. The blanket in the right is the related gene symbol of the hot map.
GABAergic and glutamatergic synapse balance are associated with cerebral dysfunction, especially in autism spectrum disorders. GABA receptors play a vital role in excitatory‐inhibited features of the brain, and autistic subjects were shown to have reduced GABAB receptor 1 (GABBR1) and GABAB receptor 2 (GABBR2) in their brain.16 Interestingly, these two factors were upregulated in SAHA‐L‐treated BJ fibroblasts (Figure 2 B). Furthermore, SAHA‐L also had a multi‐target capability to modulate the GABA and glutamate receptor signaling processes, which are known to be associated with all neurotransmitter‐related defects.17 Besides, the CREB (cAMP response element binding protein) is a TF that works not only in cell proliferation, differentiation, and process outgrowth, but also in brain activity such as learning, memory, and plasticity.18 Therefore, chemical control of CREB signaling could have an influence on the whole‐brain functional network.
Accordingly, in‐depth analysis of the upregulated nervous system genes showed that up to 128 and 103 genes were related to neuron development and synaptic transmission, respectively (Table S2). The heat map of the compiled primary neural system related genes clearly showed that the therapeutically important genes, including CNTNAP2, NRXN1, and others (Figure 2 B), were notably increased in SAHA‐L and not in DMSO‐ or l‐ββAc‐treated BJ fibroblasts. Analysis of genetic interactions by using GENEMANIA showed that the up‐regulated genes have a significant connection with each other (Figure S3) and the common gene network associated with the central nervous system.19 Collectively, the functional analysis of SAHA‐L‐upregulated genes validates the remarkable activation of selective transcripts associated human neural system and brain synapses neurotransmitter signaling. It is important to note here that neither l‐ββAc lacking SAHA nor SAHA alone had the bioefficacy of SAHA‐L. According to our previous reports,14 alteration of acetylation status at specific gene loci corresponding to the key binding 5‘‐WWGGGW‐3‘ sequence of the PIP is considered to be the mechanism behind the unusual domino effect like multiple gene‐activating capability displayed by SAHA‐L.
Although SAHA‐L was shown to have a remarkable effect on the fibroblasts, where the developmental genes are epigenetically silenced, their effect on human pluripotent stem cell (PSC) lines and whether they retain their bioactivity in PSCs was unknown. As SAHA‐L operates by binding specific DNA sequences and altering the local chromatin architecture, they are expected to have similar bioefficacy in different cell types. To this end, we evaluated the effect SAHA‐L in human‐induced pluripotency stem cells (201B7 iPS cells) along with the controls DMSO and l‐ββAc. Based on the initial optimization studies, we found 10 μm and 96 h as the optimal treatment conditions. Microarray analysis and subsequent comparative studies of SAHA‐L versus DMSO showed that, among the top 50 upregulated genes, 19 genes were common in both 201B7 iPS cells and BJ fibroblasts (Figure 3 A). Gene ontology analysis showed that the identical genes belonged to nervous system development and synaptic receptor signaling. As canonical pathway analysis indicated synaptic formation as the top pathway, we carried out real‐time PCR analysis of the key therapeutic genes including CNTNAP2, REELIN, NOS1AP, KALIRIN, DISC1, and NRXN1 after SAHA‐L treatment to both 201B7‐iPS cells and BJ fibroblasts (Figure 3 B–I). In agreement with the microarray results, SAHA‐L, and not the control DMSO, significantly (P<0.05 or P<0.01) induced the endogenous expression of CNTNAP2, NOS1 AP, REELIN, and KALIRIN in BJ fibroblasts (Figure 3 B–E) and 201B7‐iPS cells (Figure 3 F–I).
Figure 3.
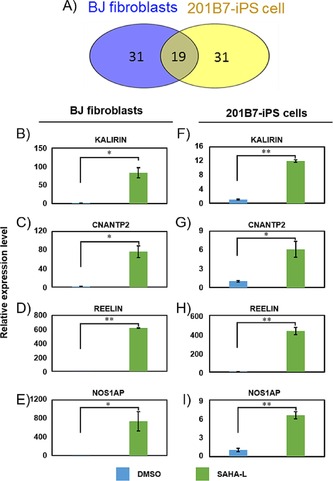
Gene expression in BJ fibroblasts and 201B7‐iPS cells. A) The common gene numbers of the top 50 genes altered by SAHA‐L in BJ fibroblasts and 201B7‐iPS cells (P<0.05). Quantitative real‐time PCR analysis of B) KALIRIN C) CNTNAP2, D) REELIN, E) NOS1AP in BJ fibroblasts and F) KALIRIN G) CNTNAP2, H) REELIN, I) NOS1AP in 201B7 iPS cells. The relative gene expression was normalized to the house‐keeping gene β‐ACTIN. Three biological replicates were performed and the means±SD are indicated; *p<0.05, **p<0.01.
The absence of CNTNAP2 is known to be responsible for neuronal migration and core autism‐related deficits.20 Likewise, DISC and REELIN are also associated with autism‐spectrum disorders like schizophrenia.21 Although SAHA‐L did not have a significant effect on the endogenous expression of DISC1 and NRXN1, the effect was still notable (Figure S4). Pluripotent stem cells and BJ fibroblasts have distinct global epigenome profiles and the acetylation status to govern the developmental genes is noticeably different in these two cell lines.22 Hence, despite the similar significance, the fold induction level of the key synaptic receptor signaling genes in SAHA‐L‐treated cells was comparatively lower in 201B7‐iPS cells with respect to BJ fibroblasts (Figure 3 B–I). Nevertheless, real‐time PCR analysis clearly demonstrates that SAHA‐L could remarkably induce the endogenous expression of synaptic receptor signaling genes in both iPS cells and BJ fibroblasts. To evaluate if the bioactivity of SAHA‐L could be retained when treated, not as conjugates, but as individual parts, we treated SAHA and PIP (l‐ββAc) individually or together in trans. Q‐RT‐PCR data showed the importance of SAHA‐L as conjugates, as no significant effect could be observed on the endogenous gene expression of any of the key brain or nervous system genes in both BJ fibroblasts and 201B7‐iPS cells (Figures S5 A and S5 B). Also, we tested the bioactivity of a control PIP‐ SAHA conjugate called SAHA‐K, where the PIP structure was altered to recognize the 5′‐WGGWWW‐3’ (W=A/T) sequence. SAHA‐K, known to selectively activate germ cell genes,15 did not induce synaptic genes (Figures S5 C and S5 D) to suggest the importance of DNA binding specificity.
As SAHA‐L induced a large number of neurodevelopmental genes, we tested whether continuous treatment of SAHA‐L could direct the differentiation of iPS cells into specific neural cell types. For this, we employed MEF‐conditional medium without β‐FGF and continuously treated the previously employed effectors in iPS cells. As expected, SAHA‐L, and not the controls (DMSO and l‐ββAc), induced relatively significant morphology changes. Marker analysis using real‐time PCR showed a significant (P<0.01) increase in the endogenous expression of essential neural cell factors like NESTIN, PAX6, and NGN2 (Figure 4 A–C). Also, SOX9 expression was suppressed with SAHA‐L treatment (Figure S6 A). It is important to note that repression of SOX9 is known to induce neurogenesis.15 However, SAHA‐L treatment has little effect on the mature neuronal gene markers MAP 2 and GDNF (Figures S6 B and S6 C) when compared to DMSO treatment. We then subjected the effector‐treated cells cultured for 15 days to immunostaining. Microscopic studies showed nestin‐positive cells in SAHA‐L‐treated cells and not in the cells treated with the control (Figure 4 D–I). Despite the alteration in treatment conditions, such as incubation time, concentration, and mode of employment, SAHA‐L did not induce the matured neural cell type. Nevertheless, the long‐term incubation of SAHA‐L in iPS cells shifted the transcriptional program from a pluripotent state to a neural progenitor state.
Figure 4.
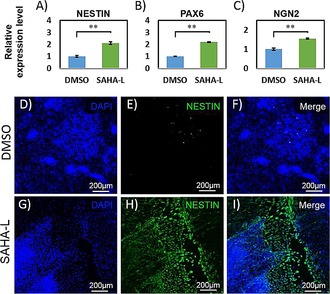
Characterization of neurogenesis in human‐induced pluripotent cells: The expression of several neural progenitor specific markers: A) NESTIN, B) PAX 6, C) NGN2 are displayed, as determined by q‐RT‐PCR analysis. The relative gene expression was normalized to the house‐keeping gene β‐ACTIN, Three biological replicates were performed and the means±SD are indicated; *p<0.05, **p<0.01. d–I: The immunostaining of neural progenitor marker NESTIN was analyzed 15 d after SAHA‐L treatment.
Recently, genetic knowledge‐based targeting of small molecules has been gaining increasing importance in therapeutic and cell‐fate gene modulation. Advanced omic and bioinformatic tools have been revealing the role of multi‐gene network and complex signaling processes in the pathological nervous system. To achieve the precise orchestration observed in the natural cellular environment, there is a need to target multiple factors. But, for clinical translation, synthetic small molecules and the employment of fewer factors are preferred. Until now, no single small molecule has been known to trigger the transcriptional activation of multiple synaptic receptors like glutamate and γ‐aminobutyric acid, which are key components of autism spectrum disorders. This is the first report that demonstrates the bioefficacy of a DNA‐based epigenetically active multi‐target small molecule to trigger the transcriptional activation of brain synapses and neurotransmitter networks in both human fibroblasts and a PSC line. Such similar bioactivity of SAHA‐L in a somatic and a pluripotent cell type, in contrast to SAHA, suggests the critical role of PIPs in directing them to a key consensus motif. At this moment, it is difficult to identify and annotate the actual mechanism and binding motif of SAHA‐L leading to the selective activation of nervous system and brain associated genes. However, analysis of binding sites and functional gene expression has shown that SAHA‐L shares about six corresponding binding sites with the ZPF521 (Figure S7). As zinc‐finger TFs, like ZFP‐521, are known to reprogram HDFs into stable self‐renewing neural stem cells,23 it is possible that SAHA‐L mimics the function of ZFP‐521 to activate neural and brain‐associated genes. Nevertheless, the remarkable activity of a single dual‐functional small molecule like SAHA‐L to activate the intricate synaptic receptor and brain development genes suggests the possibility of advancing them as brain‐cell gene switches.
Although SAHA‐L induced neural progenitor program in 201B7‐iPS cells, the bioactivity was relatively small. However, the tunability of PIP conjugates offers the prospect of advancing them to coax pluripotent stem cells in a step‐wise manner and attain the desired neural cell types. This proof‐of‐concept study, showing the similar bioactivity of SAHA‐L in entirely different cell lines, suggests their potential use as transgene‐free tools for both stem cell differentiation and transdifferentiation. Complex neurological disorders including autism and schizophrenia often involve dysregulated synaptic receptor signaling. As SAHA‐L is capable of activating critical genes known to suppress nervous system disorders, their derivatives could be developed as targeting small molecules to regulate therapeutically important synapse gene(s) of interest. Advancing this synthetic epigenetic program‐editing strategy could be useful in developing multiple variants of clinically useful bioactive ligands.
Experimental details are shown in the Supporting Information.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported by JSPS KAKENHI (grant number 24225005 to H.S. and 24310155 to T.B.) and the Basic Science and Platform Technology Program for Innovative Biological Medicine from Japan Agency for Medical Research and Development (AMED to H.S.). We also thank challenging exploratory research grant for support to G.N.P.
Y. Wei, G. N. Pandian, T. Zou, J. Taniguchi, S. Sato, G. Kashiwazaki, T. Vaijayanthi, T. Hidaka, T. Bando, H. Sugiyama, ChemistryOpen 2016, 5, 517.
References
- 1.
- 1a. Chesler E. J., Lu L., Shou S., Qu Y., Gu J., Wang J., Hsu H. C., Mountz J. D., Baldwin N. E., Langston M. A., Threadgill D. W., Manly K. F., Williams R. W., Nature Genet. 2005, 37, 233–242; [DOI] [PubMed] [Google Scholar]
- 1b. Fagiolini M., Jensen C. L., Champagne F. A., Curr. Opin. Neurobiol. 2009, 19, 207–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kern J. K., Geier D. A., Sykes L. K., Geier M. R., Translational Neurodegeneration 2013, 2, 17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jaenisch R., Bird A., Nat. Genet. 2003, 33, 245–254. [DOI] [PubMed] [Google Scholar]
- 4. Humphrey G. W., Wang Y. H., Hirai T., Padmanabhan R., Panchision D. M., Newell L. F., McKay R. D., Howard B. H., Differentiation 2008, 76, 348–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hsieh J., Nakashima K., Kuwabara T., Mejia E., Gage F. H., Proc. Natl. Acad. Sci. USA 2004, 101, 16659–16664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yang J., Tang Y., Liu H., Guo F., Ni J., Le W. D., BMC Biol. 2014, 12, 95–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kim H.-J., Bae S.-C., Am. J. Transl. Res. 2011, 3, 166–179. [PMC free article] [PubMed] [Google Scholar]
- 8. Wu Y. L., Pandian G. N., Ding Y. P., Zhang W., Tanaka Y., Sugiyama H., Chem. Biol. 2013, 20, 1311–1322. [DOI] [PubMed] [Google Scholar]
- 9. Sepers M. D., Raymond L. A., Drug Discovery Today 2014, 19, 990–996. [DOI] [PubMed] [Google Scholar]
- 10. Dervan P. B., Burli R. W., Curr. Opin. Chem. Biol. 1999, 3, 688–693. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Taylor R. D., Kawamoto Y., Hashiya K., Bando T., Sugiyama H., Chem. Asian J. 2014, 9, 2527–2533; [DOI] [PubMed] [Google Scholar]
- 11b. Syed J., Pandian G. N., Sato S., Taniguchi J., Chandran A., Hashiya K., Bando T., Sugiyama H., Chem. Biol. 2014, 21, 1370–1380. [DOI] [PubMed] [Google Scholar]
- 12. Pandian G. N., Shinohara K. i., Ohtsuki A., Nakano Y., Masafumi M., Bando T., Nagase H., Yamada Y., Watanabe A., Terada N., Sato S., Morinaga H., Sugiyama H., ChemBioChem 2011, 12, 2822–2828. [DOI] [PubMed] [Google Scholar]
- 13. Pandian G. N., Nakano Y., Sato S., Morinaga H., Bando T., Nagase H., Sugiyama H., Sci. Rep. 2012, 2, 544–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pandian G. N., Taniguchi J., Junetha S., Sato S., Han L., Saha A., AnandhaKumar C., Bando T., Nagase H., Vaijayanthi T., Taylor R. D., Sugiyama H., Sci. Rep. 2014, 4, 3843–3851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.
- 15a. Pandian G. N., Sato S., Anandhakumar C., Taniguchi J., Takashima K., Syed J., Han L., Saha A., Bando T., Nagase H., Sugiyama H., ACS Chem. Biol. 2014, 9, 2729–2736; [DOI] [PubMed] [Google Scholar]
- 15b. Syed J., Chandran A., Pandian G. N., Taniguchi J., Sato S., Hashiya K., Kashiwazaki G., Bando T., Sugiyama H., ChemBioChem 2015, 16, 1497–1501; [DOI] [PubMed] [Google Scholar]
- 15c. Han L., Pandian G. N., Junetha S., Sato S., Anandhakumar C., Taniguchi J., Saha A., Bando T., Nagase H., Sugiyama H., Angew. Chem. Int. Ed. 2013, 52, 13410–13413; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 13652–13655. [Google Scholar]
- 16. Fatemi S. H., Folsom T. D., Reutiman T. J., Thuras P. D., Cerebellum 2009, 8, 64–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Moult P. R., Biochem. Soc. Trans. 2009, 37, 1317–1322. [DOI] [PubMed] [Google Scholar]
- 18. Lonze B. E., Ginty D. D., Neuron 2002, 35, 605–623. [DOI] [PubMed] [Google Scholar]
- 19. Roth R. B., Hevezi P., Lee J., Willhite D., Lechner S. M., Foster A. C., Zlotnik A., Neurogenetics 2006, 7, 67–80. [DOI] [PubMed] [Google Scholar]
- 20. Peñagarikano O., Abrahams B. S., Herman E. I., Winden K. D., Gdalyahu A., Dong H., Sonnenblick L. I., Gruver R., Almajano J., Bragin A., Golshani P., Trachtenberg J. T., Peles E., Geschwind D. H., Cell 2011, 147, 235–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.
- 21a. Kilpinen H., Ylisaukko-Oja T., Hennah W., Palo O. M., Varilo T., Vanhala R., Nieminen-von Wendt T., von Wendt L., Paunio T., Peltonen L., Mol. Psychiatry 2008, 13, 187–196; [DOI] [PubMed] [Google Scholar]
- 21b. Akbarian S., Dialogues in Clinical Neuroscience 2014, 16, 405–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Watanabe A., Yamada Y., Yamanaka S., Philos. Trans. R. Soc. London Ser. B 2013, 368, 20120292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.
- 23a. Shahbazi E., Moradi S., Nemati S., Satarian L., Basiri M., Gourabi H., Zare Mehrjardi N., Gunther P., Lampert A., Handler K., Hatay F. F., Schmidt D., Molcanyi M., Hescheler J., Schultze J. L., Saric T., Baharvand H., Stem Cell Rep. 2016, 6, 539–551; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23b. Persikov A. V., Singh M., Nucleic Acids Res. 2014, 42, 97–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary