

Abstract
Objective
Recent sequencing studies of head and neck squamous cell carcinomas (HNSCCs) have identified the phosphatidylinositol 3-kinase (PI3K) pathway as the most frequently mutated, oncogenic pathway in this cancer type. Despite the frequency of activating genomic alterations in PIK3CA (the gene encoding the catalytic subunit of PI3K, targeted inhibitors of PI3K have not shown clinical efficacy as monotherapies. We hypothesized that co-dependent pathways, including the Ras-MEK-ERK pathway, may still be functional in the presence of PI3K inhibitors and might serve as mediators of this resistance.
Methods
We assessed the hypothesis using resazurin cell viability and trypan blue exclusion assays. We also used Western blot to characterize Ras-MEK-ERK pathway activity.
Study Design
We evaluated this hypothesis in six PIK3CA-amplified, PI3K inhibitor-resistant HNSCC cell lines following treatment with pan and alpha-isoform selective PI3K inhibitors (BKM120 and HS-173 respectively). We also tested the effect of combination treatment with PI3K inhibitor HS-173 and MEK inhibitor trametinib or EGFR inhibitor gefitinib.
Results
Our results displayed maintenance of Ras-MEK-ERK pathway activity in 4 of 6 HNSCC cell lines after PI3K inhibitor treatment. We also found that UM-SCC-69 and UM-SCC-108 cells display synergistic responses to dual therapy.
Conclusion
This study suggests that inhibition of the PI3K and Ras-MEK-ERK pathways might be effective in some HNSCC patients; however, it also prompts the study of additional resistance mechanisms to identify synergistic combination therapies for tumors resistant to these di-therapies.
Keywords: Head and Neck Cancer, PIK3CA, Targeted Therapy, Personalized Medicine, Compensatory Resistance
Introduction
Head and neck squamous cell carcinoma (HNSCC) is the sixth most common form of cancer by incidence worldwide and represents ~3% of cancer cases in the United States. Common modalities used to treat HNSCC patients include surgery, radiation, and cytotoxic chemotherapy. Despite the use of these regimens, however, patient prognosis is poor and recurrence and metastasis are very common. Five year survival rates for HNSCC are only 40-50% and have remained unchanged for many years 1.
An increased understanding of the genetic alterations in HNSCC may guide the use of targeted therapies and improve patient survival 2. Recent sequencing studies have identified mutations and copy number changes in genes within a number of cellular pathways in HNSCCs. These genetic changes may contribute to cancer development or progression, and learning more about them will guide personalized medicine protocols for this cancer type, which seek to match patients with an effective treatment option given the specific genetic signature of their disease 3-6. Among the most comprehensive genetic analyses of HNSCCs is the The Cancer Genome Atlas (TCGA) dataset, which profiles somatic mutations and copy number alterations in 279 HNSCC patients. This study and others have identified the phosphatidylinositol 3-kinase (PI3K) pathway as the most frequently mutated, oncogenic targetable pathway in this cancer type 7, 8. Of the genes in the PI3K pathway, PIK3CA, which encodes the catalytic subunit and alpha isoform of PI3K, is the most frequently altered. Activating mutation and/or amplification of this gene were observed in 36.9% of the TCGA HNSCC cohort 7. These aberrations lead to increased cell growth and viability, may drive tumor progression, and are more commonly observed in advanced stage disease 9, 10.
Inhibitors of PI3K have been developed and tested in clinical trials for HNSCC as well as other cancer types. Despite frequent activation of the PI3K pathway and its importance in HNSCC; however, these therapies have shown limited efficacy in unmatched clinical trials to date. In one recent trial, for example, HNSCC patients were given docetaxel alone or in combination with PX-866, an oral pan-PI3K inhibitor. The addition of PX-866 did not improve progression free survival in this cohort 11.
Resistance to treatment with PI3K inhibitors is poorly understood, although some data has suggested that these compounds are more effective in patients with alterations in PIK3CA or loss of PTEN. (PTEN is a tumor suppressor gene that acts as a “brake” on PI3K function and is inactivated in 10% of HNSCC patients according to TCGA data.) While an analysis of early clinical trials for PI3K inhibitors showed that PI3K altered patients were more responsive to PI3K inhibitors than patients without PIK3CA mutation or PTEN loss, this study also indicated only an 18% overall response rate within the PI3K altered molecular subgroup 12. These findings suggest that important resistance mechanisms to PI3K inhibitors are frequently present, even in PI3K altered HNSCCs.
PI3K inhibitor resistance may be due to activation of a compensatory pathway, which cells utilize to grow and divide even in the absence of PI3K signaling. The Ras-MEK-ERK pathway, as an important contributor to cell proliferation and growth, is a likely candidate for codependence in cases of PI3K inhibitor resistance. Previous studies have demonstrated that PI3K and MEK inhibitors are synergistic in some HNSCCs 13-15, as well as in a variety of other cancer types 16-19. In addition, based on preclinical evidence and frequent genetic alterations in HNSCC, trials for pan PI3K inhibitor BKM120 and alpha-isoform specific PI3K inhibitor BYL719 are ongoing (examples include: NCT02537223, NCT02051751 and NCT01602315). These agents are being tested in patients not only as monotherapies but also in combination with anti-EGFR antibody cetuximab. Inhibiting a receptor tyrosine kinase such as EGFR blocks Ras-MEK-ERK signaling and has shown efficacy in other cancer models 20. However, the specific patients that are responsive to mono- and combination therapies cannot currently be identified—each patient's tumor has a unique genetic signature and there is to date a lack of useful biomarkers to stratify and predict responses to treatment with PI3K inhibitor combination therapies.
In this study, we explore the sensitivity of several models with PIK3CA genetic alterations to combination therapies being considered for HNSCC personalized medicine trials. We sought to identify the relationships between drug sensitivity and resistance mechanisms in these models in order to begin to understand what percentages of patients would respond to each proposed combination therapy. We examined activation of the Ras-MEK-ERK pathway as a mechanism for resistance to PI3K inhibitors in PI3K altered HNSCC. To do this, we tested six HNSCC cell lines, each of which displayed both amplification of PIK3CA and resistance to PI3K inhibitor monotherapy treatment, for compensation through this pathway in the presence of PI3K inhibitors.
Materials and Methods
Cell Culture
UM-SCC cells (University of Michigan) are derived from human head and neck squamous cell carcinoma patient tumor samples and were cultured in a humidified incubator at 37 °C with 5% (vol/vol) CO2 as previously described 21. Cells were cultured in DMEM with 10% FBS, 1X Pen/Strep, 1X NEAA. Details of DNA copy number analysis are being submitted as a separate manuscript. All cell lines were confirmed to contain wild type PIK3CA as previously reported from Nimblegen V2 exome capture based experiments 22.
Chemicals
BKM120, HS-173, trametinib, and gefitinib were purchased from Selleck Chemicals. All compounds were initially dissolved in 100% DMSO to 10 mM and then diluted to the indicated concentrations for studies in vitro.
Western Blotting
Cells at 70-80% confluency were treated with DMSO or inhibitor(s) for six hours prior to harvesting and lysing in radioimmunoprecipitation assay buffer. Ten micrograms of each cell harvest was used, and standard Western blot protocols were followed as previously described 23. Primary antibodies against pERK1/2 (Thr202/Tyr204) (1:1000, catalog No. 4370; Cell Signaling Technology), ERK (1:1000, catalog No. 4695; Cell Signaling Technology), pAKT (Ser473) (1:1000, catalog No. 4060; Cell Signaling Technology), AKT (1:1000, catalog No. 4685; Cell Signaling Technology), and HSP90 (1:2000, catalog No. 4877; Cell Signaling Technology) were incubated overnight at 4°C or for 1 hour at room temperature, followed by a goat anti-rabbit horseradish peroxidase (catalog No. 111-035-045; Jackson ImmunoResearch) secondary antibody at room temperature for 1 hour. The blots were then visualized with chemiluminescence and imaged. 300dpi or greater images were retained from all Westerns and representative blots are shown.
Trypan Blue Assay
To test for cell membrane integrity and access cell viability, 32,000 cells per well were seeded into 24-well cell culture plates. After 24 hours, cells were exposed to DMSO or inhibitor in a multipoint dose-response. After 72 hour exposure, cells were disaggregated in 50 μL of medium.10 μL of the suspension was mixed with 10 μL of trypan blue (0.4% Invitrogen) and viability and total cell count were measured using Countess Automated Cell Counter (Invitrogen).
Resazurin Assay
To study relative cell viability, 2,000 cells per well were seeded (in 50 μL volume) in 84-well microplates using a Multiflo liquid handling dispensing system. After 24 hours, cells were treated with inhibitor or DMSO in a 10-point two-fold dilution series in quadruplicate. 96-well plates were prepared with inhibitors in 200X concentration and then diluted to 10X concentration in media in a second 96-well plate using the Agilent Bravo Automated Liquid Handling Platform and VWorks Automation Control Software. These inhibitors were then used to treat the cells with the desired compound concentration, again using liquid handling robotics. Cells were stained with 10 μL of 440 μM resazurin (Sigma) in PBS for 12-24 hours before fluorescent signal intensity was quantified. Quantification occured 72 hours after treatment using the Cytation3 fluorescence plate reader at excitation and emission wavelengths of 540 and 612 nM, respectively.
Statistical Analysis
Statistical analyses were performed using GraphPad Prism 6 software. Unpaired, two-tailed Student's t-tests were conducted to compare total cell counts following DMSO and 1 μM trametinib treatment from trypan blue exclusion assays with p<0.05 considered statistically significant. For resazurin assays, IC50 values were determined from the mean and standard deviation of at least quadruplicate measurements for each treatment and cell line.
Results
PIK3CA Alteration in HNSCC Cell Lines
We first identified a panel of cell lines that displayed amplification of PIK3CA to comprise our HNSCC panel. Copy number amplification for the six cell lines ranged from 2.67 to 6, with UM-SCC-69 and -108 exhibiting the highest level of amplification with 6 and 4 copies of PIK3CA, respectively. UM-SCC-2 and -14A had lower levels of PIK3CA amplification, each with 2.67 copies of the gene as shown in Figure 1A. None of the six cell lines displayed mutations in PIK3CA 22.
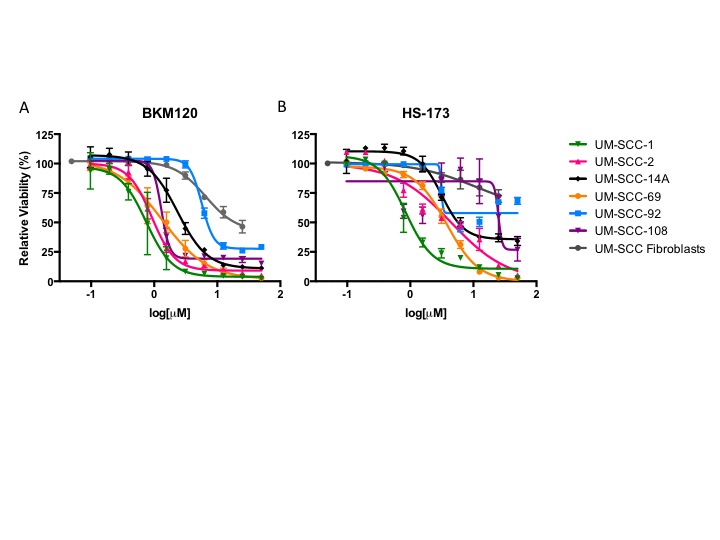
Figure 1.
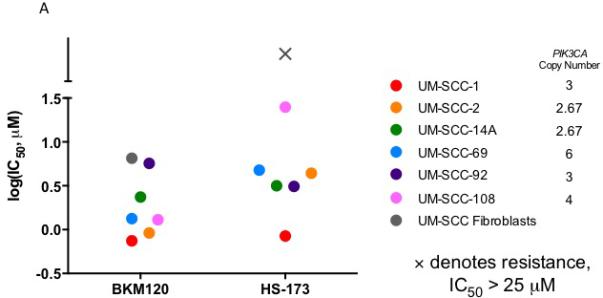
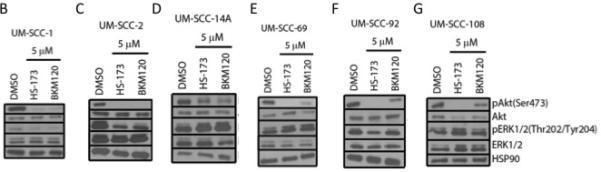
IC50 via resazurin assay (A) and Western blot analysis of PI3K and Ras-MEK-ERK pathway activation in UM-SCC-1 (B), 2 (C), 14A (D), 69 (E), 92 (F), and 108 (G) following treatment with HS-173 or BKM120.
PIK3CA Amplified HNSCC Sensitivity to PI3K Inhibitors
To then determine the sensitivity of the PIK3CA amplified HNSCC cell lines in our panel to PI3K inhibition, we used a resazurin cell viability assay to determine the IC50 value for each cell line in response to alpha-isoform specific PI3K inhibitor HS-173 and the pan PI3K inhibitor BKM120. We identified similar sensitivity to these agents, consistent with common alterations in PIK3CA and the important role of the alpha isoform in HNSCC. UM-SCC-1 cells were the most sensitive, with IC50 less than 1 μM for both inhibitors. UM-SCC-108 displayed the greatest resistance to a PI3K inhibitor, with an IC50 close to 25 μM for HS-173 (Figure 1A, S1). Despite the moderate responses to these treatments, IC50 values of these magnitudes are indicative of at least partial PI3K inhibitor resistance, suggesting the opportunity to assess combination therapies that are being advanced for clinical trials in these models.
PI3K inhibitor resistant HNSCC cell lines display differential activation of the Ras-MEK-ERK pathway
Thus, to learn more about the mechanisms of resistance of PI3K inhibitors in HNSCC, we first examined the activation of downstream PI3K and Ras-MEK-ERK pathway members after PI3K inhibitor treatment in each of the cell lines in our panel. We hypothesized that Ras-MEK-ERK pathway activity would be maintained if the pathway represents an important compensatory mechanism. We treated each cell line with 5 μM HS-173 or BKM120 and used Western blotting to assess phosphorylated and total levels of Akt and ERK (Figure 1B-G). In each cell line, Akt phosphorylation was reduced after treatment with HS-173 or BKM120, consistent with the effect of these drugs on downstream members of the PI3K pathway. In the UM-SCC-1 and UM-SCC-2 cells, phosphorylation of ERK was also reduced by 5 μM PI3K inhibitor treatment (Figure 1B, C). In the other four cell lines, ERK phosphorylation was maintained (Figure 1D-G). This maintenance of Ras-MEK-ERK pathway activity suggests that this pathway may be a co-dependent with the PI3K pathway in the UM-SCC-14A, 69, 92, and 108 cells.
Potential Synergy of PI3K and MEK Inhibitors in HNSCC
To explore the hypothesis that MEK signaling maintains viability in the presence of PIK3CA inhibition in some models, we then tested the combination of PI3K inhibitor HS-173 and MEK inhibitor trametinib in the panel of HNSCC cells. If Ras-MEK-ERK pathway activation is a mechanism of resistance to PI3K inhibitor treatment, adding trametinib as a second inhibitor might sensitize the cells to the inhibitors and result in a synergistic reduction in cell viability. Using a resazurin cell viability assay, we observed at synergistic effects of these two agents in UM-SCC-69 and UM-SCC-108 cells (Figure 2). Lack of benefit from the combination with MEK inhibitor in other HNSCC cell lines was not due to the inability of trametinib to block downstream Ras-MEK-ERK pathway activity, as 5 μM treatment resulted in complete inhibition of ERK phosphorylation via Western blotting (Figure 2, insets). We also performed trypan blue exclusion assays to examine the effect of this drug concentration on absolute cell viability in the two cell lines in which PI3K and MEK inhibitors displayed greater than additive effects (Figure 3). Trypan blue exclusion assays demonstrated that while trametinib monotherapy inhibited cell proliferation, it did not reduce cell viability (p<0.05). Combining PI3K and MEK inhibitors led to further reductions in cell viability.
Figure 2.

Cell viability via resazurin assay and Ras-MEK-ERK pathway activation via Western blot analysis (insets) for UM-SCC-1 (A), 2 (B), 14A (C), 69 (D), 92 (E), and 108 (F) after treatment with trametinib and/or HS-173.
Figure 3.
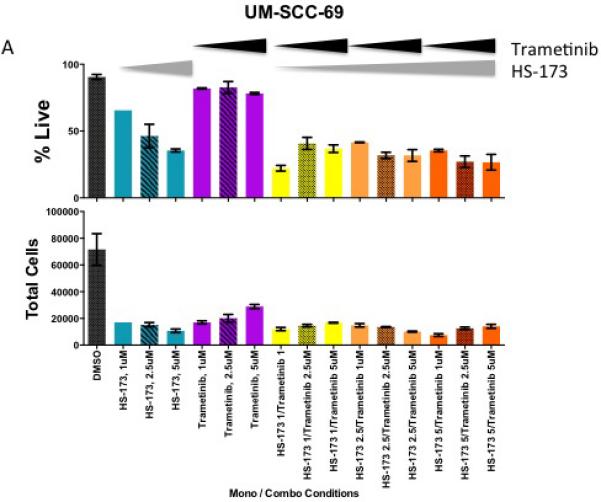

Live and total UM-SCC-69 (A) and UM-SCC-108 (B) cells after 72 hour treatment with increasing concentrations of trametinib and/or HS-173, as measured using a trypan blue exclusion assay.
Potential Synergy of PI3K and EGFR Inhibitors in HNSCC
Ras-MEK-ERK pathway activity can also be inhibited more broadly using an agent targeted against a receptor tyrosine kinase such as the epidermal growth factor receptor (EGFR). EGFR is frequently amplified in HNSCC and cetuximab, a monoclonal EGFR antibody is the only FDA-approved targeted therapy for this cancer type. Currently, several clinical trials are exploring PI3K inhibitors in combination with cetuximab. We hypothesized that blocking EGFR and the downstream Ras-MEK-ERK pathway in combination with PI3K might be a more effective treatment than using a PI3K and MEK inhibitor combination in some models. To explore this possibility, we treated the cell lines displaying Ras-MEK-ERK pathway activity after PI3K inhibitor treatment (UM-SCC-14A, 69, 92, and 108) with HS-173 in combination with gefitinib, a small molecule inhibitor of EGFR. Results of a resazurin cell viability assay for HS-173 in combination with gefitinib again indicated potential synergy in UM-SCC-69, but not in UM-SCC-14A, 92, or 108 (Figure 4). We also treated these four cell lines with vehicle, 1 μM gefitnib, 1 μM HS-173, or a combination of the inhibitors at 1 μM each. Western blot analysis of each cell line indicated that ERK phosphorylation was reduced by gefitinib treatment and Akt phosphorylation was reduced by HS-173 treatment. These results are consistent with the effects of these inhibitors on downstream members of the Ras-MEK-ERK and PI3K pathways, respectively. Treatment with both drugs led to decreased phosphorylation of both ERK and Akt, indicating that this combination caused the inhibition of expected targets (Figure 5).
Figure 4.

Cell viability for UM-SCC-14A (A), −69 (B), −92 (C), and −108 (D) after 72 hour treatment with increasing concentrations of gefitinib and/or HS-173, as measured using a resazurin assay.
Figure 5.
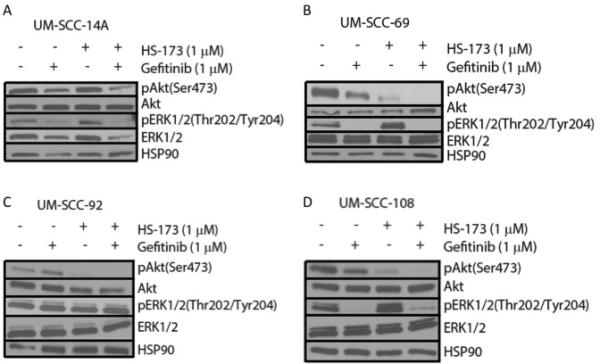
Western blot analysis of downstream PI3K and RAS-MEK-ERK pathway activation in UM-SCC-14A (A), −69 (B), −92 (C), and −108 (D) following 6 hour treatment with 1 μM gefitinib and/or 1 μM HS-173.
Discussion
In summary, our PIK3CA amplified HNSCC cell lines show intermediate to strong resistance to PI3K inhibitors suggesting that matched PI3K targeted therapies may not be effective as monotherapies in this cancer type. While some of the more sensitive UM-SCC cell lines in our panel display similar sensitivity to PI3K inhibitors as compared to PIK3CA mutant or PTEN deleted cancer cell lines with IC50 of approximately 1 μM 24, 25, other resistant models (particularly UM-SCC-92) have IC50 values closer those of fibroblast cells. This data is consistent with a phase 1/2 study of PX-866 and docetaxel in patients with solid tumors (NCT01204099), in which only a few HNSCC patients with PIK3CA amplification responded to the monotherapy despite pharmacodynamics experiments showing on target effect of the drug 11. As many groups have postulated that compensation occurs through either EGFR signaling or directly though alternative pathways activating Ras-MEK-ERK signaling, we also assessed response to dual inhibitor therapies targeting PIK3CA and EGFR or MEK/ERK signaling. These experiments showed 2/6 (33%) of the cell lines displayed additive to synergistic effects of alpha-isoform specific PI3K inhibitor HS-173 and MEK inhibitor trametinib or EGFR inhibitor gefitinib; there was no benefit of the addition of the Ras-MEK-ERK inhibitor in the other models. This data is promising and supports the preclinical data for combination trials simultaneously inhibiting both of these pathways 13-15. Our data predict that these treatments will lead to response in a subset of tumors, but also suggests that additional unknown compensatory mechanisms are driving PIK3CA inhibitor resistance in other HNSCCs.
Thus, the data presented here suggest that multiple different pathways drive PI3K inhibitor resistance and additional work is needed to understand the frequencies with which each pathway is utilized and the biomarkers predicting which combination therapy would most benefit individual patients. While we didn't fully assess all combinations that have been suggested in the literature, our data provides the foundation for future studies in HNSCC that leverage unbiased approaches. Further testing of additional HNSCC cell lines, might identify other PIK3CA amplified models with more significant responses to PI3K monotherapy or to combinations that are advancing in clinical trials. Likewise, systematic discovery based approaches to identify novel combinations that inhibit the growth of models resistant to both the Ras-MEK-ERK + PI3K and EGFR + PI3K therapies are needed to understand the additional pathways driving resistance. These studies might also improve cell kill in an even more complex therapeutic setting (e.g. tri-therapy or cycled di-therapies). For example, exploring other PIK3CA amplified or mutated HNSCC models might allow us to stratify responses based on additional genetic alterations in the PI3K, Ras-MEK-ERK, and other cellular pathways. We could then assess genetic biomarkers (personalized medicine) to predict which patients might be most and least sensitive to a specific PI3K-based combination regimen. Indeed, focusing on developing therapies for the most highly recurrent compensatory pathways may be one approach to improving therapy.
We have entered an exciting time in the HNSCC field. Several institutions have initiated personalized medicine protocols, such as the MiOTOseq trial, which aim to characterize the molecular genetics of every consenting HNSCC patient that enters the clinic. These studies are likely to lead to the development of complex genetic databases and, hopefully, increase the enrollment of HNSCC patients on appropriate interventional trials. As noted above, personalized trials have increased the overall survival of patients with HNSCC 26, 27, but the overall rates of improvement have been underwhelming. As the field moves forward, we need to begin to understand how HNSCCs respond to matched targeted therapies in order to take the next step towards improving overall response. Here, we focused on understanding the resistance pathways to inhibitors of the most recurrently altered oncogenic pathway in HNSCC. Our data indicates a complex and differential response to matched therapy; it also suggests the value of future work utilizing unbiased approaches to nominate co-dependent pathways driving this resistance. Developing an improved understanding of resistance to matched therapies in HNSCC as well as the frequencies with which each resistance mechanism is observed represents one key step to improving the overall survival of patients enrolling in personalized medicine trials.
Supplementary Material
{kind=link}
Acknowledgments
Grant Support: Dr. J. Chad Brenner received funding from NIH Grants U01DE025184 and P30:CA046592 S1. Nicole Michmerhuizen was supported by the NSF Graduate Research Fellowship Program.
References
- 1.Massano J, Regateiro FS, Januario G, Ferreira A. Oral squamous cell carcinoma: review of prognostic and predictive factors. Oral Surg. Oral Med. Oral Pathol. 2006;102(1):67–76. doi: 10.1016/j.tripleo.2005.07.038. [DOI] [PubMed] [Google Scholar]
- 2.Ludwig ML, Birkeland AC, Hoesli R, Swiecicki P, Spector ME, Brenner JC. Changing the paradigm: the potential for targeted therapy in laryngeal squamous cell carcinoma. Cancer Biol Med. 2016;13(1):87–100. doi: 10.28092/j.issn.2095-3941.2016.0010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Birkeland AC, Ludwig ML, Meraj TS, Brenner JC, Prince ME. The Tip of the Iceberg: Clinical Implications of Genomic Sequencing Projects in Head and Neck Cancer. Cancers (Basel) 2015;7(4):2094–109. doi: 10.3390/cancers7040879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Birkeland AC, Uhlmann WR, Brenner JC, Shuman AG. Getting personal: Head and neck cancer management in the era of genomic medicine. Head Neck. 2015 doi: 10.1002/hed.24132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Birkeland AC, Brenner JC. Personalizing medicine in head and neck squamous cell carcinoma: The rationale for combination therapies. Med Res Arch. 2015:3. doi: 10.18103/mra.v0i3.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tillman BNYM, Birkeland AC, Liu C, Hovelson DH, Cani AK, Palanisamy N, Carskadon S, Carey TE, Bradford CR, Tomlins S, McHugh JB, Spector ME, Brenner JC. Targeted sequencing of an epidemiologically low risk patient defines Fibroblast Growth Factor Receptor family aberrations as a putative driver of head and neck squamous cell carcinoma. Head Neck. 2015 doi: 10.1002/hed.24292. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. 2015;517(7536):576–82. doi: 10.1038/nature14129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lui VW, Hedberg ML, Li H, et al. Frequent mutation of the PI3K pathway in head and neck cancer defines predictive biomarkers. Cancer Discov. 2013;3(7):761–9. doi: 10.1158/2159-8290.CD-13-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Osaki M, Oshimura M, Ito H. PI3K-Akt pathway: its functions and alterations in human cancer. Apoptosis. 2004;9(6):667–76. doi: 10.1023/B:APPT.0000045801.15585.dd. [DOI] [PubMed] [Google Scholar]
- 10.Isaacsson Velho PH, Castro G, Jr., Chung CH. Targeting the PI3K Pathway in Head and Neck Squamous Cell Carcinoma. Am Soc Clin Oncol Educ Book. 2015:123–8. doi: 10.14694/EdBook_AM.2015.35.123. [DOI] [PubMed] [Google Scholar]
- 11.Jimeno A, Bauman JE, Weissman C, et al. A randomized, phase 2 trial of docetaxel with or without PX-866, an irreversible oral phosphatidylinositol 3-kinase inhibitor, in patients with relapsed or metastatic head and neck squamous cell cancer. Oral Oncol. 2015;51(4):383–8. doi: 10.1016/j.oraloncology.2014.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Janku F, Hong DS, Fu S, Piha-Paul SA, Naing A, Falchook GS, et al. Assessing PIK3CA and PTEN in early-phase trials with PI3K/AKT/mTOR inhibitors. Cell Rep. 2014;6(2):377–87. doi: 10.1016/j.celrep.2013.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wirtz ED, Hoshino D, Maldonado AT, Tyson DR, Weaver AM. Response of head and neck squamous cell carcinoma cells carrying PIK3CA mutations to selected targeted therapies. JAMA Otolaryngol Head Neck Surg. 2015;141(6):543–9. doi: 10.1001/jamaoto.2015.0471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mohan S, Vander Broek R, Shah S, et al. MEK Inhibitor PD-0325901 Overcomes Resistance to PI3K/mTOR Inhibitor PF-5212384 and Potentiates Antitumor Effects in Human Head and Neck Squamous Cell Carcinoma. Clin Cancer Res. 2015;21(17):3946–56. doi: 10.1158/1078-0432.CCR-14-3377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mazumdar T, Byers LA, Ng PK, et al. A comprehensive evaluation of biomarkers predictive of response to PI3K inhibitors and of resistance mechanisms in head and neck squamous cell carcinoma. Mol Cancer Ther. 2014;13(11):2738–50. doi: 10.1158/1535-7163.MCT-13-1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Inaba K, Oda K, Aoki K, et al. Synergistic antitumor effects of combination PI3K/mTOR and MEK inhibition (SAR245409 and pimasertib) in mucinous ovarian carcinoma cells by fluorescence resonance energy transfer imaging. Oncotarget. 2016 doi: 10.18632/oncotarget.8807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sunayama J, Matsuda K, Sato A, et al. Crosstalk between the PI3K/mTOR and MEK/ERK pathways involved in the maintenance of self-renewal and tumorigenicity of glioblastoma stem-like cells. Stem Cells. 2010;28(11):1930–9. doi: 10.1002/stem.521. [DOI] [PubMed] [Google Scholar]
- 18.Xing Y, Hogge DE. Combined inhibition of the phosphoinosityl-3-kinase (PI3Kinase) P110delta subunit and mitogen-extracellular activated protein kinase (MEKinase) shows synergistic cytotoxicity against human acute myeloid leukemia progenitors. Leuk Res. 2013;37(6):697–704. doi: 10.1016/j.leukres.2013.03.003. [DOI] [PubMed] [Google Scholar]
- 19.Ayub A, Yip WK, Seow HF. Dual treatments targeting IGF-1R, PI3K, mTORC or MEK synergize to inhibit cell growth, induce apoptosis, and arrest cell cycle at G1 phase in MDA-MB-231 cell line. Biomed Pharmacother. 2015;75:40–50. doi: 10.1016/j.biopha.2015.08.031. [DOI] [PubMed] [Google Scholar]
- 20.Serra V, Scaltriti M, Prudkin L, et al. PI3K inhibition results in enhanced HER signaling and acquired ERK dependency in HER2-overexpressing breast cancer. Oncogene. 2011;30(22):2547–57. doi: 10.1038/onc.2010.626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brenner JC, Graham MP, Kumar B, et al. Genotyping of 73 UM-SCC head and neck squamous cell carcinoma cell lines. Head Neck. 2010;32(4):417–26. doi: 10.1002/hed.21198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu J, Pan S, Hsieh MH, et al. Targeting Wnt-driven cancer through the inhibition of Porcupine by LGK974. Proc Natl Acad Sci U S A. 2013;110(50):20224–9. doi: 10.1073/pnas.1314239110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Birkeland AC, Yanik M, Tillman BN, et al. Identification of Targetable ERBB2 Aberrations in Head and Neck Squamous Cell Carcinoma. JAMA Otolaryngol Head Neck Surg. 2016 doi: 10.1001/jamaoto.2016.0335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mueller A, Bachmann E, Linnig M, et al. Selective PI3K inhibition by BKM120 and BEZ235 alone or in combination with chemotherapy in wild-type and mutated human gastrointestinal cancer cell lines. Cancer Chemother Pharmacol. 2012;69(6):1601–15. doi: 10.1007/s00280-012-1869-z. [DOI] [PubMed] [Google Scholar]
- 25.Koul D, Fu J, Shen R, et al. Antitumor activity of NVP-BKM120--a selective pan class I PI3 kinase inhibitor showed differential forms of cell death based on p53 status of glioma cells. Clin Cancer Res. 2012;18(1):184–95. doi: 10.1158/1078-0432.CCR-11-1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tsimberidou AM, Wen S, Hong DS, et al. Personalized medicine for patients with advanced cancer in the phase I program at MD Anderson: validation and landmark analyses. Clin Cancer Res. 2014;20(18):4827–36. doi: 10.1158/1078-0432.CCR-14-0603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chau NG, Li YY, Jo VY, et al. Incorporation of Next-Generation Sequencing into Routine Clinical Care to Direct Treatment of Head and Neck Squamous Cell Carcinoma. Clin Cancer Res. 2016 doi: 10.1158/1078-0432.CCR-15-2314. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.