

Abstract
The use of mitochondrial transfer as a clinic procedure is drawing closer to reality. Here we provide a detailed overview of mitochondrial transfer techniques – both established and recent – including pronuclear, spindle, ooplasmic and blastomere transfer. Reasons as to why some techniques are more suitable for the prevention of mitochondrial DNA disease than others, as well as the advantages and disadvantages of each methodology, are discussed. The possible clinical introduction of these techniques has raised concerns about the adverse effects they may have on resultant embryos and offspring. Success rates of each technique, embryo viability and developmental consequences post mitochondrial transfer are addressed through analysis of evidence obtained from both animal and human studies. Counterarguments against potential mitochondrial-nuclear genome incompatibility are also provided. Additional clinical applications of mitochondrial transfer techniques are discussed. These include the rescue or enhancement of fertility in women of advanced maternal age or those suffering from diabetes. An alternative to using mitochondrial DNA transfer for germ line therapies is the therapeutic use of somatic cell nuclear transfer for the generation of personalised stem cells. Although ethically challenging, this method could offer patients already suffering from mitochondrial DNA diseases a novel treatment option.
1. Introduction
Mitochondria are the energy-producing powerhouses of the cell. Their function is dependent on proteins transcribed from nuclear and mitochondrial DNA (mtDNA). Mitochondrial DNA is circular in structure and contains 37 genes. mtDNA diseases are inherited by all offspring irrespective of gender, as all mitochondria in the embryo originate from oocyte cytoplasm (Giles et al., 1980). With progressive genetic technologies in detecting disease-associated mtDNA mutations, over 300 rRNA/tRNA mutations (http://www.mitomap.org/foswiki/bin/view/MITOMAP/MutationsRNA) and over 300 coding/control region point mutations have now been identified (http://www.mitomap.org/foswiki/bin/view/MITOMAP/MutationsCodingControl).
Disruption of essential metabolic pathways in persons suffering from mtDNA disease, especially in high energy-demanding organs, leads to severe disability and early death. At present, no preventative treatments are available to prospective parents who are afflicted and wish to conceive (Pfeffer et al., 2012). Clinical diagnosis is challenging due to varying symptoms and phenotypes, even within a single family. This is due to mitochondrial heteroplasmy, where mutational load at the cellular level determines phenotype: low mutational load will result in asymptomatic phenotypes or low severity, but once the number of affected mitochondria exceed a certain threshold, patients become increasingly symptomatic. There may also be unequal distribution of mutated mtDNA between organs depending on mitochondrial segregation patterns during fetal development, giving rise to various phenotypes depending on the levels of mutated mtDNA present and the organs involved (Larsson and Clayton, 1995).
This review focuses on “mtDNA disease”, not “mitochondrial disease”, the latter referring to disorders that are caused by mutations in both mtDNA and nuclear DNA. In the case of nuclear DNA mutations, Mendelian genetics are applicable (Angelini et al., 2009), whereas mtDNA mutations are inherited maternally (Giles et al., 1980). Only around 20% of disorders involving defective mitochondrial oxidative phosphorylation (OXPHOS) are accounted for by mutations in mtDNA (Darin et al., 2001).
Research in both animals and humans has offered hope for the prevention of mtDNA disease via assisted reproductive technology (ART) techniques involving micromanipulation. This review explores techniques that could be used for the prevention of mtDNA disease transmission, their additional potential uses in the clinical setting and the concern of mitochondrial-nuclear genome mismatch.
2. Mitochondrial transfer techniques
2.1. Pronuclear transfer
Pronuclear transfer (Fig. 1A) involves the transfer of pronuclei from one zygote to another (Craven et al., 2010). This technique first requires fertilisation of healthy donated egg/s (provided by the mitochondrial donor) with the intending male parent sperm. Simultaneously, the intending mother's affected oocytes are fertilised with the intending father's sperm. Both sets of fertilised oocytes are allowed to develop to the early zygote stage where the pronuclei are visible. Using micromanipulation equipment, the pronuclei of zygotes formed from donated oocytes are removed within a karyoplast, and discarded. Therapeutic pronuclear transfer involves the movement of two pronuclei from the affected zygotes (also in the form of a karyoplast), into the enucleated healthy zygotes. The resulting zygotes contain nuclear DNA from each of the intending parents and a donor's mtDNA.
Fig. 1.

Methods for prevention of mtDNA disease transmission: (A) pronuclear and (B) spindle transfer. ICSI – intracytoplasmic sperm injection; mtDNA – mitochondrial DNA; nDNA – nuclear DNA; IVF – in vitro fertilisation.
Reproduced from Reznichenko et al., 2015, with permission from SAJBL.
Using this method, Craven et al. (2010) have demonstrated less than 2.0% carryover of mtDNA between abnormally fertilised zygotes that possessed either one or three pronuclei. They also demonstrated that pronuclear transfer was compatible with onward in vitro embryo development. Blastocyst development rate was approximately half (17%) of that found amongst normally fertilised embryos (32%). This can be explained by the lack of a reliable morphological factor that can be used to differentiate between male and female pronuclei in zygotes. When pronuclei were transferred from the abnormally fertilised zygotes in this study, the authors could not be certain whether they were reconstructing zygotes with one female and one male pronucleus. If the resultant zygote is deficient in one or the other genome, development will be impaired.
A disadvantage from an ethical point of view is that half of the embryos (affected zygotes which will be enucleated and have their nuclear genetic material transferred) will be destroyed. However, unlike spindle transfer, in vitro fertilisation (IVF) can be performed if the sperm parameters so permit, and intracytoplasmic sperm injection (ICSI) is not necessarily required. Another advantage of pronuclear over spindle transfer is that pronuclei can be more easily visualised than spindles, using a conventional inverted microscope. During the process of nuclear transfer, the karyoplast is placed within the perivitelline space and then allowed to fuse to the donor cytoplast using the inactivated viral vector SeV (Sendai virus, also known as HVJ-E) (Tachibana et al., 2009, Craven et al., 2010). This raises concern about the possible effects of a viral vector on an embryo or its development. Tachibana et al. (2009) demonstrated the lack of SeV genetic material in reconstructed embryos by reverse transcription (RT)-PCR analysis, indicating that the viral vector does not pose any potential threats in the offspring or further generations. As this is a novel technique, pronuclear transfer has only been applied to abnormally fertilised zygotes, which possessed either one or three pronuclei (Craven et al., 2010). This method needs to be assessed in a primate animal model, as has been done with spindle transfer, in order to prove its efficacy and safety (Tachibana et al., 2009).
2.2. Spindle transfer
Spindle transfer (ST) (Fig. 1B) aims to achieve the same results as pronuclear transfer – inheritance of parental nuclear DNA and donor mtDNA in the offspring. In this method, the transfer of parental nuclear DNA occurs before fertilisation (Tachibana et al., 2009). This technique involves excision of the metaphase II spindle from the donor oocyte, which will provide the cytoplasmic constituent (including mitochondria) to the embryo. Again, contained within a karyoplast, the donor nuclear DNA will be discarded. The chromosome-spindle complex will then be removed from the oocyte of the intending mother, and transferred to the enucleated healthy donor oocyte (Tachibana et al., 2009).
A rhesus macaque study using this technique resulted in the birth of three healthy offspring (Tachibana et al., 2009). The presence of nuclear DNA originating from spindle donors and mtDNA from recipient oocytes was established though genetic analysis. No spindle donor mtDNA was observed in the offspring, demonstrating efficient mitochondrial replacement. Postnatal follow up analysis into adulthood (from birth to 3 years old) has shown that routine blood and body weight measurements were comparable to age-matched controls (Tachibana et al., 2013).
The same research group has also implemented this technique in human oocytes to study the feasibility of fertilisation, embryo development and embryonic stem cell (ESC) yield (Tachibana et al., 2013). Fertilisation rate in ST oocytes was comparable to controls (73% versus 75%, respectively). Although a substantial number of ST zygotes (52%) contained abnormal numbers of pronuclei, showing abnormal fertilisation, blastocyst development (62%) in normally fertilised zygotes was similar to controls. ESC isolation rates from ST blastocysts (38%) were also similar to controls, and all cell lines exhibited the presence of donor mtDNA only and normal karyotypes. The relatively high incidence of abnormal fertilisation was surprising, as it was not observed previously in the 2009 primate study. Possibly, human meiotic spindles were prematurely activated during the ST procedure resulting in impaired continuation of meiosis post fertilisation. Analysis after immunolabeling with α- and β-tubulins revealed that some ST oocyte spindles had prematurely progressed to late anaphase II (Tachibana et al., 2013). ESCs derived from the abnormally fertilised zygotes exhibited triploidy, stemming from the presence of two sets of the maternal genome. This finding showed that some oocytes failed to complete meiosis and the chromosomes failed to segregate into the second polar body, probably resulting from premature activation. This study highlights the efficacy and potential use of ST for the prevention of mtDNA disease in humans. If the technique is performed successfully, ST replaces affected recipient mitochondria and does not inhibit embryo development or the derivation of stable stem cell lines.
ST has also been performed in mature metaphase II (MII) human oocytes which were subsequently parthenogenically activated instead of fertilised (Paull et al., 2013). This work too showed blastocyst development post ST, and mtDNA carryover of less than 1%. mtDNA carryover decreased in blastocysts and stem cells spindle donor mtDNA became undetectable, and this situation persisted after passaging for more than one year, clonal expansion, differentiation into neurons, cardiomyocytes or β-cells, and after cellular reprogramming. Mitochondrial function in ESCs and their differentiated phenotypes as assessed by rates of respiratory chain enzyme activity and oxygen consumption was determined to be comparable to controls (Paull et al., 2013). This finding illustrates that metabolic function is not disrupted post introduction of donor mitochondria that are foreign to the nuclear genome.
A limitation of spindle transfer is that the donor oocyte must be denuded before the procedure due to the need for zona pellucida thinning and laser assisted zona drilling. Subsequently, the oocyte can only be fertilised through ICSI. Another limitation is that this procedure is more costly due to the fact that the spindle can only be visualised using specialised equipment that is coupled to an imaging system that uses polarised light birefringence. The chromosome-spindle complex is a fragile structure that needs to be handled with utmost care, as the DNA is not as stable as in pronuclei. Recently, another group has advised that the first polar body be removed along with the spindle from the donor oocyte before the procedure, as the polar body can be resorbed during later fusion of the karyoplast, and cause polyploidy (Greggains et al., 2014). Another disadvantage is that chromosome scattering can occur during metaphase II, and this evidence of spindle damage has previously been found to occur as a result of temperature fluctuation (Almeida and Bolton, 1995) and advanced maternal age (Battaglia et al., 1996). Craven et al. (2011) recommend that spindle transfer should be performed under highly regulated environmental conditions with minimal risk of damage, that otherwise could result in aneuploidy during manipulation of the spindle.
2.3. Ooplasmic transfer
Also referred to as cytoplasmic transfer, this form of mitochondrial transfer entails movement of some ooplasm, containing wild-type mitochondria, from a healthy donor oocyte into an affected one (Flood et al., 1990) (Fig. 2). Theoretically, the consequent heteroplasmy in the resulting embryo should lead to dilution of mutant mitochondria and rescue of the defective mtDNA phenotype, at least to some extent. Success however depends on the unpredictable nature of mitochondrial segregation during fetal development and which organs will eventually have levels of mutant mtDNA above the disease threshold. Ooplasmic transfer has been reported in patients that had previously experienced failed embryonic development, where the donated portion of ooplasm is thought to provide mitochondria and other factors that support developmental competency (Van Blerkom et al., 1998, Huang et al., 1999, Jacobs et al., 2006). Of concern is the finding in some studies of an increased incidence of chromosomal abnormalities and birth defects post ooplasmic transfer (Brown et al., 2006, Jacobs et al., 2006). This raises doubt about the clinical use of this technique for women at risk of transmitting mtDNA disease to their offspring.
Fig. 2.
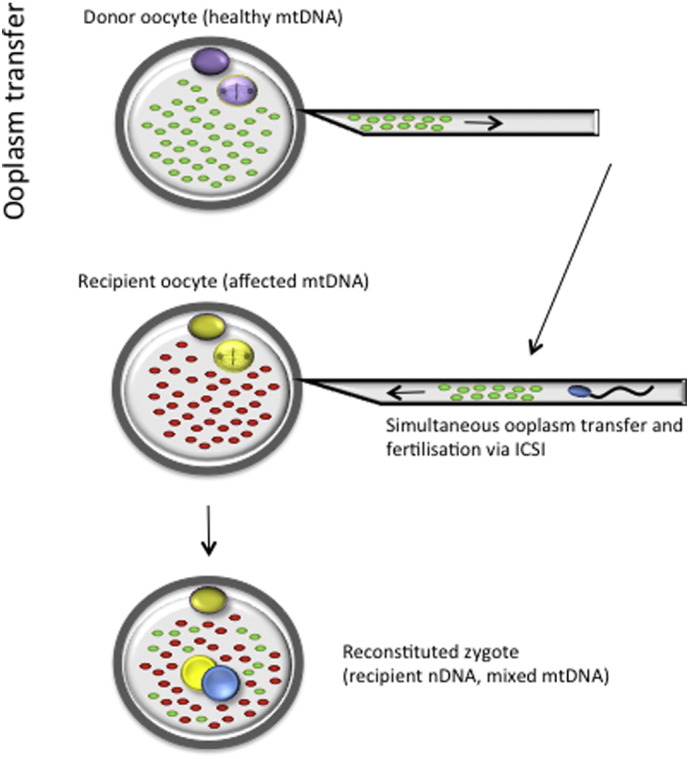
Ooplasmic transfer. Donor ooplasm is transferred to a recipient oocyte. Sperm is injected simultaneously during the transfer to fertilise the denuded recipient oocyte.
Similarly to ST, ooplasmic transfer requires oocyte denudation before the procedure, which results in fertilisation only being possible via ICSI. The sperm and donor ooplasm are introduced simultaneously in one injection (Fig. 2). While ooplasmic transfer could provide a significant improvement in embryo development for patients who have experienced repeated IVF failure, this methodology does not necessarily provide the best approach to the prevention of mtDNA disease transmission and carries many risks related to abnormal embryo development. Only small volumes of donor ooplasm can be transferred (10–15% of the entire oocyte) and it is believed that this would provide insufficient amounts of donor mitochondria to “override” the mutant mtDNA in the affected recipient oocyte (Thorburn et al., 2001, Taylor and Turnbull, 2005, Brown et al., 2006, Jacobs et al., 2006). Furthermore, it is uncertain whether larger volumes could be transferred and what effect this will have on the ultrastructure of the recipient oocyte. For these reasons, this technique has not been the primary focus of research for the prevention of mtDNA disease.
2.4. Blastomere transfer
Blastomere transfer involves transplantation of a blastomere from an affected embryo into an enucleated healthy donor oocyte (Fig. 3). Similar to other methods, the embryo will inherit mtDNA from the healthy donor oocyte cytoplasm, but it is unclear whether this technique can successfully prevent mtDNA disease in the resulting offspring. An entire cell (a blastomere) is fused to the recipient oocyte, instead of merely a karyoplast containing mostly nuclear material (with very little mitochondrial carryover). Indeed, bovine studies have revealed high levels of mtDNA heteroplasmy in the resulting embryos (Steinborn et al., 2000, Ferreira et al., 2007). This implies that the resulting offspring will be heteroplasmic and therefore the same issues of mitochondrial segregation and threshold effect will apply as discussed previously in ooplasmic transfer. This questions the use of this technique for the prevention of mitochondrial disorders. This methodology has not been applied clinically and no studies have been reported in humans, as it is closely aligned to reproductive cloning and involves the same micromanipulation procedures, using postzygotic nuclear transplantation.
Fig. 3.

Blastomere transfer. A blastomere from an affected mother's embryo is transferred to a healthy donor oocyte.
In cases of reproductive cloning in other mammalian species (Bordignon et al., 2013, Hajian et al., 2013, Kamimura et al., 2013), although the techniques have been perfected, manipulated embryos undergo developmental failure and the resulting offspring are found to succumb to early death, which is presumed to be coupled to unanticipated epigenetic events. Regarding somatic cell nuclear transfer (SCNT), a genome-wide study revealed that although an oocyte has the ability to epigenetically reprogram transplanted DNA, it cannot fully replicate the DNA demethylation which occurs during natural fertilisation. Consequently, atypical methylation patterns in promoter and repetitive element regions cause disruption of characteristic developmental events and cell growth regulation (Peat and Reik, 2012). A similar phenomenon could possibly occur following the transfer of blastomere nuclei, although blastomeres are not differentiated (unlike somatic cells). Roberts (1999) hypothesised that the resulting embryo may have impaired developmental potential.
3. Concern regarding mitochondrial-nuclear genome incompatibility
The interaction between nuclear and mitochondrial gene products ensures successful cell function through essential metabolic pathways. Many nuclear-encoded proteins are transported to mitochondria to perform their respective functions within the electron transport chain. With the imminent emergence of mitochondrial replacement therapy, there is increasing concern over the possible incompatibility that this could create in the communication network that exists between the nucleus and mitochondria, which could have deleterious effects on the metabolomics of a cell (Chinnery et al., 2014).
Although the mitochondrial genome is highly conserved amongst all human populations, it has been proposed that during human evolution mtDNA missense mutations accumulated in non-African populations during their migration to the northern hemisphere (Wallace, 2005). This phenomenon served as an adaptation of mitochondrial bioenergetics to more northern climates with the selected mtDNA variants providing superior reproductive advantages under the pressure of the novel environmental conditions (Ruiz-Pesini et al., 2004, Wallace, 2005). Taken together with the presence of various mtDNA haplotypes amongst different population groups today, it may therefore be advantageous to select mitochondrial donors from the same ethnic group as the affected recipient. This approach will increase the chance of similar mitochondrial genome haplotypes and therefore compatibility with the nuclear genome (Burgstaller et al., 2015).
Several murine studies have demonstrated the lack of any mitonuclear incompatibility as normal healthy offspring are obtained when breeding genetically distant mice (Battersby and Shoubridge, 2001, Gregorová et al., 2008, Cannon et al., 2011). ST in rhesus macaques was performed purposefully using very divergent nuclear and mtDNA haplotypes, and long-term follow up of these animals demonstrated absence of any metabolic malfunction or adverse health and development (Tachibana et al., 2009, Tachibana et al., 2013). Research in this animal model, the closest relative to man, has demonstrated that mitochondrial transfer should not compromise the functionality of mitonuclear interactions. Normal development and differentiation was also observed in human embryos and their derived ESCs after mitochondrial transfer using “unmatched” mtDNA (Craven et al., 2010, Tachibana et al., 2013).
One must consider the many children born to interracial parents with divergent haplotype groups. All of the offspring inherit maternal mitochondria that would then theoretically be “incompatible” with half of their nuclear DNA. Human outbreeding has introduced extensive mitochondrial and nuclear genome mixing without obvious adverse effects to offspring. Admittedly, potential effects may for the moment remain hidden, and there is therefore a need for further understanding of mitonuclear mismatch. However, mitochondrial transfer currently appears to be a mere extension of the natural genome mixing that is common amongst humans today.
4. Clinical applications of mitochondrial transfer
4.1. Prevention of mtDNA disease
Mitochondrial transfer techniques have the potential to provide patients with novel reproductive options for the prevention of mtDNA disease. To date, women who are at risk of transmitting mtDNA disease to their children have to turn to donor oocytes or pre-implantation genetic diagnosis (PGD). However, both options have their limitations. The use of donor oocytes results in the intending mother being genetically unrelated to her child. PGD on the other hand is relatively unsuccessful in detecting mtDNA mutations, especially in women who carry a high mutational load in their oocytes and low mutational load in cells outside the ovaries. Typically, PGD work up is conducted using maternal lymphocytes to detect the mutation and optimise the PGD test. In addition, PGD does not offer any therapeutic or preventative solutions if all the embryos obtained are affected (and therefore untransferrable). Consequently, it is important rather to focus on eliminating mutant mtDNA completely from the embryo, possibly through mitochondrial transfer.
4.2. Aging oocytes
Mitochondria are the primary source of intracellular production of reactive oxygen species (ROS). The association of ROS, aging and metabolic decline is a theory that was established 60 years ago (Harman, 1956). Increased ROS levels lead to decreased metabolic function and energy production, stemming from mutated mitochondrial respiratory chain proteins which cause partial uncoupling of the respiratory chain. Continuous accumulation of mutations arising from age-related ROS also affects the potential of the mtDNA genome to replicate. Therefore, aging begins at the molecular level and then progresses to cell, tissue and finally organ dysfunction (Kirkwood, 2005, Kirkwood, 2008). The mutation rate of mtDNA is approximately 15 times that of the nuclear genome. The mitochondrial genome replicates much more often and independently from the nucleus. Furthermore, in comparison to the nuclear genome, mtDNA lacks protective histones and the DNA repair mechanisms are deficient (Richter, 1995, Mecocci et al., 1999).
Reproductive decline due to women postponing childbirth beyond their 30s is becoming increasingly common. Female reproductive physiology has not kept up with relatively recent social and cultural tendencies resulting in age-related infertility, aneuploidy and fetal defects in women of advanced maternal age (Hook, 1981, Huang et al., 2008, Allen et al., 2009, Bentov et al., 2011). Key causative factors are mitochondrial dysfunction from accumulation of mtDNA mutations and accumulation of oxygen radicals (Reynier et al., 2001, Bartmann et al., 2004, Bentov et al., 2011). Aneuploidy can be explained by the fact that resumption of meiosis during oocyte maturation requires a significant amount of energy, sourced from mitochondrial oxidative phosphorylation. Furthermore, fertilisation potential of oocytes and developmental potential of embryos are heavily dependent on adenosine 5-triphosphate (ATP), mtDNA content and mtDNA copy number (Reynier et al., 2001, Van Blerkom, 2011). Infertility can also be accounted for by poor implantation rates, another energy-demanding process (Bartmann et al., 2004). Moreover, mitochondrial copy number in oocytes of patients where the cause of fertilisation failure was unknown was decreased when compared to oocytes from patients who experienced IVF failure stemming from severe sperm defects. This group predicted that a low mtDNA copy number results from faulty ooplasm maturation.
Through mitochondrial transfer, all of these infertility issues could potentially be overcome via introduction of large amounts of healthy mitochondria, which will be able to supply the necessary amount of ATP required for fertilisation and normal embryo development. Novel techniques such as ST will offer women, who have experienced repeated IVF failure unrelated to severe sperm defects, have unexplained fertility or are of advanced maternal age, an increased chance of conceiving a healthy child.
4.3. Diabetic mothers-to-be
Some studies have revealed that women suffering from diabetes mimic older mothers-to-be in their poor reproductive outcomes, experiencing increased rates of aneuploidies, miscarriages and birth defects. These factors have been attributed to mitochondrial dysfunction (Bentov and Casper, 2013). In a diabetic patient this is believed to be related to high oxidative stress (Maritim et al., 2003), which leads to alterations in mitochondrial ultrastructure, increased mtDNA copy number and reduced levels of ATP and tricarboxylic acid (TCA) cycle metabolites (Calabrese et al., 2005). The increase in mtDNA copy number is surprising and suggests that maternal diabetes either stimulates mitochondrial biogenesis or inhibits degradation in oocytes (Wang et al., 2009). Studies in mice have confirmed that ovulated oocytes in diabetic mice present with an elevated frequency of aneuploidies, caused by spindle defects and chromosome misalignment during meiosis (Wang et al., 2009). These researchers also concluded that oocytes develop abnormally due to the toxic diabetic environment, which triggers mitochondrial damage. This correlation between mitochondrial dysfunction and insulin resistance has led to the belief that offspring developing from oocytes containing defective mitochondria may be predisposed to obesity and insulin resistance. Abnormal mitochondrial metabolism may in this way be responsible for inter-generational transmission of metabolic disease (Turner and Robker, 2015). By introducing the nuclear genome of affected mothers into healthy oocytes that possess undamaged mitochondria, diabetic women would hypothetically have an improved chance of falling pregnant with a healthy fetus.
5. Conluding remarks and prospectives
On the basis of a detailed technical and ethical feasibility assessment, only pronuclear and spindle transfer has been considered for human clinical trials. They are the techniques that have been mentioned both in an ethical review of mtDNA disease prevention by the Nuffield Council on Bioethics (2012), as well as in the Human Fertilisation and Embryology Authority (HFEA) public debates and review reports (summarised in an updated 2014 HFEA review, which can be accessed at:
http://www.hfea.gov.uk/docs/Third_Mitochondrial_replacement_scientific_review.pdf).
In February 2015, the UK Parliament voted in favour of amendment to the Human Fertilisation and Embryology Act 2008 to legalise human mitochondrial replacement and commence human clinical application. The Human Fertilisation and Embryology (Mitochondrial Donation) Regulations 2015 came into force on 29th October 2015 and can be accessed at:
http://www.legislation.gov.uk/uksi/2015/572/pdfs/uksi_20150572_en.pdf
However, only one case of human pronuclear transfer has been reported, and it was performed in abnormally fertilised zygotes (Craven et al., 2010). Spindle transfer has been explored more extensively, with studies performed in rhesus macaques, resulting in the birth of healthy offspring which presented with normal growth curves into adulthood and extremely low levels of mtDNA carryover. The macaque studies were conducted for three years after birth and no further follow up has been reported. Human spindle transfer has yielded blastocyst development and ESC isolation with the passaging of karyotypically normal differentiated cell lines (Tachibana et al., 2009, Tachibana et al., 2013). Potential concerns with pronuclear and spindle transfer include the impact these procedures might have on the risk of mtDNA carryover during karyoplast transfer, embryogenesis, epigenetics and genome integrity (Craven et al., 2011). These issues need to be carefully assessed through more in-depth research.
Although spindle transfer has resulted in the birth of healthy primate offspring, unforeseen difficulties arose when the same group of researchers applied the technique to mature human oocytes (Tachibana et al., 2009, Tachibana et al., 2013). Further investigation is needed to establish long-term safety and efficacy of human pronuclear and spindle transfer for clinical applications. Rhesus macaque studies have provided promising results with the absence of any abnormalities into adulthood (Tachibana et al., 2013). Future clinical trials should focus on optimisation of the techniques to reduce mutated mtDNA carryover to less than the current 1% and to minimise the rate of abnormal fertilisation (Amato et al., 2014). Moreover, the possible effects of exposing chromosomes and spindles to polarised light birefringence need to be examined. Aneuploidy also needs to be reduced, as abberant segregation of chromosomes during maturation (metaphase II) is more frequent in aged oocytes (Battaglia et al., 1996). Other difficulties include achieving synchronised ovarian stimulation of the donor and recipient women as oocyte aspiration will need to take place on the same day. Clinical application of ST will most likely need to utilise cryopreserved oocytes due to these logistical hurdles, for which the techniques will need to be optimised. Cryopreservation of the spindle donor oocyte (affected) rather than the mitochondrial donor oocyte is advised, as primate studies have demonstrated that cytoplasts are more susceptible to damage post vitrification than are spindles (Tachibana et al., 2013).
Understandably, any novel clinical treatment requires constructive criticism and an evaluation of associated risks. Mitochondrial transfer will surely progress to human clinical trials for the prevention of mtDNA disease in the relatively near future and spread beyond the borders of the UK. This conclusion is based on the success that has been achieved in animal models, human preimplantation embryos and the lack of mitonuclear incompatibility in mixed race humans. In addition, donor mtDNA may be matched to the affected mother's haplotype to further eliminate concern of mitonuclear incompatibility (Burgstaller et al., 2015).
One alternative to using mtDNA transfer for germ line therapies is the use of therapeutic cloning (a technically similar micromanipulation technique) for the generation of personalised stem cells. Although ethically challenging, this method could offer patients already suffering from mtDNA disease a novel treatment option. Since induced pluripotent stem cells (iPSCs) from these patients would still contain mutated mtDNA, stem cells could be harvested from embryos produced through somatic cell nuclear transfer to donor oocytes, which carry healthy mitochondria (Greggains et al., 2014). Using adult skin and amnion fibroblasts, a recent report has demonstrated that human nuclear transfer (NT) embryos contain very low levels of somatic cell mtDNA, with heteroplasmy levels below those found in MII spindle transfer human embryos (Greggains et al., 2014). This minimal level of mtDNA carryover highlights the suitability of SCNT for therapeutic use in mtDNA disease patients. However, this type of therapy would only treat the affected person and not offer prevention of transmission of mutated mtDNA to consecutive generations.
Another emerging technique that has been applied in a murine model is the use of mitochondria-targeted nucleases that can selectively eliminate mutated germ line mtDNA. Similarly to therapeutic cloning, this approach avoids the need for mtDNA transfer as it induces a shift in the mtDNA heteroplasmy thereby preventing transmission of the mutant haplotypes to offspring (Reddy et al., 2015).
Mitochondrial transfer, though yet to be perfected, will prospectively offer affected mothers-to-be the opportunity to conceive a genetically related healthy child that only carries donor mtDNA. These techniques introduce promising reproductive options to patients who at the moment have to rely on the use of donor oocytes or unreliable and relatively unsuccessful PGD. Mitochondrial transfer can also be of clinical benefit to mothers-to-be suffering from diabetes, women of advanced maternal age, or patients that have suffered IVF failure that is not attributable to a male factor. The novel methods discussed in this paper also have the potential to expand research avenues in personalised stem cell therapies.
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the review.
Funding
This work was supported by grants from the South African Medical Research Council Flagship Awards Project SAMRC-RFA-UFSP-01-2013/STEM CELLS, the SAMRC Extramural Unit for Stem Cell Research and Therapy and the Institute for Cellular and Molecular Medicine of the University of Pretoria.
References
- Allen E.G., Freeman S.B., Druschel C., Hobbs C.A., O'Leary L.A., Romitti P.A., Royle M.H., Torfs C.P., Sherman S.L. Maternal age and risk for trisomy 21 assessed by the origin of chromosome nondisjunction: a report from the Atlanta and National Down Syndrome Projects. Hum. Genet. 2009;125:41–52. doi: 10.1007/s00439-008-0603-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida P.A., Bolton V.N. The effect of temperature fluctuations on the cytoskeletal organisation and chromosomal constitution of the human oocyte. Zygote. 1995;3:357–365. doi: 10.1017/s0967199400002793. [DOI] [PubMed] [Google Scholar]
- Amato P., Tachibana M., Sparman M., Mitalipov S. Three-parent in vitro fertilization: gene replacement for the prevention of inherited mitochondrial diseases. Fertil. Steril. 2014;101:31–35. doi: 10.1016/j.fertnstert.2013.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelini C., Bello L., Spinazzi M., Ferrati C. Mitochondrial disorders of the nuclear genome. Acta Myologica. 2009;28:16–23. [PMC free article] [PubMed] [Google Scholar]
- Bartmann A.K., Romao G.S., da Silveira R.E., Ferriani R.A. Why do older women have poor implantation rates? A possible role of the mitochondria. J. Assist. Reprod. Genet. 2004;21:79–83. doi: 10.1023/B:JARG.0000027018.02425.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battaglia D.E., Goodwin P., Klein N.A., Soules M.R. Influence of maternal age on meiotic spindle assembly in oocytes from naturally cycling women. Hum. Reprod. 1996;11:2217–2222. doi: 10.1093/oxfordjournals.humrep.a019080. [DOI] [PubMed] [Google Scholar]
- Battersby B.J., Shoubridge E.A. Selection of a mtDNA sequence variant in hepatocytes of heteroplasmic mice is not due to differences in respiratory chain function or efficiency of replication. Hum. Mol. Genet. 2001;10:2469–2479. doi: 10.1093/hmg/10.22.2469. [DOI] [PubMed] [Google Scholar]
- Bentov Y., Casper R.F. The aging oocyte - can mitochondrial function be improved? Fertil. Steril. 2013;99:18–22. doi: 10.1016/j.fertnstert.2012.11.031. [DOI] [PubMed] [Google Scholar]
- Bentov Y., Yavorska T., Esfandiari N., Jurisicova A., Casper R.F. The contribution of mitochondrial function to reproductive aging. J. Assist. Reprod. Genet. 2011;28:773–783. doi: 10.1007/s10815-011-9588-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordignon V., El-Beirouthi N., Gasperin B.G., Albornoz M.S., Martinez-Diaz M.A., Schneider C., Laurin D., Zadworny D., Agellon L.B. Production of cloned pigs with targeted attenuation of gene expression. PLoS One. 2013;8 doi: 10.1371/journal.pone.0064613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown D.T., Herbert M., Lamb V.K., Chinnery P.F., Taylor R.W., Lightowlers R.N., Craven L., Cree L., Gardner J.L., Turnbull D.M. Transmission of mitochondrial DNA disorders: possibilities for the future. Lancet. 2006;368:87–89. doi: 10.1016/S0140-6736(06)68972-1. [DOI] [PubMed] [Google Scholar]
- Burgstaller J.P., Johnston I.G., Poulton J. Mitochondrial DNA disease and developmental implications for reproductive strategies. Mol. Hum. Reprod. 2015;21:11–22. doi: 10.1093/molehr/gau090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese V., Lodi R., Tonon C., D'Agata V., Sapienza M., Scapagnini G., Mangiameli A., Pennisi G., Stella A.M.G., Butterfield D.A. Oxidative stress, mitochondrial dysfunction and cellular stress response in Friedreich's ataxia. J. Neurol. Sci. 2005;233:145–162. doi: 10.1016/j.jns.2005.03.012. [DOI] [PubMed] [Google Scholar]
- Cannon M.V., Dunn D.A., Irwin M.H., Brooks A.I., Bartol F.F., Trounce I.A., Pinkert C.A. Xenomitochondrial mice: investigation into mitochondrial compensatory mechanisms. Mitochondrion. 2011;11:33–39. doi: 10.1016/j.mito.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinnery P.F., Craven L., Mitalipov S., Stewart J.B., Herbert M., Turnbull D.M. The challenges of mitochondrial replacement. PLoS Genet. 2014;10 doi: 10.1371/journal.pgen.1004315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craven L., Tuppen H.A., Greggains G.D., Harbottle S.J., Murphy J.L., Cree L.M., Murdoch A.P., Chinnery P.F., Taylor R.W., Lightowlers R.N. Pronuclear transfer in human embryos to prevent transmission of mitochondrial DNA disease. Nature. 2010;465:82–85. doi: 10.1038/nature08958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craven L., Elson J.L., Irving L., Tuppen H.A., Lister L.M., Greggains G.D., Byerley S., Murdoch A.P., Herbert M., Turnbull D. Mitochondrial DNA disease: new options for prevention. Hum. Mol. Genet. 2011;20:R168–R174. doi: 10.1093/hmg/ddr373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darin N., Oldfors A., Moslemi A.-R., Holme E., Tulinius M. The incidence of mitochondrial encephalomyopathies in childhood: clinical features and morphological, biochemical, and DNA abnormalities. Ann. Neurol. 2001;49:377–383. [PubMed] [Google Scholar]
- Ferreira C.R., Meirelles F.V., Yamazaki W., Chiaratti M.R., Méo S.C., Perecin F., Smith L.C., Garcia J.M. The kinetics of donor cell mtDNA in embryonic and somatic donor cell-derived bovine embryos. Cloning and Stem Cells. 2007;9:618–629. doi: 10.1089/clo.2006.0082. [DOI] [PubMed] [Google Scholar]
- Flood J.T., Chillik C.F., van Uem J.F., Iritani A., Hodgen G.D. Ooplasmic transfusion: prophase germinal vesicle oocytes made developmentally competent by microinjection of metaphase II egg cytoplasm. Fertil. Steril. 1990;53:1049–1054. [PubMed] [Google Scholar]
- Giles R.E., Blanc H., Cann H.M., Wallace D.C. Maternal inheritance of human mitochondrial DNA. Proc. Natl. Acad. Sci. U. S. A. 1980;(77):6715–6719. doi: 10.1073/pnas.77.11.6715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greggains G.D., Lister L.M., Tuppen H.A.L., Zhang Q., Needham L.H., Prathalingam N., Hyslop L.A., Craven L., Polanski Z., Murdoch A.P. Therapeutic potential of somatic cell nuclear transfer for degenerative disease caused by mitochondrial DNA mutations. Sci. Report. 2014:4. doi: 10.1038/srep03844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregorová S., Divina P., Storchova R., Trachtulec Z., Fotopulosova V., Svenson K.L., Donahue L.R., Paigen B., Forejt J. Mouse consomic strains: exploiting genetic divergence between Mus M. musculus and Mus M. domesticus subspecies. Genome Res. 2008;18:509–515. doi: 10.1101/gr.7160508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajian M., Kiani M., Hosseini M.S., Ostadhosseini S., Forouzanfar M., Afrough M., Nasr-Esfahani M.H. Specific activation requirements of zona-free sheep oocytes before and after somatic cell nuclear transfer. Cell Rep. 2013;15:247–257. doi: 10.1089/cell.2012.0089. [DOI] [PubMed] [Google Scholar]
- Harman D. Aging: a theory based on free radical and radiation chemistry. J. Gerontol. 1956;11:298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- Hook E.B. Rates of chromosome abnormalities at different maternal ages. Obstet. Gynecol. 1981;58:282–285. [PubMed] [Google Scholar]
- Huang C.C., Cheng T.C., Chang H.H., Chang C.C., Chen C.I., Liu J., Lee M.S. Birth after the injection of sperm and the cytoplasm of tripronucleate zygotes into metaphase II oocytes in patients with repeated implantation failure after assisted fertilization procedures. Fertil. Steril. 1999;72:702–706. doi: 10.1016/s0015-0282(99)00309-x. [DOI] [PubMed] [Google Scholar]
- Huang L., Sauve R., Birkett N., Fergusson D., van Walraven C. Maternal age and risk of stillbirth: a systematic review. Can. Med. Assoc. J. 2008;178:165–172. doi: 10.1503/cmaj.070150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs L.J.A.M., de Wert G., Geraedts J.P.M., de Coo I.F.M., Smeets H.J.M. The transmission of OXPHOS disease and methods to prevent this. Hum. Reprod. Update. 2006;12:119–136. doi: 10.1093/humupd/dmi042. [DOI] [PubMed] [Google Scholar]
- Kamimura S., Inoue K., Ogonuki N., Hirose M., Oikawa M., Yo M., Ohara O., Miyoshi H., Ogura A. Mouse cloning using a drop of peripheral blood. Biol. Reprod. 2013;89:24. doi: 10.1095/biolreprod.113.110098. [DOI] [PubMed] [Google Scholar]
- Kirkwood T.B.L. Understanding the odd science of aging. Cell. 2005;120:437–447. doi: 10.1016/j.cell.2005.01.027. [DOI] [PubMed] [Google Scholar]
- Kirkwood T.B.L. A systematic look at an old problem. Nature. 2008;451:644–647. doi: 10.1038/451644a. [DOI] [PubMed] [Google Scholar]
- Larsson N.G., Clayton D.A. Molecular genetic aspects of human mitochondrial disorders. Annu. Rev. Genet. 1995;29:151–178. doi: 10.1146/annurev.ge.29.120195.001055. [DOI] [PubMed] [Google Scholar]
- Maritim A.C., Sanders R.A., Watkins J.B., 3rd Diabetes, oxidative stress, and antioxidants: a review. J. Biochem. Mol. Toxicol. 2003;17:24–38. doi: 10.1002/jbt.10058. [DOI] [PubMed] [Google Scholar]
- Mecocci P., Fanó G., Fulle S., MacGarvey U., Shinobu L., Polidori M.C., Cherubini A., Vecchiet J., Senin U., Beal M.F. Age-dependent increases in oxidative damage to DNA, lipids, and proteins in human skeletal muscle. Free Radic. Biol. Med. 1999;26:303–308. doi: 10.1016/s0891-5849(98)00208-1. [DOI] [PubMed] [Google Scholar]
- Nuffield Council on Bioethics . Nuffield Council of Bioethics; London: 2012. Report – Novel Techniques for the prevention of mitochondrial DNA disorders: an ethical review. [Google Scholar]
- Paull D., Emmanuele V., Weiss K.A., Treff N., Stewart L., Hua H., Zimmer M., Kahler D.J., Goland R.S., Noggle S.A. Nuclear genome transfer in human oocytes eliminates mitochondrial DNA variants. Nature. 2013;493:632–637. doi: 10.1038/nature11800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peat J.R., Reik W. Incomplete methylation reprogramming in SCNT embryos. Nat. Genet. 2012;44:965–966. doi: 10.1038/ng.2393. [DOI] [PubMed] [Google Scholar]
- Pfeffer G., Majamaa K., Turnbull D.M., Thorburn D., Chinnery P.F. Treatment for mitochondrial disorders. Cochrane Database Syst. Rev. 2012;4 doi: 10.1002/14651858.CD004426.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy P., Ocampo A., Suzuki K., Luo J., Bacman S.R., Williams S.L., Sugawara A., Okamura D., Tsunekawa Y., Wu J. Selective elimination of mitochondrial mutations in the germline by genome editing. Cell. 2015;161:459–469. doi: 10.1016/j.cell.2015.03.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynier P., May-Panloup P., Chrétien M.F., Morgan C.J., Jean M., Savagner F., Barrière P., Malthièry Y. Mitochondrial DNA content affects the fertilizability of human oocytes. Mol. Hum. Reprod. 2001;7:425–429. doi: 10.1093/molehr/7.5.425. [DOI] [PubMed] [Google Scholar]
- Reznichenko A., Huyser C., Pepper M.S. Mitochondrial transfer: ethical, legal and social implications in assisted reproduction. S. Afr. J. Bioeth. Law. 2015;8:32–35. [Google Scholar]
- Richter C. Oxidative damage to mitochondrial DNA and its relationship to ageing. Int. J. Biochem. Cell Biol. 1995;27:647–653. doi: 10.1016/1357-2725(95)00025-k. [DOI] [PubMed] [Google Scholar]
- Roberts R.M. Prevention of human mitochondrial (mtDNA) disease by nucleus transplantation into an enucleated donor oocyte. Am. J. Med. Genet. 1999;87:265–266. doi: 10.1002/(sici)1096-8628(19991126)87:3<265::aid-ajmg14>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- Ruiz-Pesini E., Mishmar D., Brandon M., Procaccio V., Wallace D.C. Effects of purifying and adaptive selection on regional variation in human mtDNA. Science. 2004;303:223–226. doi: 10.1126/science.1088434. [DOI] [PubMed] [Google Scholar]
- Steinborn R., Schinogl P., Zakhartchenko V., Achmann R., Schernthaner W., Stojkovic M., Wolf E., Müller M., Brem G. Mitochondrial DNA heteroplasmy in cloned cattle produced by fetal and adult cell cloning. Nat. Genet. 2000;25:255–257. doi: 10.1038/77000. [DOI] [PubMed] [Google Scholar]
- Tachibana M., Sparman M., Sritanaudomchai H., Ma H., Clepper L., Woodward J., Li Y., Ramsey C., Kolotushkina O., Mitalipov S. Mitochondrial gene replacement in primate offspring and embryonic stem cells. Nature. 2009;461:367–372. doi: 10.1038/nature08368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachibana M., Amato P., Sparman M., Woodward J., Sanchis D.M., Ma H., Gutierrez N.M., Tippner-Hedges R., Kang E., Lee H.-S. Towards germline gene therapy of inherited mitochondrial diseases. Nature. 2013;493:627–631. doi: 10.1038/nature11647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor R.W., Turnbull D.M. Mitochondrial DNA mutations in human disease. Nat. Rev. Genet. 2005;6:389–402. doi: 10.1038/nrg1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorburn D.R., Dahl H.H., Singh K.K. The pros and cons of mitochondrial manipulation in the human germ line. Mitochondrion. 2001;1:123–127. doi: 10.1016/s1567-7249(01)00013-7. [DOI] [PubMed] [Google Scholar]
- Turner N., Robker R.L. Developmental programming of obesity and insulin resistance: does mitochondrial dysfunction in oocytes play a role? Mol. Hum. Reprod. 2015;21:23–30. doi: 10.1093/molehr/gau042. [DOI] [PubMed] [Google Scholar]
- Van Blerkom J. Mitochondrial function in the human oocyte and embryo and their role in developmental competence. Mitochondrion. 2011;11:797–813. doi: 10.1016/j.mito.2010.09.012. [DOI] [PubMed] [Google Scholar]
- Van Blerkom J., Sinclair J., Davis P. Mitochondrial transfer between oocytes: potential applications of mitochondrial donation and the issue of heteroplasmy. Hum. Reprod. 1998;13:2857–2868. doi: 10.1093/humrep/13.10.2857. [DOI] [PubMed] [Google Scholar]
- Wallace D.C. The mitochondrial genome in human adaptive radiation and disease: on the road to therapeutics and performance enhancement. Gene. 2005;354:169–180. doi: 10.1016/j.gene.2005.05.001. [DOI] [PubMed] [Google Scholar]
- Wang Q., Ratchford A.M., Chi M.M.-Y., Schoeller E., Frolova A., Schedl T., Moley K.H. Maternal diabetes causes mitochondrial dysfunction and meiotic defects in murine oocytes. Mol. Endocrinol. 2009;23:1603–1612. doi: 10.1210/me.2009-0033. [DOI] [PMC free article] [PubMed] [Google Scholar]