

Abstract
The effects of Notch signaling are context-dependent and both oncogenic and tumor-suppressive functions have been described. Notch signaling in melanoma is considered oncogenic, but clinical trials testing Notch inhibition in this malignancy have not proved successful. Here, we report that expression of the constitutively active intracellular domain of Notch4 (N4ICD) in melanoma cells triggered a switch from a mesenchymal-like parental phenotype to an epithelial-like phenotype. The epithelial-like morphology was accompanied by strongly reduced invasive, migratory, and proliferative properties concomitant with the downregulation of epithelial–mesenchymal transition markers Snail2 (SNAI2), Twist1, vimentin (VIM), and MMP2 and the reexpression of E-cadherin (CDH1). The N4ICD-induced phenotypic switch also resulted in significantly reduced tumor growth in vivo. Immunohistochemical analysis of primary human melanomas and cutaneous metastases revealed a significant correlation between Notch4 and E-cadherin expression. Mechanistically, we demonstrate that N4ICD induced the expression of the transcription factors Hey1 and Hey2, which bound directly to the promoter regions of Snail2 and Twist1 and repressed gene transcription, as determined by EMSA and luciferase assays. Taken together, our findings indicate a role for Notch4 as a tumor suppressor in melanoma, uncovering a potential explanation for the poor clinical efficacy of Notch inhibitors observed in this setting.
Introduction
During recent years, emerging evidence suggests that Notch signaling plays an important role in the development and progression of several cancers including melanoma (1–4). The majority of these studies report an oncogenic function of Notch1 while a thorough understanding of other Notch receptors is still sparse. Considering the profound evidence about the oncogenic role of Notch in melanoma, a phase II trial using broad Notch signaling inhibitors surprisingly only showed minimal clinical activity against metastatic melanoma (5). Therefore, more in-depth knowledge about the detailed functions of Notch signaling in melanoma is required to assess the potential of Notch as a therapeutic target. In this study, we report a novel tumor-suppressive function of Notch4 in melanoma by triggering a mesenchymal–epithelial–like transition that leads to a low motile, proliferative, and invasive phenotype that shows reduced tumorigenicity in vivo.
Materials and Methods
Cell culture
The primary human melanoma cell line WM35, and metastatic cell lines WM9, WM164, and 451Lu were kindly provided by Dr. Meenhard Herlyn (The Wistar Institute, Philadelphia, PA) and grown in RPMI1640 medium (Sigma-Aldrich), supplemented with 2% FBS (PPA) and 2% L-glutamine (PPA). All cells were maintained at 37° C in a humidified atmosphere containing 5% CO2. Cells were harvested for individual experiments after washing with PBS (pH 7.4; Life Technologies) using trypsin (Life Technologies). Cell lines received in 2005 were tested for authenticity in 2011 (WM9 and WM35), 2012 (WM164), and 2014 (451Lu) using short tandem repeat (STR) genotyping.
In vivo study
Female mutant inbred, 3- to 8-week-old C.B-17/IcrHanHsdArcPrkdcscid mice were purchased from the Animal Resources Centre (Canning Vale, Australia) and randomly separated into two groups of 5 mice and injected with 1 × 106 451Lu cells transduced with pLNCX-empty vector or pLNCX-N4ICD. Tumor growth was monitored twice weekly for 4 weeks, measured using a caliper. Tumor volume was calculated according to the formula: (width × length × height)/2.
Plasmid and siRNA transfection
Transfection experiments were performed as described previously (6) using 150 pmol siRNAs or 2 μg of plasmid DNA (Supplementary Table S1).
Viral vector production and transduction
Experiments were performed as described previously (6) using vectors pLNCX-empty vector and pLNCX-N4ICD.
Immunoblotting
Immunoblotting was performed as described previously (6) using specific primary and corresponding peroxidase-conjugated secondary antibodies (Supplementary Table S2). Protein lysates from frozen tissue samples were generated using 500 μL RIPA buffer per 10 mg of tissue and sonicated at 180 watts 10 × 10 seconds.
qRT-PCR
qRT-PCR was performed as described previously (7) using gene-specific primers (Supplementary Table S3). The qPCR array (PAHS-090Z, Qiagen) was performed according to the manufacturer’s protocol using mRNA extracted from WM9.
Electrophoretic mobility shift assay
Electrophoretic mobility shift assay (EMSA) was performed as described previously (6) using Cy3-labeled oligonucleotides (Supplementary Table S4).
Luciferase assay
Luciferase assay was performed as described previously (6) in WM164 using the respective plasmids (Supplementary Tables S1 and S5) cotransfected with pSV-β-Galactosidase control vector for normalization.
Migration assay
Migration assay was performed as described previously (6) using pLNCX-empty vector or pLNCX-N4ICD–transduced cells.
Invasion assay
Previously described migration assay (6) was modified by coating FluoroBlok cell culture inserts with Matrigel (Corning).
Wound healing assay
Wound healing assay was performed as described previously (6) using pLNCX-empty vector or pLNCX-N4ICD–transduced cells.
Proliferation assay
Proliferation assay was performed as described previously (6) using pLNCX-empty vector or pLNCX-N4ICD–transduced cells.
Adhesion assay
Adhesion assay was performed as described previously (6) using pLNCX-empty vector or pLNCX-N4ICD–transduced cells and E-cadherin–expressing keratinocytes (HaCaT).
Chromatin immunoprecipitation assay
Chromatin immunoprecipitation (ChIP) was performed as described previously (6) using specific antibodies (Supplementary Table S2) and promoter-specific primers (Supplementary Table S3).
IHC
Formalin-fixed, paraffin-embedded tissues from the archives of the Department of Dermatology and the Department of Pathology, University Hospital St. Poelten, Karl Landsteiner University of Medical Sciences (St. Poelten, Austria) were deparaffinized and stained using the BenchMark XT automated immune-staining platform and the Ultra View Universal DAB Detection System (Ventana Medical Systems). The collection and storage of samples were performed according to the local ethical guidelines (EK-Votum number: GS4-EK-3/063–2011). Antigen retrieval was performed before using prediluted antibodies (Supplementary Table S2). Scoring of tissue slides was performed independently by two investigators (0, no staining; 1+, weak positive staining; 2+, moderate positive staining; 3+, strong positive staining). The stained sections were examined with an Olympus BX53 microscope and photographed with an Olympus DP73 camera (Olympus Electronics).
Statistical analysis
Unpaired t test was used to determine statistical significance (not significant, P > 0.05; *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001). Spearman ρ was calculated using IBM SPSS Statistics, version 20.
Results
Overexpression of the constitutively active N4ICD results in an epithelial-like phenotypic switch in human melanoma cells
Overexpression of intracellular domain of Notch4 (N4ICD), but not empty vector, caused a phenotypic switch from a parental mesenchymal-like to an epithelial-like phenotype (Fig. 1A and Supplementary Fig. S1A). These epithelial-like cells exhibited strongly reduced chemokinesis (Fig. 1B, left), reduced migration (Fig. 1B, right and Supplementary Fig. S1B), invasiveness (Fig. 1C and Supplementary Fig. S1C), and proliferation (Supplementary Fig. S1D), but increased adhesion to keratinocytes (Supplementary Fig. S1E). Subcutaneous injection of 1 × 106 empty vector control or N4ICD-transduced 451Lu cells into the right flank of C. B-17/IcrHanHsdArcPrkdcscid mice resulted in palpable tumor formation of empty vector cells 14 days after injection. In comparison, N4ICD-transduced cells showed a delayed tumor onset of more than 4 days. Tumors generated by parental cells resulted in significantly higher tumor volume throughout the observation period (Fig. 1D). Taken together, the observed alterations of cell mobility following N4ICD overexpression combined with the significantly reduced tumor growth in vivo suggest a possible tumor-suppressive role of Notch4 in melanoma.
Figure 1.
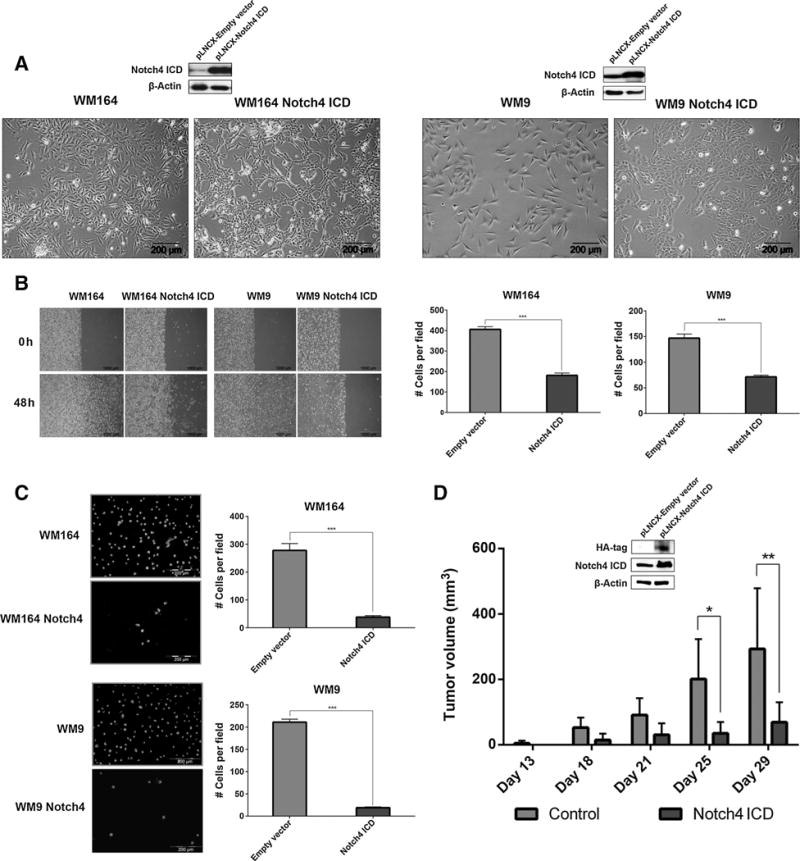
Phenotypic switch following N4ICD overexpression in human melanoma. A, morphology of N4ICD-transduced cells. Expression of N4ICD was confirmed by immunoblotting. β-Actin was used as a loading control. B, chemokinesis (left) and migration (right) of N4ICD-transduced cells. C, invasion of N4ICD-transduced cells. D, tumor volume induced by N4ICD or empty vector–transduced 451Lu. Expression of N4ICD was confirmed by immunoblotting. β-Actin was used as a loading control. n.s., not significant; *, P ≤ 0.05; ***, P ≤ 0.01; ***, P ≤ 0.001.
Notch4-mediated molecular changes resemble a mesenchymal–epithelial transition involving noncanonical Notch signaling
On the molecular level, the phenotypic switch is accomplished by changes of several characteristic markers of epithelial–mesenchymal transition. Investigating the expression pattern of 50 EMT-related genes in N4ICD-transduced WM9 using a qPCR array revealed 17 significantly changed genes (Supplementary Table S6), of which 11 showed an expression pattern corresponding to a MET (Fig. 2A). Changes of selected EMT markers, including E-cadherin (CDH1), vimentin (VIM), MMP-2 (MMP2), Snail2 (SNAI2), and Twist1 (TWIST1) were confirmed by immunoblotting and qRT-PCR (Fig. 2B and Supplementary Fig. S2A). Immunohistochemical analyses of 30 primary human melanomas and 30 cutaneous and subcutaneous metastases revealed a significant positive correlation between Notch4 and E-cadherin expression (Fig. 2C, top; Supplementary Table S7). Interestingly, areas, especially melanoma nests, with high Notch4 staining matched with E-cadherin positivity (Fig. 2C, bottom and Supplementary Fig. S2B), but Notch4 was also found in E-cadherin–negative tissue. Snail2 and Twist1 are downregulated in N4ICD-overexpressing cells and both have been shown to be activated by Notch ICD through its main DNA-bound mediator CSL (RBPJ; refs. 8,9). Silencing experiments using sequence-specific siRNA targeting CSL showed that both EMTRs are upregulated following CSL knockdown on the protein and mRNA level (Fig. 2D and Supplementary Fig. S3A), suggesting that CSL acts as a transcriptional repressor. Analysis of the promoter regions of Snail2 and Twist1 revealed the presence of potential CSL-binding sites (Supplementary Fig. S3B; as reported by refs. 10–12). EMSA and ChIP assays showed binding of CSL to these sequences on Snail2 and Twist1 promoters (Supplementary Fig. S3C and S3D). Considering canonical Notch signaling, in which binding of Notch ICD to the DNA-bound transcriptional repressor CSL results in target gene activation (13), these results indicate that Notch4 induced suppression of Snail2 and Twist1 is mediated in a noncanonical fashion, despite CSL binding to promoters of Snail2 and Twist1.
Figure 2.
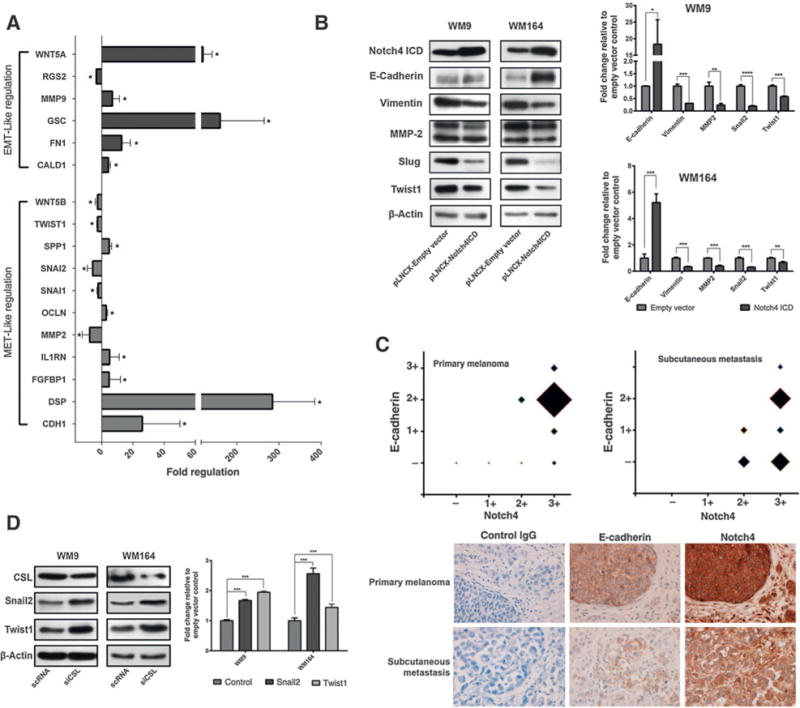
Notch4 regulates EMT markers. A, mRNA levels of WM9 transduced with pLNCX-empty vector or pLNCX-N4ICD were analyzed using a qPCR array. Significant results are relative to empty vector–transduced cells and plotted as fold increase. B, immunoblots (left) and mRNA levels (right) of WM9 and WM164 transduced with pLNCX-empty vector or pLNCX-N4ICD C, E-cadherin and Notch4 staining of primary (n = 30) and metastatic (n = 30) melanoma samples plotted against each other (top). The size of the square indicates the number of samples, showing the respective expression levels. IHC of a primary melanoma and a subcutaneous metastasis for E-cadherin and Notch4 and unspecific secondary IgG antibody (×40; bottom). D, immunoblots (left) and mRNA levels (right) of WM9 and WM164 transfected with siRNA targeting CSL. The error bars represent SDs from the mean. n.s., not significant; *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001.
Notch downstream targets Hey1 and Hey2 regulates Snail2 and Twist1
Next, we confirmed the specificity of the observed changes after N4ICD overexpression by silencing Notch4, which led to the upregulation of Snail2 and Twist1, suggestive of Notch4 acting as a transcriptional repressor (Supplementary Fig. S4A and S4B). In search of a noncanonical Notch signal transduction mechanism, we proposed that the Notch target genes Hey1 (HEY1) and Hey2 (HEY2) might be transcriptional repressors of Snail2 and Twist1. As expected, both Hey proteins are regulated by Notch4 (Fig. 3A and Supplementary Fig. S4C). Silencing and overexpression experiments identified Hey1 as a transcriptional repressor of Snail2 and Twist1 (Fig. 3B). Knockdown and overexpression experiments showed that Hey2 regulates Snail2 but not Twist1 (Fig. 3C). These observations were confirmed in another melanoma cell line (Supplementary Fig. S4D and S4E). Taken together, these results demonstrate that Hey1 and Hey2 as downstream targets of Notch4 are involved in the regulation of Snail2 and Twist1.
Figure 3.
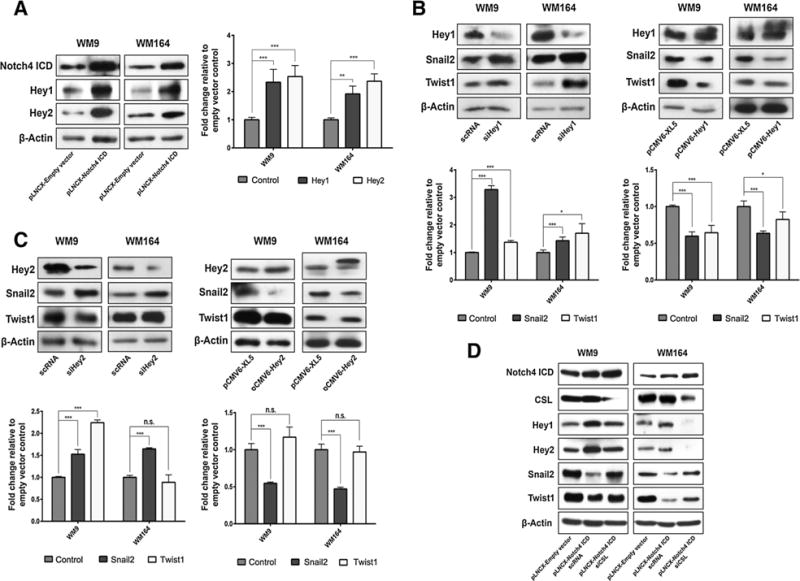
Notch effector genes Hey1/2 suppress Snail2 and Twist1 expression. A, immunoblots (left) and mRNA levels (right) of WM9 and WM164 transduced with pLNCX-empty vector or pLNCX-N4ICD. B, immunoblots (top) and mRNA (bottom) of WM9 and WM164 transfected with siRNA-targeting Hey1 (left) or plasmids encoding Hey1 cDNA (right). C, immunoblots (top) and mRNA levels (bottom) of WM9 and WM164 transfected with siRNA targeting Hey2 (left) or plasmids encoding Hey2 cDNA (right). D, immunoblots of WM9 and WM164 transduced with pLNCX-empty vector, pLNCX-N4ICD, or pLNCX-N4ICD and siRNA targeting CSL. Error bars, SDs from the mean. n.s., not significant; *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001.
Hey1 and Hey2 directly bind to and suppress Snail2 and Twist1 promoter activity
To investigate the hierarchy of N4 signal transduction, we performed double transfection experiments. Silencing of CSL in N4ICD-overexpressing cells resulted in decreased expression of Hey1 and Hey2, confirming that both Hey proteins are direct targets of Notch (14,15). In addition, CSL silencing in N4ICD-overexpressing cells led to reexpression of Snail2 and Twist1, which can be attributed to decreased expression of Hey1/2 and the diminished suppressive activity of CSL itself (Fig. 3D). Furthermore, melanoma cells were simultaneously transiently transfected with specific siRNA targeting Notch4 and plasmids encoding Hey1 or Hey2 cDNA, respectively. Cells overexpressing Hey1 showed suppressed expression levels of Snail2 and Twist1 despite a significant decrease in Notch4 protein levels (Fig. 4A, top), indicating that Hey1 overrules Notch4. Cotransfection of siRNA targeting Notch4 and Hey2 cDNA resulted in the suppression of Snail2 but upregulated Twist1 (Fig. 4A, bottom), which again indicates that Hey2 overrides Notch4 in the regulation of Snail2 but has no effect on Twist1. Silencing of Hey1/2 in Notch4-overexpressing cells confirmed these findings (Supplementary Fig. S5A). Sequence analysis of Snail2 and Twist1 promoter regions revealed several possible Hey protein–binding motives (E-Boxes as reported by ref. 16). EMSA of two randomly chosen E-Boxes (Supplementary Fig. S5B) showed that Hey1 and Hey2 bind to these sequences while showing reduced binding to mutated control sequences (Fig. 4B). To corroborate the direct regulation of Snail2 and Twist1 by Hey proteins, we performed luciferase assays. Overexpression of Hey1 caused a strongly reduced luciferase activity of Snail2 or Twist1 promoter constructs while Hey2 only reduced the luciferase activity of Snail2 constructs (Fig. 4C). The luciferase activity was recovered by mutating E-Boxes in the promoter regions. To conclude, these experiments show that Hey1 and Hey2 directly suppress Snail2 and Hey1 Twist1 expression.
Figure 4.
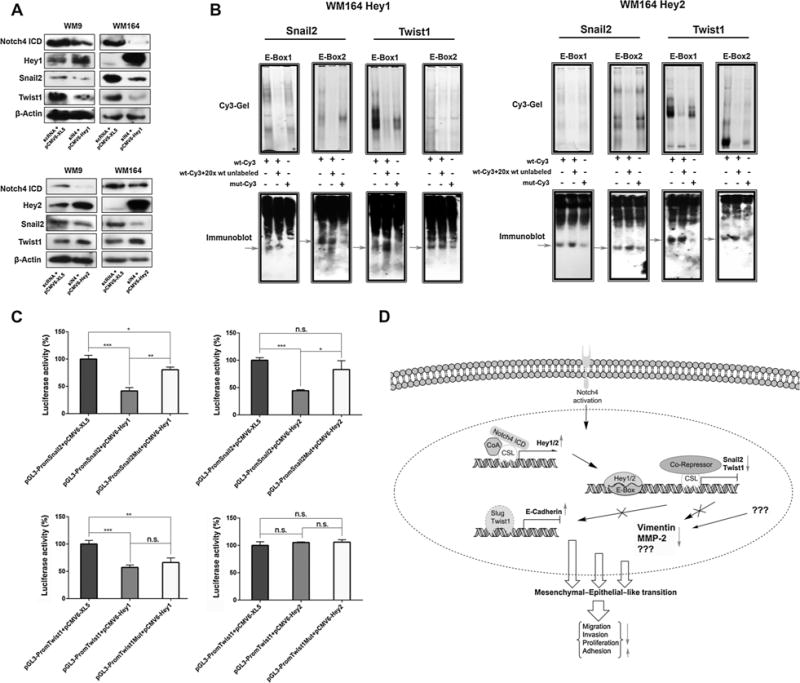
Notch4-mediated activation of Hey1/2 suppress Snail2 and Twist1. A, immunoblots of WM9 and WM164 cotransfected with siRNA targeting Notch4 and plasmids encoding Hey1 or Hey2 cDNA. B, EMSA of WM164 incubated with Cy3-labeled promoter fragments and probed for Hey1 or Hey2. C, luciferase assay of WM164 cells cotransfected with the indicated plasmids. Error bars, SDs from the mean. n.s., not significant; *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001. D, proposed model of Notch4 signal transduction leading to a MET-like transition.
Discussion
Overexpression of the constitutively active N4ICD results in the emergence of an epithelial-like phenotype characterized by strongly reduced migratory, invasive, and proliferative properties in vitro. MET is described by increased E-cadherin expression, whereas several mesenchymal markers like vimentin, MMP-2, Snail2, and Twist1 are downregulated, which, at least partially, is mediated through Hey1 and Hey2 (Fig. 4D). E-cadherin is well known to have tumor-suppressive functions and reexpression in melanoma has been shown to reduce invasive behavior (17). Opposed to E-cadherin, vimentin (18), MMP-2 (19), Snail2 (20), and Twist1 (21) are reported to facilitate migration, invasiveness, and metastasis formation, suggesting that the observed molecular changes following N4ICD overexpression correspond to the observed alterations in cellular functionality. Moreover, N4ICD overexpression resulted in a significant increase of epithelial markers such as DSP, OCLN, SPP1, FGFBP-1, and IL1RN indicating the induction of an epithelial gene signature. These observations combined with reduced tumor growth of N4ICD-overexpressing cells in vivo strongly suggest a tumor-suppressive function of Notch4 in melanoma by promoting the formation of a less aggressive epithelial-like phenotype. This finding is further strengthened by immunohistochemical analyses of primary human melanoma samples showing a significant positive correlation between E-cadherin and Notch4 in melanoma nests and areas of E-cadherin expression in cutaneous and subcutaneous metastases. Nevertheless, the detailed mechanisms and circumstances that determine the subset of Notch-activated genes are highly context dependent and still need to be examined in more detail. One of the possible consequences of a mesenchymal–epithelial transition, the reexpression of E-cadherin, has been linked to cell survival at metastatic sites in breast carcinoma (22) and several studies reported the involvement of MET in promoting metastatic colonization (reviewed in ref. 23). Taken together, our data identified Notch4 as a tumor suppressor in melanoma, suggesting a possible explanation for the clinical ineffectiveness of γ-secretase inhibitors for the treatment of metastatic melanoma. However, it cannot be ruled out that the observed phenotypic switch results in dormant cells with capabilities of increased survival. More detailed understanding of the mechanisms how active Notch4 induces a MET-like phenotype in melanoma could open new avenues to interfere with tumor dormancy contributing to overcome drug resistance.
Supplementary Material
Acknowledgments
The retroviral vector pLNCX containing the Notch4 ICD and the contributing empty vector were generously provided by Dr. Aly Karsan (Departments of Experimental Medicine, Pathology and Laboratory Medicine, University of British Columbia and British Columbia Cancer Agency, Department of Medical Biophysics, British Columbia Cancer Agency, Vancouver, British Columbia, Canada). The authors thank the staff of the Center for Medical Research (ZMF), Graz, for technical assistance, Nina Lengger (Department of Pathophysiology and Allergy Research, Medical University of Vienna) and Michela Karner (Department of Pathology, University Hospital St. Poelten), and Karl Landsteiner University of Health Sciences (St. Poelten, Austria) for their help in preparing histopathologic specimens.
Grant Support
This work was supported by the Austrian Science Fund (grants no. P18630-B05 and P21156-B18 to H. Schaider), the EU Marie Curie Initial Training Network (ITN; Biomedical engineering for cancer and brain disease diagnosis and therapy development, EngCaBra. Proj.no.PITN-GA-2010-264417 to H. Bergler and C. Hafner), and the PhD program “Molecular Medicine” of the Medical University of Graz (D. Ravindran Menon).
Footnotes
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Authors’ Contributions
Conception and design: E. Bonyadi Rad, H. Hammerlindl, C. Wels, H. Bergler, H. Schaider
Development of methodology: E. Bonyadi Rad, H. Hammerlindl, C. Wels, M. Kitzwoegerer, H. Bergler, H. Schaider
Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): E. Bonyadi Rad, H. Hammerlindl, C. Wels, U. Popper, H. Breiteneder, M. Kitzwoegerer, C. Hafner, H. Schaider
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): E. Bonyadi Rad, H. Hammerlindl, D. Ravindran Menon, H. Breiteneder, M. Kitzwoegerer, C. Hafner, H. Schaider
Writing, review, and/or revision of the manuscript: E. Bonyadi Rad, H. Hammerlindl, C. Wels, C. Hafner, H. Schaider
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): E. Bonyadi Rad, H. Hammerlindl, D. Ravindran Menon, C. Hafner, M. Herlyn, H. Schaider
Study supervision: H. Schaider
References
- 1.Massi D, Tarantini F, Franchi A, Paglierani M, Di Serio C, Pellerito S, et al. Evidence for differential expression of Notch receptors and their ligands in melanocytic nevi and cutaneous malignant melanoma. Mod Pathol. 2006;19:246–54. doi: 10.1038/modpathol.3800526. [DOI] [PubMed] [Google Scholar]
- 2.Balint K, Xiao M, Pinnix CC, Soma A, Veres I, Juhasz I, et al. Activation of Notch1 signaling is required for beta-catenin-mediated human primary melanoma progression. J Clin Invest. 2005;115:3166–76. doi: 10.1172/JCI25001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu ZJ, Xiao M, Balint K, Smalley KS, Brafford P, Qiu R, et al. Notch1 signaling promotes primary melanoma progression by activating mitogen-activated protein kinase/phosphatidylinositol 3-kinase-Akt pathways and up-regulating N-cadherin expression. Cancer Res. 2006;66:4182–90. doi: 10.1158/0008-5472.CAN-05-3589. [DOI] [PubMed] [Google Scholar]
- 4.Bedogni B, Warneke JA, Nickoloff BJ, Giaccia AJ, Powell MB. Notch1 is an effector of Akt and hypoxia in melanoma development. J Clin Invest. 2008;118:3660–70. doi: 10.1172/JCI36157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee SM, Moon J, Redman BG, Chidiac T, Flaherty LE, Zha Y, et al. Phase 2 study of RO4929097, a gamma-secretase inhibitor, in metastatic melanoma: SWOG 0933. Cancer. 2015;121:432–40. doi: 10.1002/cncr.29055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wels C, Joshi S, Koefinger P, Bergler H, Schaider H. Transcriptional activation of ZEB1 by Slug leads to cooperative regulation of the epithelial-mesenchymal transition-like phenotype in melanoma. J Invest Dermatol. 2011;131:1877–85. doi: 10.1038/jid.2011.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.RavindranMenon D, Das S, Krepler C, Vultur A, Rinner B, Schauer S, et al. A stress-induced early innate response causes multidrug tolerance in melanoma. Oncogene. 2015;34:4448–59. doi: 10.1038/onc.2014.372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leong KG, Niessen K, Kulic I, Raouf A, Eaves C, Pollet I, et al. Jagged1-mediated Notch activation induces epithelial-to-mesenchymal transition through Slug-induced repression of E-cadherin. J Exp Med. 2007;204:2935–48. doi: 10.1084/jem.20071082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tian Y, Xu Y, Fu Q, Chang M, Wang Y, Shang X, et al. Notch inhibits chondrogenic differentiation of mesenchymal progenitor cells by targeting Twist1. Mol Cell Endocrinol. 2015;403:30–8. doi: 10.1016/j.mce.2015.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lam LT, Bresnick EH. Identity of the beta-globin locus control region binding protein HS2NF5 as the mammalian homolog of the notch-regulated transcription factor suppressor of hairless. J Biol Chem. 1998;273:24223–31. doi: 10.1074/jbc.273.37.24223. [DOI] [PubMed] [Google Scholar]
- 11.Tun T, Hamaguchi Y, Matsunami N, Furukawa T, Honjo T, Kawaichi M. Recognition sequence of a highly conserved DNA binding protein RBP-J kappa. Nucleic Acids Res. 1994;22:965–71. doi: 10.1093/nar/22.6.965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Persson LM, Wilson AC. Wide-scale use of Notch signaling factor CSL/RBP-Jkappa in RTA-mediated activation of Kaposi’s sarcoma-associated herpesvirus lytic genes. J Virol. 2010;84:1334–47. doi: 10.1128/JVI.01301-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gordon WR, Arnett KL, Blacklow SC. The molecular logic of Notch signaling–a structural and biochemical perspective. J Cell Sci. 2008;121(Pt 19):3109–19. doi: 10.1242/jcs.035683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Iso T, Kedes L, Hamamori Y. HES and HERP families: multiple effectors of the Notch signaling pathway. J Cell Physiol. 2003;194:237–55. doi: 10.1002/jcp.10208. [DOI] [PubMed] [Google Scholar]
- 15.Meier-Stiegen F, Schwanbeck R, Bernoth K, Martini S, Hieronymus T, Ruau D, et al. Activated Notch1 target genes during embryonic cell differentiation depend on the cellular context and include lineage determinants and inhibitors. PLoS One. 2010;5:e11481. doi: 10.1371/journal.pone.0011481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fischer A, Gessler M. Delta-Notch–and then? Protein interactions and proposed modes of repression by Hes and Hey bHLH factors. Nucleic Acids Res. 2007;35:4583–96. doi: 10.1093/nar/gkm477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hsu MY, Meier FE, Nesbit M, Hsu JY, Van Belle P, Elder DE, et al. E-cadherin expression in melanoma cells restores keratinocyte-mediated growth control and down-regulates expression of invasion-related adhesion receptors. Am J Pathol. 2000;156:1515–25. doi: 10.1016/S0002-9440(10)65023-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rodriguez MI, Peralta-Leal A, O’Valle F, Rodriguez-Vargas JM, Gonzalez-Flores A, Majuelos-Melguizo J, et al. PARP-1 regulates metastatic melanoma through modulation of vimentin-induced malignant transformation. PLoS Genet. 2013;9:e1003531. doi: 10.1371/journal.pgen.1003531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Luca M, Huang S, Gershenwald JE, Singh RK, Reich R, Bar-Eli M, et al. Expression of interleukin-8 by human melanoma cells up-regulates MMP-2 activity and increases tumor growth and metastasis. Am J Pathol. 1997;151:1105–13. [PMC free article] [PubMed] [Google Scholar]
- 20.Fenouille N, Tichet M, Dufies M, Pottier A, Mogha A, Soo JK, et al. The epithelial-mesenchymal transition (EMT) regulatory factor SLUG (SNAI2) is a downstream target of SPARC and AKT in promoting melanoma cell invasion. PLoS One. 2012;7:e40378. doi: 10.1371/journal.pone.0040378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weiss MB, Abel EV, Dadpey N1, Aplin AE. FOXD3 modulates migration through direct transcriptional repression of TWIST1 in melanoma. Mol Cancer Res. 2014;12:1314–23. doi: 10.1158/1541-7786.MCR-14-0170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chao YL, Shepard CR, Wells A. Breast carcinoma cells re-express E-cadherin during mesenchymal to epithelial reverting transition. Mol Cancer. 2010;9:179. doi: 10.1186/1476-4598-9-179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gunasinghe NP, Wells A, Thompson EW, Hugo HJ. Mesenchymal-epithelial transition (MET) as a mechanism for metastatic colonisation in breast cancer. Cancer Metastasis Rev. 2012;31:469–78. doi: 10.1007/s10555-012-9377-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.