

Abstract
The prenyl group is an important component in bioactive compounds. Herein, we report the assembly of prenylated heteroarenes through a cascade Minisci reaction and acid‐promoted dehydration sequence. The use of potassium (3‐hydroxy‐3‐methylbut‐1‐yl)trifluoroborate as a new coupling reagent allows the direct introduction of prenyl and 3‐hydroxy‐3‐methylbutyl groups to a wide variety of electron‐deficient heteroarenes. Synthetic application is also demonstrated.
Keywords: dehydration, heteroarenes, Minisci reaction, prenylation, radical addition
The prenyl group is prevalent as a key pharmacophore element in numerous naturally occurring bioactive compounds.1 For example, many prenylated flavonoids1b and indole alkaloids2 have been identified to exhibit diverse bioactivities (Figure 1). It was evidenced that the prenyl group has good binding affinity with proteins, and its incorporation could usually enhance membrane permeability, thereby improving the bioactivity and bioavailability of the corresponding prenylated compounds.1b On the other hand, aromatic N‐heterocycles are widely occurring in drugs.3 It is, therefore, of paramount interest to develop synthetic methods for the assembly of prenylated aromatic N‐heterocycles, which may find potential applications in drugs, but are nevertheless synthetically challenging to chemists.
Figure 1.
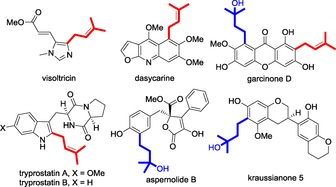
Representative bioactive compounds containing prenyl (red) and/or 3‐hydroxy‐3‐methylbutyl (blue) groups.
In nature, prenyl groups are introduced through enzymatic reactions with prenyl pyrophosphate.4 The necessity to use specific substrates may limit their synthetic utilities. Alternatively, different synthetic approaches are available to access prenylated arenes in the literature, among which the most common strategy relies on metal‐catalyzed cross‐coupling reactions (Scheme 1 a).5 The limitation of this strategy is that pre‐functionalized arenes and noble‐metal catalysts are often needed. Friedel–Crafts‐type prenylation of electron‐rich arenes in the presence of different Lewis acids offers another straightforward route, with poor regioselectivity and overreaction typically observed (Scheme 1b).2a, 6 In addition, Claisen rearrangement of allyl ethers is amenable to synthesize prenylated phenols (Scheme 1 c).7 Recently, metal‐catalyzed direct prenylations of aryl C−H bonds have been developed,8 yet the employment of directing groups necessary for reactivity represents a major drawback (Scheme 1 d). Moreover, prenylations through pyran‐ring annulation followed by reductive ring opening were also disclosed recently.9 Of note, though elegant, few of the above‐mentioned protocols are applicable to the prenylation of aromatic N‐heterocycles, especially electron‐deficient ones.5f–5h
Scheme 1.
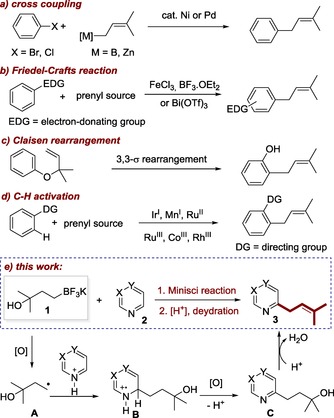
Different approaches toward prenylated arenes.
The past years have witnessed the great power of the Minisci reaction for the direct functionalization of heteroarenes, wherein an in situ‐generated, nucleophilic, carbon‐centered radical reacts with an electron‐deficient N‐aromatic compound, providing a simple and effective method for late‐stage modification of complex heteroarene structures.10 For instance, in 2010, Baran and co‐workers disclosed that aryl radicals generated from arylboronic acids could add to heterocycles at ambient temperature.10c Shortly after that, Molander et al. showed that trifluoroborates served as good radical precursors to realize the direct alkylation of various heterocyles.10d To develop a general and practical protocol for the synthesis of prenylated heteroarenes, we were drawn to the possibility of using a radical‐based transformation. However, the direct use of prenylboron as the prenyl source might be problematic, owing to the propensity of radicals to react with the double bond.11 To obviate this possibility, we envisioned that potassium (3‐hydroxy‐3‐methylbut‐1‐yl)trifluoroborate 1 bearing a hydroxyl group might be well suited for this purpose (Scheme 1 e). We reasoned that the tertiary hydroxyl group, which is expected to be stable under the radical conditions, could serve as a precursor of the double bond. Thus, in our reaction design, 1 is oxidized in situ to generate a nucleophilic alkyl radical A, which attacks the protonated heteroarene to form a radical cation B. Upon oxidation and deprotonation, B is aromatized to provide the alkylated heteroarene C. Once the 3‐hydroxy‐3‐methylbutyl side chain was introduced, an acid‐promoted dehydration of the corresponding tertiary alcohol would give the final prenylated product. Challenges might exist, because the presence of a proximal hydroxyl will attenuate the nucleophilicity of A.10d It should be noted that the 3‐hydroxy‐3‐methylbutyl group itself is also a frequently encountered substituent in natural products (Figure 1, in blue).1f, 12
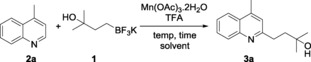
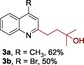
Our investigation began with the preparation of boronate 1. Trifluoroborate 1 could be synthesized through an iridium‐catalyzed hydroboration13 of 2‐methyl‐3‐buten‐2‐ol and a follow‐up addition of aqueous KHF2. The two‐step synthesis ensures a 66 % overall yield for gram‐scale preparation [Eq. (1)]. Of note, 1 is air and moisture stable, which increases the ease with which it can be handled. With a reliable method for the preparation of 1 in hand, we set out to investigate the key Minisci reaction by using lepidine 2 a as a model substrate. Interestingly, when the reaction was conducted under the conditions of Mn(OAc)3⋅2 H2O (2.5 equiv), TFA (1.0 equiv), in AcOH:H2O (1:1 v/v) at 50 °C, and under air, no corresponding alkylation product was observed (Table 1, entry 1).10d Realizing that generated radical might be quenched by atmospheric oxygen, we replaced the air atmosphere with argon. Indeed, the desired product 3 a was formed, albeit in low yield (entry 2). Other oxidants such as K2S2O8 and PhI(OAc)2 were ineffective for the reaction (entries 3 and 4). Different solvents were then screened (entries 5–13). A mixture of TFE/AcOH (4:1, v/v) gave a significantly better result, providing 3 a in a 1H NMR yield of 32 %. The elevation of temperature to 60 °C shortened the reaction time and increased the yield to 45 % (entries 14 and 15). The use of TFA as an additive was beneficial for the reactivity, as its omission gave a decreased yield (entry 16). To fulfill a better conversion, increased loadings of 1 (5.0 equiv) and oxidant (6.5 equiv) were employed, and a higher yield of 68 % was obtained (entries 17–19). The use TFE/AcOH (1:1) as the solvent ensures better reproducibility, owing to an improved solubility of the oxidant (entry 20). (Eqn. (1)
Table 1.
Optimization of the reaction conditions.[a]
| ||||
|---|---|---|---|---|
| Entry | Mn[OAc]3⋅2 H2O [equiv] | Solvent [v/v] | Temp. [°C] | Yield [%][b] |
| 1 | 2.5 | AcOH:H2O (1:1) | 50 | 0 |
| 2 | 2.5 | AcOH:H2O (1:1) | 50 | 6 |
| 3 | 2.5[c] | AcOH:H2O (1:1) | 50 | 0 |
| 4 | 2.5[d] | AcOH:H2O (1:1) | 50 | 0 |
| 5 | 2.5 | toluene | 50 | 2 |
| 6 | 2.5 | AcOH | 50 | 0 |
| 7 | 2.5 | CH3CN | 50 | 0 |
| 8 | 2.5 | MeOH | 50 | 0 |
| 9 | 2.5 | DMF | 50 | 0 |
| 10 | 2.5 | acetone | 50 | 0 |
| 11 | 2.5 | THF | 50 | 0 |
| 12 | 2.5 | TFE | 50 | 12 |
| 13 | 2.5 | TFE:AcOH (4:1) | 50 | 32 |
| 14[e] | 2.5 | TFE:AcOH (4:1) | 30 | 0 |
| 15[e] | 2.5 | TFE:AcOH (4:1) | 60 | 45 |
| 16[e, f] | 2.5 | TFE:AcOH (4:1) | 60 | 21 |
| 17[e, g] | 3.0 | TFE:AcOH (4:1) | 60 | 61 |
| 18[e, h] | 4.0 | TFE:AcOH (4:1) | 60 | 62 |
| 19[e, i] | 6.5 | TFE:AcOH (4:1) | 60 | 68 |
| 20[e, i] | 6.5 | TFE:AcOH (1:1) | 60 | 67 (62)[j] |
[a] Reaction conditions: 2 a (0.2 mmol, 1.0 equiv), 1 (1.0 equiv), Mn(OAc)3⋅2 H2O (2.5 equiv), TFA (1.0 equiv), solvent (2.0 mL), 18 h. [b] 1H NMR yield. [c] K2S2O8 as oxidant. [d] PhI(OAc)2 as oxidant. [e] 4 h. [f] Without TFA. [g] 1 (3.0 equiv). [h] 1 (4.0 equiv). [i] 1 (5.0 equiv). [j] Isolated yield.
(1).

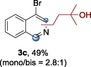
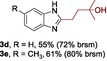
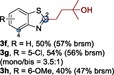
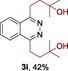
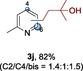
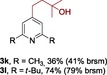
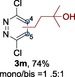
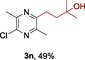
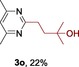
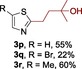
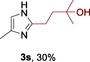
The scope of the reaction was then evaluated on a broad range of nitrogen‐containing heteroarenes. As shown in Table 2, 4‐bromoquinoline could also be converted to the desired product in 50 % yield (3 b). Isoquinoline 2 c gave the mono‐ and bis‐ alkylated products in a combined yield of 49 %. In addition, benzimidazoles (2 d, 2 e) and benzothiazoles (2 f–h) were also suitable for this transformation. Interestingly, when 5‐chloro‐benzothiazole 2 g was applied, the bis‐alkylated product at both the C2 and C4 positions was also observed, probably owing to the electron‐withdrawing nature of the chloro substituent. Furthermore, phthalazine 2 i underwent the reaction smoothly to afford the bis‐alkylated product 3 i in moderate yield. In accordance with the previous observations,10c, 10f the reaction of pyridines gave a regioisomeric mixture (3 j) with alkylation taking place predominantly at the electron‐deficient C2 and C4 positions. The use of 2,6‐disubstituted pyridine rendered the reaction selective at the C4 position (3 k, 3 l). Pyridazine (2 m), pyrazine (2 n), and pyrimidine (2 o) bearing two heteroatoms in the aromatic ring all delivered the corresponding products successfully. Five‐membered heteroarenes such as thiazole (2 p–r) and imidazole (2 s) were amenable to alkylation as well. It should be noted that halogen functional groups were tolerated in several cases (3 b, 3 c, 3 g, 3 m, 3 n, and 3 q), thus providing good handles for further derivation of the products.
Table 2.
Introduction of 3‐hydroxy‐3‐methylbutyl group to heteroarenes through the Minisci reaction.

| ||
|---|---|---|
|
|
|
|
|
|
|
|
|
|
|
|
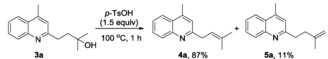
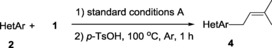
Having successfully established the method for the introduction of 3‐hydroxy‐3‐methylbutyl group, we turned our attention to the follow‐up dehydration reaction. Upon treatment with p‐toluenesulfonic acid (1.5 equiv) in toluene at 100 °C, 3 a could be smoothly dehydrated to give the desired prenylated product 4 a in 87 % yield, along with a minor (11 %) terminal olefin 5 a [Eq. (2)]. This result encouraged us to test the viability of a telescoping synthesis of 4 a. Thus, starting from quinoline 2 a without the isolation of the intermediate 3 a, a decent yield (59 %) of 4 a was obtained for two steps [Eq. (3)].
(2).
(3).

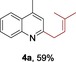
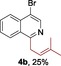
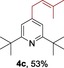
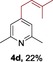
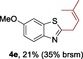
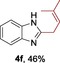
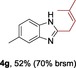
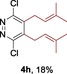
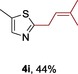
The protocol for the telescoping synthesis of prenylated heteroarenes could also be extended to other heterocyclic substrates (Table 3). It was found that isoquinoline (4 b), pyridines (4 c, 4 d), benzothiazoles (4 e), benzimidazoles (4 f, 4 g), pyridazine (4 h), and thiazole (4 i) all successfully delivered the corresponding prenylated products. Although low yields were obtained in certain cases, the ability for straightforward and late‐stage modification of heteroarenes still make this protocol valuable in medicinal chemistry.
Table 3.
Telescoping synthesis of prenylated heteroarenes through the Minisci reaction and dehydration.
| ||
|---|---|---|
|
|
|
|
|
|
|
|
|
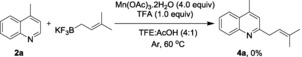
The direct Minisci‐type prenylation with potassium prenyl trifluoroborate as the coupling partner gave no prenylated product 4 a, verifying our hypothesis that a tri‐substituted alkene is not compatible with our radical conditions [Eq. (4)]. To demonstrate the potential utility of our protocol in medicinal chemistry, camptothecin,14 an antitumor drug bearing several vulnerable functional groups, was subjected to telescoping synthesis. Direct prenylation at the C7 position was accomplished without the need of functional‐group protection, albeit in a low yield of 15 % [Eq. (5)].
(4).
(5).

In conclusion, to realize the prenylation of N‐heteroarenes by using the Minisci reaction, we have designed and synthesized a new coupling reagent, potassium (3‐hydroxy‐3‐methylbut‐1‐yl)trifluoroborate 1. The reaction of 1 enables the direct introduction of 3‐hydroxy‐3‐methylbutyl and the prenyl group, both of which are frequently encountered in bioactive compounds. Owing to the importance of N‐heteroarenes in medicinal chemistry, we anticipate this protocol will find application in the drug‐discovery process.
Experimental Section
General Procedure for the Synthesis of 3
Under an atmosphere of argon, manganese(III) acetate (3.2 mmol, 6.5 equiv), potassium (3‐hydroxy‐3‐methylbut‐1‐yl) trifluoroborate 1 (2.5 mmol, 5.0 equiv), heteroarene 2 (0.5 mmol, 1.0 equiv), trifluoroacetic acid (0.5 mmol, 1.0 equiv), and a 1:1 mixture of trifluoroethanol/acetic acid (5 mL) were added in turn to a 15 mL Schlenk tube charged with a magnetic stirring bar. The Schlenk tube was stirred at 60 °C for 4 h. The mixture was then allowed to cool to room temperature. The solvent was removed under vacuum and the residue was slowly added to a saturated aqueous solution of NaHCO3 (10 mL). The aqueous layer was extracted with EtOAc (3×10 mL). The organic layers were combined and washed with brine and dried over Na2SO4. After being concentrated under reduced pressure, the residues were purified by flash column chromatography on silica with an appropriate eluent to afford the pure product 3.
General Procedure for the Synthesis of 4
Under an atmosphere of argon, manganese(III) acetate (3.2 mmol, 6.5 equiv), 1 (2.5 mmol, 5.0 equiv), heteroarene 2 (0.5 mmol, 1.0 equiv), trifluoroacetic acid (0.5 mmol, 1.0 equiv), and a 1:1 mixture of trifluoroethanol/acetic acid (5 mL) were added in turn to a 15 mL Schlenk tube charged with a magnetic stirring bar. The Schlenk tube was stirred at 60 °C for 4 h. The mixture was then allowed to cool to room temperature. The solvent was removed under vacuum and the residue was slowly added to a saturated aqueous solution of NaHCO3 (10 mL). The aqueous layer was extracted with EtOAc (3×10 mL). The organic layers were combined and washed with saturated EDTA‐2Na and brine, successively. The organic phase was dried over Na2SO4 and concentrated. The residual was dissolved in 5.0 mL of toluene and p‐toluenesulfonic acid (1.0 mmol, 2.0 equiv) was added. The mixture was reacted at 100 °C for 0.5–1 h under an atmosphere of argon. After cooling to room temperature, the mixture was quenched by slow addition of saturated NaHCO3 (10 mL). The aqueous layer was then extract with EtOAc (3×10 mL) and washed with brine. The organic layers were dried over Na2SO4, filtered, and concentrated. Purification was made achieved flash column chromatography by using an appropriate eluent to give the pure product 4.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We are grateful for the support of this work by The National Key Research and Development Program of China (2016YFA0602900), “1000‐Youth Talents Plan”, a Start‐up Grant from Sun Yat‐sen University and National Natural Science Foundation of China (Nos. 81402794, 21472250 and 21502242).
D.-H. Tan, Y.-F. Zeng, Y. Liu, W.-X. Lv, Q. Li, H. Wang, ChemistryOpen 2016, 5, 535.
References
- 1.
- 1a. Papageorgiou V. P., Assimopoulou A. N., Couladouros E. A., Hepworth D., Nicolaou K. C., Angew. Chem. Int. Ed. 1999, 38, 270; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 1999, 111, 280; [Google Scholar]
- 1b. Botta B., Vitali A., Menendez P., Misiti D., Delle Monache G., Curr. Med. Chem. 2005, 12, 713; [DOI] [PubMed] [Google Scholar]
- 1c. Yazaki K., Sasaki K., Tsurumaru Y., Phytochemistry 2009, 70, 1739; [DOI] [PubMed] [Google Scholar]
- 1d. Alhassan A. M., Abdullahi M. I., Uba A., Umar A., Trop. J. Pharm. Res. 2014, 13, 307; [Google Scholar]
- 1e. Visconti A., Solfrizzo M., Food Addit. Contam. 1995, 12, 515; [DOI] [PubMed] [Google Scholar]
- 1f. Wang S., Li Q., Jing M., Alba E., Yang X., Sabaté R., Han Y., Pi R., Lan W., Yang X., Chen J., Neurochem. Res. 2016, 41, 1806; [DOI] [PubMed] [Google Scholar]
- 1g. Cui C. B., Kakeya H., Okada G., Onose R., Osada H., J. Antibiot. 1996, 49, 527. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Li S.-M., Nat. Prod. Rep. 2010, 27, 57; [DOI] [PubMed] [Google Scholar]
- 2b. Lindel T., Marsch N., Adla S. K., Top. Curr. Chem. 2012, 309, 67. [DOI] [PubMed] [Google Scholar]
- 3.For selected reviews on aromatic N-heterocycles in drugs, see:
- 3a. Pitt W. R., Parry D. M., Perry B. G., Groom C. R., J. Med. Chem. 2009, 52, 2952; [DOI] [PubMed] [Google Scholar]
- 3b. McGrath N. A., Brichacek M., Njardarson J. T., J. Chem. Educ. 2010, 87, 1348; [Google Scholar]
- 3c. Baumann M., Baxendale I. R., Ley S. V., Nikbin N., Beilstein J. Org. Chem. 2011, 7, 442; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3d. Baumann M., Baxendale I. R., Beilstein J. Org. Chem. 2013, 9, 2265; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3e. Taylor R. D., MacCoss M., Lawson A. D. G., J. Med. Chem. 2014, 57, 5845. [DOI] [PubMed] [Google Scholar]
- 4. Kuzuyama T., Noel J. P., Richard S. B., Nature 2005, 435, 983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.For selected metal-catalyzed cross-coupling reactions, see:
- 5a. Lipshutz B. H., Kim S. K., Mollard P., Blomgren P. A., Stevens K. L., Tetrahedron 1998, 54, 6999; [Google Scholar]
- 5b. Kaiser F., Schmalz H.-G., Tetrahedron 2003, 59, 7345–7355; [Google Scholar]
- 5c. Gerbino D. C., Mandolesi S. D., Schmalz H.-G., Podestá J. C., Eur. J. Org. Chem. 2009, 3964–3972; [Google Scholar]
- 5d. Iwasaki M., Yorimitsu H., Oshima K., Bull. Chem. Soc. Jpn. 2009, 82, 249–253; [Google Scholar]
- 5e. Anderson K., Calo F., Pfaffeneder T., White A. J. P., Barrett A. G. M., Org. Lett. 2011, 13, 5748–5750; [DOI] [PubMed] [Google Scholar]
- 5f. Farmer J. L., Hunter H. N., Organ M. G., J. Am. Chem. Soc. 2012, 134, 17470; [DOI] [PubMed] [Google Scholar]
- 5g. Yang Y., Mustard T. J. L., Cheong P. H. Y., Buchwald S. L., Angew. Chem. Int. Ed. 2013, 52, 14098; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 14348; [Google Scholar]
- 5h. Yang Y., Buchwald S. L., J. Am. Chem. Soc. 2013, 135, 10642; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5i. Chen T.-Y., Krische M. J., Org. Lett. 2013, 15, 2994–2997; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5j. Thomas C., Kataeva O., Schmidt A. W., Knölker H.-J., Org. Biomol. Chem. 2014, 12, 872–875; [DOI] [PubMed] [Google Scholar]
- 5k. Xu L., Liu Z., Dong W., Song J., Miao M., Xu J., Ren H., Org. Biomol. Chem. 2015, 13, 6333; [DOI] [PubMed] [Google Scholar]
- 5l. Zhang Z., Xu L., Chen Z., Liu Z., Miao M., Song J., Ren H., Synlett 2015, 26, 2784; [Google Scholar]
- 5m. Ellwart M., Knochel P., Angew. Chem. Int. Ed. 2015, 54, 10662; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 10808. [Google Scholar]
- 6.For selected Lewis acid catalyzed prenylation, see:
- 6a. Araki S., Manabe S., Butsugan Y., Bull. Chem. Soc. Jpn. 1984, 57, 1433; [Google Scholar]
- 6b. Ramachandra M. S., Subbaraju G. V., Synth. Commun. 2006, 36, 3723; [Google Scholar]
- 6c. Judd K. E., Caggiano L., Org. Biomol. Chem. 2011, 9, 5201; [DOI] [PubMed] [Google Scholar]
- 6d. Jäger S. N., Porta E. O. J., Labadie G. R., Mol. Diversity 2016, 20, 407; [DOI] [PubMed] [Google Scholar]
- 6e. Villani-Gale A. J., Eichman C. C., Eur. J. Org. Chem. 2016, 2925. [Google Scholar]
- 7.For selected Claisen rearrangement induced prenylation, see:
- 7a. Murray R. D. H., Ballantyne M. M., Mathai K. P., Tetrahedron Lett. 1970, 11, 243; [Google Scholar]
- 7b. Takamatsu N., Inoue S., Kishi Y., Tetrahedron Lett. 1971, 12, 4661; [Google Scholar]
- 7c. Murray R. D. H., Sutcliffe M., Hasegawa M., Tetrahedron 1975, 31, 2966; [Google Scholar]
- 7d. Kyogoku K., Hatayama K., Yokomori S., Seki T., Tanaka I., Agric. Biol. Chem. 1975, 39, 667; [Google Scholar]
- 7e. Daskiewicz J. B., Bayet C., Barron D., Tetrahedron Lett. 2001, 42, 7241; [Google Scholar]
- 7f. Gester S., Metz P., Zierau O., Vollmer G., Tetrahedron 2001, 57, 1015; [Google Scholar]
- 7g. Mali R. S., Joshi P. P., Sandhu P. K., Manekar-Tilve A. J., Chem. Soc. Perkin Trans. 1 2002, 371; [Google Scholar]
- 7h. Kawamura T., Hayashi M., Mukai R., Terao J., Nemoto H., Synthesis 2012, 44, 1308–1314. [Google Scholar]
- 8.For selected metal-catalyzed C−H prenylation reactions, see:
- 8a. Zhang Y. J., Skucas E., Krische M. J., Org. Lett. 2009, 11, 4248; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8b. Zeng R., Fu C. J., Ma S., J. Am. Chem. Soc. 2012, 134, 9597; [DOI] [PubMed] [Google Scholar]
- 8c. Wang H., Schröder N., Glorius F., Angew. Chem. Int. Ed. 2013, 52, 5386; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 5495; [Google Scholar]
- 8d. Cong X., Li Y., Wei Y., Zeng X., Org. Lett. 2014, 16, 3926; [DOI] [PubMed] [Google Scholar]
- 8e. Kim M., Sharma S., Mishra N. K., Han S., Park J., Kim M., Shin Y., Kwak J. H., Han S. H., Kim I. S., Chem. Commun. 2014, 50, 11303; [DOI] [PubMed] [Google Scholar]
- 8f. Kuhl N., Schröder N., Glorius F., Adv. Synth. Catal. 2014, 356, 1443; [Google Scholar]
- 8g. Suzuki Y., Sun B., Sakata K., Yoshino T., Matsunaga S., Kanai M., Angew. Chem. Int. Ed. 2015, 54, 9944; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 10082; [Google Scholar]
- 8h. Bunno Y., Murakami N., Suzuki Y., Kanai M., Yoshino T., Matsunaga S., Org. Lett. 2016, 18, 2216; [DOI] [PubMed] [Google Scholar]
- 8i. Jo H., Han S., Park J., Choi M., Han S. H., Jeong T., Lee S. Y., Kwak J. H., Jung Y. H., Kim I. S., Tetrahedron 2016, 72, 571; [Google Scholar]
- 8j. Kumar G. S., Kapur M., Org. Lett. 2016, 18, 1112; [DOI] [PubMed] [Google Scholar]
- 8k. Liu W., Richter S. C., Zhang Y., Ackermann L., Angew. Chem. Int. Ed. 2016, 55, 7747; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 7878. [Google Scholar]
- 9.
- 9a. Hesse R., Kataeva O., Schmidt A. W., Knölker H.-J., Chem. Eur. J. 2014, 20, 9504–9509; [DOI] [PubMed] [Google Scholar]
- 9b. Hesse R., Schmidt A. W., Knölker H.-J., Tetrahedron 2015, 71, 3485–3490. [Google Scholar]
- 10.For a review, see:
- 10a. Duncton M. A., J. Med. Chem. Commun. 2011, 2, 1135. For representative examples, see: [Google Scholar]
- 10b. Minisci F., Vismara E., Fontana F., Heterocycles 1989, 28, 489; [Google Scholar]
- 10c. Seiple I. B., Su S., Rodriguez R. A., Gianatassio R., Fujiwara Y., Sobel A. L., Baran P. S., J. Am. Chem. Soc. 2010, 132, 13194; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10d. Molander G. A., Colombel V., Braz V. A., Org. Lett. 2011, 13, 1852; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10e. Fujiwara Y., Domingo V., Seiple I. B., Gianatassio R., Del Bel M., Baran P. S., J. Am. Chem. Soc. 2011, 133, 3292; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10f. Presset M., Fleury-Bregeot N., Oehlrich D., Romobouts F., Molander G. A., J. Org. Chem. 2013, 78, 4615; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10g. Antonchick A. P., Burgmann L., Angew. Chem. Int. Ed. 2013, 52, 3267; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 3349; [Google Scholar]
- 10h. O'Hara F., Blackmond D. G., Baran P. S., J. Am. Chem. Soc. 2013, 135, 12122; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10i. DiRocco D. A., Dykstra K., Krska S., Vachal P., Conway D. V., Tudge M., Angew. Chem. Int. Ed. 2014, 53, 4802; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 4902; [Google Scholar]
- 10j. Jin J., MacMillan D. W. C., Nature 2015, 525, 87; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10k. Jin J., MacMillan D. W. C., Angew. Chem. Int. Ed. 2015, 54, 1565; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 1585. [Google Scholar]
- 11.
- 11a. Brill Z. G., Grove H. K., Maimone T. J., Science 2016, 352, 1078; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11b. Chu X. Q., Meng H., Zi Y., Xu X. P., Ji S. J., Chem. Eur. J. 2014, 20, 17198; [DOI] [PubMed] [Google Scholar]
- 11c. Peng H., Yu J. T., Jiang Y., Cheng J., Org. Biomol. Chem. 2015, 13, 10299. [DOI] [PubMed] [Google Scholar]
- 12. Yuan L., Huang W., Zhou K., Wang Y., Dong W., Du G., Gao X., Ma Y., Hu Q., Nat. Prod. Res. 2015, 29, 1914. [DOI] [PubMed] [Google Scholar]
- 13. Bungard C. J., Hartman G. D., Manikowski J. J., Perkins J. J., Bai C., Brandish P. E., Euler D. H., Hershey J. C., Schmidt A., Fang Y., Norcross R. T., Rushmore T. H., Thompson C. D., Meissner R. S., Bioorg. Med. Chem. 2011, 19, 7374. [DOI] [PubMed] [Google Scholar]
- 14. Hsiang Y. H., Hertzberg R., Hecht S., Liu L. F., J. Biol. Chem. 1985, 260, 14873. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary