

Abstract
Microbially derived surfactants, so‐called biosurfactants, have drawn much attention in recent years and are expected to replace current petrochemical surfactants, owing to their environmental and toxicological benefits. One strategy to support that goal is to reduce production costs by replacing relatively expensive sugars with cheaper raw materials, such as short‐chain alkanes. Herein, we report the successful one‐pot total synthesis of rhamnolipids, a class of biosurfactants with 12 stereocenters, from butane as sole carbon and energy source through the design of a tailored whole‐cell biocatalyst.
Keywords: alkanes, biocatalysis, biosurfactants, biotransformation, rhamnolipids
Surfactants are an important class of chemicals that are used in daily life. They are used not only in personal and household care applications, like shampoos and cleansers, but also in industrial applications, such as paints or adhesives. With more than 13 million metric tons produced annually, surfactants also represent one of the chemical industry's most important product classes.1 The majority of commercially available surfactants are based on petrochemicals, such as linear alkylbenzenesulphonate (LAB) or sodium laurethe sulphate (SLS). In addition, most of them show poor environmental and health properties, such as low biodegradability, toxicity, or skin irritation, and are produced by energy‐intensive chemical processes.
Recently, the demand for mild, non‐toxic, bio‐based surfactants has grown significantly, not only driven by consumer demand, but also to improve the environmental footprint of many industrial applications such as enhanced oil recovery. Among bio‐based surfactants, the so‐called “biosurfactants” have drawn much attention over the past decades. They are produced by microorganisms, usually by converting natural raw materials, such as sugars, fatty acids, or glycerol, into sophisticated surfactant molecules with beneficial environmental and toxicological properties, using low‐energy‐demanding fermentation processes. The most prominent examples are sophorolipids2 and rhamnolipids (Scheme 1).3
Scheme 1.
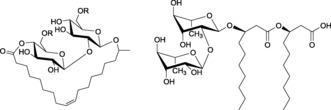
Structure of sophorolipids (left, R=H or Ac) and rhamnolipids (right, shown is the major component).
Although well‐known for many years, the large‐scale industrial usage of these compounds has been limited by high production costs, owing to low achievable yields, titers, and productivities on the one hand, and due to the safety issues of some of the microorganisms used on the other (e.g. the natural producer of rhamnolipids, P. aeruginosa, is known to induce pneumonia and other diseases). Progress in genetic and molecular biology methods as well as advanced expertise in bioprocessing and fermentation technologies have recently changed that picture. A significant body of work has described the process optimization for sophorolipids,4 and its commercialization in larger capacities has already been initiated.5 Regarding rhamnolipids, several reports can be found on the design of rhamnolipid‐producing cells in non‐pathogenic strains such as P. putida KT2440.6 In this case, the ability of P. putida to synthesize activated rhamnose is combined with the integration of the genes rhlA, rhlB, (and in some cases rhlC) originating from P. aeruginosa, which are known to be responsible for rhamnolipid biosynthesis from activated rhamnose and intermediates from the fatty‐acid biosynthesis pathway.7
Although these processes will surely be competitive in yielding products that are suitable as secondary surfactants, it will still be challenging to meet the cost targets for their use as primary surfactants and to replace a significant fraction of the existing surfactants. One key aspect is the relatively high cost contribution from fermentation feedstocks, especially from sugar. Therefore, the utilization of cheaper raw materials in the production of biosurfactants might be a viable option to decrease their manufacturing costs. As can be seen in Figure 1, the use of simple alkanes is an economically interesting option. While the cost per weight is much higher for alkanes than for sugars (data not shown), the cost per energy content is significant lower. Assuming the full theoretical yield can be achieved for the different raw materials, the energy content is reasonable and offers a proper value for comparison according to simple physicochemical considerations based on Hess's law. Looking into the price history of raw sugar and naphtha (as an alkane equivalent), it can be seen that over the last 25 years alkanes were always significantly cheaper than sugars, thus providing a potential cost benefit while still yielding the same mild, non‐toxic, and biodegradable surfactants.
Figure 1.
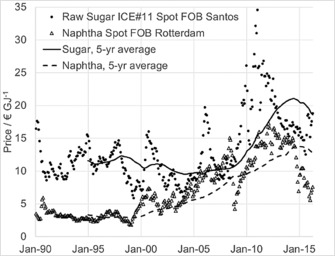
Price history of naphtha (open triangles) and raw sugar (filled circles) in € per GJ energy content from 1990 until 2016. The spot prices FOB (free on board) as well as the 5‐year averages for naphtha (dotted line) and sugar (filled line) are given. Source: Evonik procurement.
Unfortunately, the chemical usage of alkanes remains a very difficult task.8 Owing to the high activation energy of a non‐activated CH bond, it requires harsh conditions to chemically modify alkanes, especially in terms of selectivity and yield.
In contrast, biocatalysis offers a mild and selective method for alkane modification. Several enzymes have been reported that can oxidize non‐activated CH bonds to the corresponding alcohols with high regioselectivity.9 One of the most useful enzyme systems in that respect is the AlkBGT system, originating from P. putida.10 It oxidizes terminal CH3 groups of alkanes and related compounds, such as fatty acids, with extraordinary regioselectivity. In contrast to at least some bacterial cytochrome P450 monooxygenases of the CYP153 family, AlkBGT has a tendency to over‐oxidize and catalyzes the further oxidation of the substrate to the carboxylate moiety. It has recently been demonstrated that fatty‐acid methyl esters can thus be transformed to the corresponding ω‐carboxy compounds or—by integration of proper transaminases—to ω‐amino fatty‐acid esters,11 which are important building blocks for polyamides.12
Recently, we have been able to show that the AlkBGT system is also capable of converting gaseous alkanes, such as n‐butane, to the corresponding acids with reasonable reaction rates, thus activating a cheap and easily available raw material.13
Therefore, we set out to evaluate if the combination of these enzymatic reaction systems, that is, the AlkBGT system for butane activation and the RhlABC system for rhamnolipid assembly, results in the rhamnolipid biosynthesis from n‐butane as the sole carbon and energy source. From a chemical point of view, this approach represents a one‐pot, >25 step, convergent total synthesis, yielding a compound with 12 stereocenters and three C−O bonds that need to be formed in a highly regio‐ and stereoselective manner.
To investigate the reliability of the concept, we started by using butyric acid as the sole carbon source for a P. putida KT2440 strain that had been equipped with rhlABC genes from P. aeruginosa as well as with alkBGT genes from P. putida GPo1. Induction of the rhlABC genes was performed under the control of the pRha promoter, as KT2440 is known to not metabolize any added rhamnose.14 The strain (BOA‐PP‐002) was grown on glucose; cells were harvested by centrifugation, washed twice with fresh buffer to remove any residual glucose or other carbon sources, and placed into a 300 mL DASGIP fermenter. Butyric acid was added (as sodium butyrate) in fresh buffer without any additional carbon source, and rhamnolipid formation was monitored over time by using HPLC. As can be seen from Figure 2 (grey circles), the strain immediately started to produce rhamnolipids, yielding a final titer of about 700 mg l −1. This confirms both butyric acid uptake and its conversion into a precursor for central metabolic pathways as well as functional expression of the rhlABC genes for rhamnolipid production.
Figure 2.
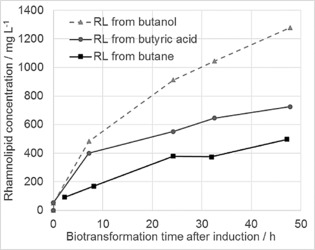
Time course of rhamnolipid biosynthesis when using either butyric acid (grey circles), 1‐butanol (grey triangles), or n‐butane (black squares) as the sole carbon and energy source with P. putida strain BOA‐PP‐002.
As a next step, we tested the functionality of the integrated AlkBGT system by replacing butyric acid with 1‐butanole, which requires two oxidation steps to achieve butyric acid prior to activation to butyryl‐CoA. Again (cf. Figure 2, grey triangles), rhamnolipid production started instantly, yielding an even higher titer of >1200 mg l −1.
Finally, after proving the functionality of RhlABC as well as that of the AlkBGT system, we took the final step by using gaseous n‐butane as the sole substrate (Scheme 2). Figure 2 (black squares) shows that, indeed, significant amounts of rhamnolipids (ca. 500 mg l −1) were produced, thus providing proof of our concept, that rhamnolipid production from cheap raw materials, such as gaseous alkanes, is possible.
Scheme 2.
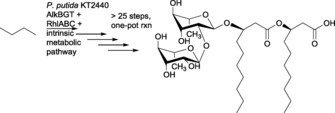
Desired reaction from n‐butane to rhamnolipid through the three‐stage oxidation of butane to butyric acid (first step), catalyzed by the AlkBGT system from P. putida, followed by conversion of butyric acid to the final product using the intrinsic metabolic pathways of P. putida as well as the RhlABC enzyme system from P. aeruginosa.
Next steps will focus on process and strain optimization to increase volumetric productivities and titers towards industrially relevant values. On the process side, this will include optimized butane transfer under explosion‐free conditions and, on the strain side, the fine‐tuning of butane oxidation and rhamnolipid production activities.
Experimental Section
Strain Construction
Bacterial Strains
E. coli 10‐beta (New England BioLabs Inc) was used for cloning and plasmid propagation. To select E. coli and P. putida transformants, antibiotics were used at the following concentrations:: kanamycin, 50 mg l −1; tetracycline, 10 mg l −1 (for E. coli) or 50 mg l −1 (for P. putida). For induction of the rhaPBAD promoter, 0.2 % (w/v) filtered‐sterilized l‐rhamnose was added to the culture media. alkBGT expression was induced by the addition of 0.025 % (v/v) dicyclopropyl ketone (DCPK).
DNA Methods
Recombinant DNA work was carried out according to standard protocols.15 A shuttle vector for replication in E. coli and P. putida was constructed from the cloning vector pACYC184. This new vector contains the pVS1‐origin for replication in P. putida, with the chloramphenicol resistance gene replaced by a kanamycin resistance gene. To produce rhamnolipids, a synthetic operon consisting of the rhlABC genes from P. aeruginosa PAO1 7c was synthesized by using DNA2.0. This operon was ligated into the new shuttle vector under the control of the rhaPBAD promoter. The correct nucleotide sequence was confirmed by DNA sequencing. The pBT10 plasmid contained the alkane monooxygenase genes (alkBFG and alkST operons) in the broad‐host range vector pCOM10 (KmR).16 The plasmids were transferred into E. coli and P. putida KT2440 (ATCC 47054) through electroporation, finally yielding strain BOA‐PP‐002.
Analytics
To determine the product concentration in the different examples, the fermentation broth was diluted immediately after sampling with acetone at a volume ratio of 1:1 and centrifuged at 21 000 g and 4 °C for 2 min. The sample supernatant was then subjected to HPLC analysis [Agilent Technologies 1200 Series; evaporative light scattering detector Sedex LT‐ELSD Model 85LT; Zorbax SB‐C8 rapid resolution column (4.6×150 mm, 3.5 μm)]. Mobile phase A: aqueous 0.1 % TFA; mobile phase B: methanol. Column oven set at 40 °C. Gradient: start with 70 % B in A to 100 % B within 15 min at a flow rate of 1 mL min−1 followed by 5 min of re‐equilibration with 70 % B in A. Reference materials were used, and the identity and purity were checked by using HPLC–MS/MS and NMR.
Production of Rhamnolipids by using BOA‐PP‐002 and Butyric Acid, 1‐Butanol, or n‐Butane
On an lysogeny broth (LB) agar plate containing 50 mg l −1 each of tetracycline and kanamycin, an inoculation loop full of glycerol cryoculture of strain P. putida BOA‐PP‐002 was streaked. The agar plate was incubated for 24 h at 30 °C.
Nine 100 mL shake flasks were filled with 25 mL of LB medium containing 50 mg l −1 each of tetracycline and kanamycin, and each was inoculated with a single colony of the overgrown agar plate and incubated in a shaking incubator for 24 h at 30 °C and 200 rpm.
Nine 1 L flasks were each used to mix 75 mL of M9 medium (composition: 15 g l −1 glucose, 6.8 g l −1 Na2PO4, 3 g l −1 KH2PO4, 0.5 g l −1 NaCl, 2 g l −1 NH4Cl, 15 g l −1 yeast extract, 0.49 g l −1 MgSO4×7 H2O, 50 mg l −1 kanamycin sulfate, 15 mL Ll−1 trace‐element solution US3 consisting of 36.5 g l −1 of 37 % strength hydrochloric acid, 11.91 g l −1 MnCl2×4 H2O, 1.87 g l −1 ZnSO4×7 H2O, 0.84 g l −1 Na‐EDTA×2 H2O, 0.3 g l −1 H3BO3, 0.25 g l −1 Na2MoO4×2 H2O, 4.7 g l −1 CaCl2×2 H2O, 17.3 g l −1 FeSO4×7 H2O, 0.15 g l −1 CuCl2×2 H2O) and the pre‐culture from the 100 mL flasks. The cultures were incubated at 30 °C and 200 rpm. After 3 h of incubation, expression of the alkBGT genes was induced by adding 0.4 mm of DCPK. The cultures were incubated for a further 16 h at 30 °C and 200 rpm.
The cultures of the nine 1 L flasks were combined and centrifuged at 5500 g at room temperature for 10 min. The supernatant was discarded and the pellet was re‐suspended in 100 mL of modified M9 medium (composition as provided above, without glucose and yeast extract). This washing step was repeated for the complete removal of glucose and other possible carbon sources.
In three 300 mL fermenters, 180 mL of modified M9 medium (composition of which is provided above, without glucose and yeast extract) as well as 50 mg l −1 each of tetracycline and kanamycin were added. The fermenters were inoculated with a large volume of pre‐culture suspension from the earlier step to reach a start OD600 of 4 for the experiments with butyrate (A) and 1‐butanol (B), or a start OD600 of 8 for the experiment with n‐butane (C). The following parameters were set during fermentation: temperature 30 °C (all), gassing with air 3 NL h−1 (A and B) or with n‐butane/air mixture (25 %/75 %) 2 NL h−1 (C), stirrer speed set at 700 rpm (A and B) or 900 rpm (C), pH value of 7.1 regulated with 1 m phosphoric acid (A) or with 5 % ammonia solution (B) or an initial pH value of 7.4, not regulated throughout the experiment (C). A continuous feed with sodium butyrate solution (A: 0.25 g l −1 h−1) or 1‐butanol solution (B: 0.17 g l −1 h−1) was applied. After 15 h (A and B) or 18 h (C), the expression of rhamnolipid production genes was induced by adding 2 g l −1 of rhamnose.
All experiments were run in duplicate and the average values for rhamnolipid titers are given.
For aeration, stainless‐steel filter/sparger tips with an outer diameter of 6.35 mm, a length of 25.4 mm, and a pore size of 10 μm were used. The used n‐butane/air mixture (25 %/75 %) ensures that the reaction always takes place under non‐explosive conditions.
Acknowledgements
We thank Anja Thiessenhusen for vector construction and cloning, Julia Niewalda for technical assistance, and Dr. Anne Jeremias and her team for running all analytics.
C. Gehring, M. Wessel, S. Schaffer, O. Thum, ChemistryOpen 2016, 5, 513.
References
- 1. Reznik G. O., Vishwanath P., Pynn M. A., Sitnik J. M., Todd J. J., Wu J., Jiang Y., Keenan B. G., Castle A. B., Haskell R. F., Smith T. F., Somasundaran P., Jarrell K. A., Appl. Microbiol. Biotechnol. 2010, 86, 1387–1397. [DOI] [PubMed] [Google Scholar]
- 2. Van Bogaert I. N. A., Saerens K., De Muynck C., Develter D., Soetaert W., Vandamme E. J., Appl. Microbiol. Biotechnol. 2007, 76, 23–34. [DOI] [PubMed] [Google Scholar]
- 3. Henkel M., Syldatk C., Hausmann R., in Biosurfactants: Production and Utilization—Processes, Technologies, and Economics (Eds. N. Kosaric, F. Vardar-Sukan), CRC Press, Florida, 2014, 83–100. [Google Scholar]
- 4. Van Bogaert I. N. A., Ciesielska K., Devreese B., Soetaert W., in Biosurfactants: Production and Utilization—Processes, Technologies, and Economics, (Eds. N. Kosaric, F. Vardar-Sukan), CRC Press, Florida, 2014, 19–36. [Google Scholar]
- 5.Evonik Industries AG, Evonik commercializes biosurfactants, retrieved from http://corporate.evonik.de/en/media/press_releases/corporate/pages/news-details.aspx?newsid=60276, 21.06.2016.
- 6. Loeschcke A., Thies S., Appl. Microbiol. Biotechnol. 2015, 99, 6197–6214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.
- 7a. Wittgens A., Tiso T., Arndt T. T., Wenk P., Hemmerich J., Mueller C., Wichmann R., Kuepper B., Zwick M., Wilhelm S., Hausmann R., Syldatk C., Rosenau F., Blank L. M., Microb. Cell Fact. 2011, 10, 80; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7b. Schaffer S., Thiessenhusen A., Wessel M., Evonik Industries AG, Germany, DE102012201360A1, 2013;
- 7c. Schaffer S., Wessel M., Thiessenhusen A., Stein N., Evonik Goldschmidt GmbH, Germany, WO2012013554A1, 2012.
- 8.
- 8a. Munz D., Strassner T., Inorg. Chem. 2015, 54, 5043–5052; [DOI] [PubMed] [Google Scholar]
- 8b. Shul′pin G. B., Catalysts 2016, 6, 50–89. [Google Scholar]
- 9. Schrewe M., Julsing M. K., Buehler B., Schmid A., Chem. Soc. Rev. 2013, 42, 6346–6377. [DOI] [PubMed] [Google Scholar]
- 10. Eggink G., Lageveen R. G., Altenburg B., Witholt B., J. Biol. Chem. 1987, 262, 17712–17718. [PubMed] [Google Scholar]
- 11. Ladkau N., Assmann M., Schrewe M., Julsing M. K., Schmid A., Buehler B., Metab. Eng. 2016, 36, 1–9. [DOI] [PubMed] [Google Scholar]
- 12.Evonik Industries AG, An Alternative raw Material for Polyamide 12: Evonik is Operating a Pilot Plant for Bio-based ω-amino Lauric Acid, retrieved from http://corporate.evonik.de/en/media/search/pages/news-details.aspx?newsid=37328, 21.06.2016.
- 13.
- 13a. Pfeffer J. C., Haas T., Thum O., Erhardt F., Wittmann E.-M., Evonik Industries AG, Germany, EP2602328A1, 2013;
- 13b. Engel P., Haas T., Pfeffer J. C., Thum O., Gehring C., Evonik Industries AG, Germany, WO2013110557A1, 2013.
- 14. Jeske M., Altenbuchner J., Appl. Microbiol. Biotechnol. 2010, 85, 1923–1933. [DOI] [PubMed] [Google Scholar]
- 15. Molecular Cloning: A Laboratory Manual, 3rd ed. (Eds.: J. F. Sambrook, D. W. Russell), Cold Spring Harbor Laboratory Press, New York, NY, 2000. [Google Scholar]
- 16. Schrewe M., Magnusson A. O., Willrodt C., Bühler B., Schmid A., Adv. Synth. Catal. 2011, 353, 3485–3495. [Google Scholar]