

Abstract
Integrin-mediated adhesion of epithelial cells to extracellular matrix (ECM) proteins induces prolonged tyrosine phosphorylation and partial activation of epidermal growth factor receptor (EGFR) in an integrin-dependent and EGFR ligand-independent manner. Integrin-mediated activation of EGFR in epithelial cells is required for multiple signal transduction events previously shown to be induced by cell adhesion to matrix proteins, including tyrosine phosphorylation of Shc, Cbl, and phospholipase Cγ, and activation of the Ras/Erk and phosphatidylinositol 3′-kinase/Akt signaling pathways. In contrast, activation of focal adhesion kinase, Src, and protein kinase C, adhesion to matrix proteins, cell spreading, migration, and actin cytoskeletal rearrangements are induced independently of EGFR kinase activity. The ability of integrins to induce the activation of EGFR and its subsequent regulation of Erk and Akt activation permitted adhesion-dependent induction of cyclin D1 and p21, Rb phosphorylation, and activation of cdk4 in epithelial cells in the absence of exogenous growth factors. Adhesion of epithelial cells to the ECM failed to efficiently induce degradation of p27, to induce cdk2 activity, or to induce Myc and cyclin A synthesis; subsequently, cells did not progress into S phase. Treatment of ECM-adherent cells with EGF, or overexpression of EGFR or Myc, resulted in restoration of late-G1 cell cycle events and progression into S phase. These results indicate that partial activation of EGFR by integrin receptors plays an important role in mediating events triggered by epithelial cell attachment to ECM; EGFR is necessary for activation of multiple integrin-induced signaling enzymes and sufficient for early events in G1 cell cycle progression. Furthermore, these findings suggest that EGFR or Myc overexpression may provoke ligand-independent proliferation in matrix-attached cells in vivo and could contribute to carcinoma development.
Integrins are a family of heterodimeric transmembrane proteins that serve as receptors for extracellular matrix proteins such as fibronectin (FN), laminins, and collagens. Integrins act as important regulators of cell function through their ability to mediate adhesion to extracellular matrices, to induce cytoskeletal rearrangements, and to activate intracellular signaling pathways. The coordinated cellular response to matrix attachment through integrins has been shown to induce a panoply of changes in cell behavior, including alterations in cell survival, proliferation, spreading and migration, gene transcription, and differentiation (15, 18, 20, 21).
Many intracellular signaling molecules are activated by integrin engagement, including components of the Ras/Raf/MEK/Erk pathway, the phosphatidylinositol 3′-kinase (PI-3K)/Akt pathway, Src and Abl tyrosine kinases, focal adhesion kinase (FAK), Rho GTPases, the scaffolding proteins Cas, Cbl, and paxillin (and associated signaling molecules), and the serine kinases protein kinase C (PKC), p21-activated kinase (PAK), integrin-linked kinase (ILK), and myosin light chain kinase (MLCK) (51). Many of the signaling molecules activated by integrins are also activated by other receptor-ligand interactions (52). This has raised important issues regarding the basis for signal specificity and whether there is coordination between distinct receptor pathways within the cell. The possibility that integrins can coordinate their activities with other receptors is supported by several recent findings demonstrating interdependence and cross talk between different classes of cellular receptors (12, 34, 52, 54).
There are several examples of cross talk between integrins and receptor tyrosine kinase (RTK) pathways (14, 34, 52, 54). Growth factors that activate RTKs can regulate integrin-mediated events such as cell adhesion, cell spreading, and cell migration through alterations in integrin localization and activation (27, 32, 58). Conversely, signals generated by integrins are required for full activation of growth factor signaling pathways. For example, extracellular matrix-mediated adhesion is required for growth factor-induced cell cycle progression in fibroblasts (2, 16, 53). Integrins contribute to fibroblast cell cycle progression by regulating cyclin D1 expression through multiple pathways involving Erk, PI-3K, and the Rho family GTPases Rac, cdc42, and Rho (16, 44). Integrins also lower the levels of the negative cell cycle regulators p21cip1 and p27kip1 (10).
More-recent observations provide evidence for a distinct type of cross talk in which integrins can activate RTKs in the absence of exogenously added receptor ligands (35, 41). Several examples of RTKs activated by integrins include epidermal growth factor receptor (EGFR), insulin receptor, platelet-derived growth factor receptor (PDGFR), hepatocyte growth factor receptor (HGFR/Met), vascular endothelial growth factor receptor (VEGFR), and Ron (17, 36, 50, 55, 56, 63). The full implications of these interactions have yet to be fully understood; however, they suggest yet another mechanism for cross talk between integrin- and growth factor-linked pathways.
To better understand the role of integrin-induced RTK activation in integrin function, we examined which events triggered by attachment to integrin ligands are dependent on EGFR activation. These studies demonstrate that FN-induced EGFR activity is required for activation of a subset of integrin-induced signaling pathways and that these events are important for regulating early cell cycle events in epithelial cells. In addition, we show that integrin engagement may lead to EGF-independent proliferation under conditions where EGFR is overexpressed.
MATERIALS AND METHODS
Cells and adhesion assays.
Cos7 cells, obtained from the American Type Culture Collection (ATCC), and the Rat1 cell line clone 6 (38) were maintained in Dulbecco's modified Eagle medium (DMEM) (Gibco) supplemented with 10% fetal bovine serum (FBS) (Gibco), 2 mM glutamine, 50 U of penicillin, and 50 μg of streptomycin/ml. CV1 cells, obtained from ATCC, were maintained in MEM-Hanks (Gibco) supplemented with 10% FBS, 1 mM sodium pyruvate, 0.1 mM NEN amino acids, 25 mM HEPES, 2 mM glutamine, 50 U of penicillin, and 50 μg of streptomycin/ml. Keratinocytes were obtained from Clonetics and cultured under the supplier's recommended conditions. Prostate epithelial cells isolated from normal human tissue were cultured as previously described (22). Adhesion assays were performed as described previously (33). FN, collagen I, and laminin were obtained from Becton Dickinson. Polylysine and tamoxifen were purchased from Sigma. The EGFR inhibitors AG1478 (1 μM; Calbiochem) and PD168393 (0.5 μM; Calbiochem) were used at doses causing more than 90% inhibition of EGFR phosphorylation. AG1478, PD168393, U0126 (10 μM; Calbiochem), or wortmannin (100 nM; Calbiochem) was added to suspended cells 15 min prior to plating. EGF (Upstate Biochemical) or HGF (Calbiochem), when used, was added at the time of plating.
Antibodies.
Monoclonal antibodies against PKCα, PKCδ, PKCɛ, EGFR, Erk, p27, and FAK and polyclonal Shc antibodies were purchased from Transduction Labs. Polyclonal antibodies against FAK, Cbl, cyclin E, cyclin A, cdk2 (M2), cdk4 (C22), PKCβI, PKCβII, and PKCδ were purchased from Santa Cruz. A second immunoprecipitating anti-EGFR monoclonal antibody was obtained from ATCC (HB-8508). Monoclonal antibodies against p21 and cyclin D1 were purchased from Becton Dickinson. A monoclonal antibody against EGFR (Ab12), used for immunoblotting, was purchased from BioMarkers. Phosphospecific antibodies against Erk1/2 (T202/Y204), Rb (Thr821), and Akt (S473) were purchased from New England Biolabs. Phosphospecific antibodies against Src (Y418), EGFR, and Rb (S807/811) were from BioSource International. Monoclonal anti-PLCγ1 and polyclonal anti-Myc antibodies were purchased from Upstate Biochemical. Monoclonal blocking antibodies against α5 integrin (BIIG2) and β1 integrin (AIIB2) were obtained from the Iowa Hybridoma Bank. The monoclonal anti-Src antibody 327 has been described previously (30). The anti-Met monoclonal antibody (D1) was obtained from George Vande Woude. The monoclonal antibody against the hemagglutinin (HA) tag was generated in cell culture from the 12CA5 hybridoma (61). The anti-phosphotyrosine monoclonal antibody 4G10 was provided by Tom Roberts, Dana Farber Cancer Institute. The antibody against the C-terminal region of FAK (FRNK) was generated by using a glutathione S-transferase (GST)-FRNK fusion protein purified from bacteria that was submitted to Covance for polyclonal antibody production. The antibody against Akt was generated against the synthetic peptide RPHFPQFSYSASGTA from the C terminus of murine Akt1 (synthesized by Charles Dahl, Department of Biochemistry and Molecular Pharmacology, Harvard Medical School). The peptide was conjugated to keyhole limpet hemocyanin by glutaraldehyde treatment as described elsewhere (4), and the conjugate was submitted to Covance for polyclonal antibody production. The final bleeds were pooled, and peptide-blocking experiments confirmed that the antibody was specific for Akt.
Constructs and plasmid transfections.
Generation of p75.EGFR.F1.HA and of the Rat1 cell line clone 6, expressing p75.EGFR.F2.HA, has been described previously (38). The myr.EGFR.F2.HA expression vector was constructed as follows. A synthetic oligonucleotide encoding the myristoylation domain of c-Src and an in-frame 3′ Xba1 site and stop codons was subcloned into the mammalian expression vector pCG (57) to generate plasmid pCM. Two copies of FKBP domains containing a C-terminal HA epitope tag were subcloned into the XbaI site of the pCM vector as an XbaI/SpeI fragment to generate plasmid pCM.F2.HA. The entire cytoplasmic domain of human EGFR (amino acids 669 to 1210) was amplified by PCR as an XbaI/SpeI fragment and subcloned into the XbaI site of pCM.F2.HA (pCM.EGFR.F2.HA). The DNA sequence of the PCR product was verified. pSVL-HA-Erk was a gift from Mike Weber, University of Virginia. pDest12.2-dnEGFR was generated by subcloning the XhoI/HindIII fragment of pMLVneo-EGFRK721M (a gift from Alan Wells, University of Pittsburgh, Pittsburgh, Pa.) into pBluescript, subcloning the resulting EGFRK721M KpnI/XbaI fragment into the Gateway entry vector pENTR3C (Gibco), and then recombining EGFRK721M into the pDEST12.2 expression vector (Gibco). Cos7 cells were transfected at 106 per 10-cm-diameter plate with 4 μg of total DNA by using LT1 lipid as described by the manufacturer (Panvera). Forty-eight hours after transfection, cells were used in adhesion assays as described above.
Virus production and infection.
pLPCX-EGFR was generated by subcloning the EGFR XhoI/HindIII fragment of pMLVneo-EGFR into pLPCX (Clontech) as described previously (43). pCLXSN-cyclin D1 was generated by subcloning the cyclin D1 EcoRI/XhoI fragment of pX-cyclin D1 (a gift from Giulio Draetta) into the EcoRI/XhoI site of pCLXSN (Imgenex). pCLXSN-MycER was generated by subcloning the MycERG525R EcoRI fragment of pBabe-MycER (a generous gift from Martin Eilers) (6, 31) into the EcoRI site of pCLXSN. The orientation was verified by sequencing. The VSV-GPG-293 retroviral packaging cell line (39) was transfected with 15 μg of an empty vector or pLPCX-GFP-FRNK, pLPCX-EGFR, pCLXSN-cyclin D1, or pCLXSN-MycER by using LT1 lipid reagent (Panvera). Five and six days later, the cell medium was harvested, pooled, filtered, and used to infect CV1 cells in the presence of 8 μg of Polybrene/ml. Seventy-two hours after infection, cells were selected in 7 μg of puromycin/ml or 400 μg of G418/ml for 2 weeks. Surviving cells were pooled, maintained in standard CV1 growth medium, and analyzed as described in Results.
siRNA and transfections.
Small interfering RNA (siRNA) to p27 was designed according to the criteria outlined by Elbashir et al. (19). The sequence targeted was AACCCGGGACTTGGAGAAGCA, a region whose sequence is conserved in humans and rodents. The siRNA was synthesized by Xerogon. The sense strand was r(CCCGGGACUUGGAGAAGCA)d(TT), and the antisense strand was r(UGCUUCUUCAAGUCCCGGG)d(TT). A scrambled siRNA recommended by Ambion was used as a control. siRNAs were transfected into 7.5 × 105 CV1 cells on 100-mm-diameter plates by using 2 μl of 20 μM siRNA and 4 μl of Oligofectamine (Gibco) according to the manufacturer's protocol. Forty-eight hours later, cells were used in adhesion assays as described above.
IPs and immunoblotting.
Cells adherent to FN or left in suspension were lysed in radioimmunoprecipitation assay (RIPA) buffer (10 mM Tris [pH 7.2], 158 mM NaCl, 1 mM EDTA, 0.1% sodium dodecyl sulfate [SDS], 1% sodium deoxycholate, 1% Triton X-100, 1 mM Na3VO4, 1 mM phenylmethylsulfonyl fluoride [PMSF], 100 U of aprotinin/ml, and 10 μg of leupeptin/ml), passed through a 25-gauge needle, and clarified by centrifugation at 13,000 × g for 10 min. Protein concentrations were determined by using the bicinchoninic acid assay (Pierce). For blotting with phosphospecific antibodies, cells were lysed in Triton-X buffer (20 mM Tris [pH 7.5], 0.5 mM EDTA, 1 mM EGTA, 0.27 M sucrose, 1% Triton X-100, 50 mM NaF, 10 mM β-glycerophosphate, 5 mM sodium pyrophosphate, 1 mM Na3VO4, 1 mM PMSF, 100 U of aprotinin/ml, and 10 μg of leupeptin/ml). Immunoprecipitation (IP) mixtures were incubated for 3 h at 4°C with protein G- or protein A-conjugated agarose beads (Pierce) to capture the complexes. All IP mixtures were washed three times with lysis buffer, resuspended in 2× SDS sample buffer, boiled, and analyzed by SDS-polyacrylamide gel electrophoresis. Gels were transferred to a polyvinylidene difluoride membrane and probed by immunoblotting. After being blocked in 5% bovine serum albumin in Tris-buffered saline containing 0.1% Tween 20 and incubated with the primary antibody, blots were incubated with a horseradish peroxidase-conjugated secondary antibody (Bio-Rad) and visualized with a chemiluminescence reagent (NEN). Blots were stripped in low-pH 2% SDS at 65°C for 60 min, rinsed, and reprobed as indicated.
Cell fractionation.
Cells were lysed in hypotonic cytosolic buffer (10 mM Tris [pH 7.4], 0.5 mM EDTA, 1 mM Na3VO4, 1 mM PMSF, 10 μg of leupeptin/ml, and 100 U of aprotinin/ml) for 20 min on ice after being washed with phosphate-buffered saline. Cells were collected and broken open by Dounce homogenization. The soluble cytosolic fraction (S30) was collected after centrifugation at 30,000 × g for 30 min. The pellets were resuspended in RIPA buffer, and supernatants (P30) were collected after centrifugation at 14,000 × g. The ratio of cytosolic protein to RIPA soluble protein was maintained when extracts were loaded onto SDS gels.
Cell cycle.
For cell cycle studies, all cells at confluency were serum starved for 48 h in DMEM containing 0.1% FBS. Cells were trypsinized, placed in suspension, pretreated with the indicated drugs, and then plated onto FN-coated tissue culture plates or plated at 2 × 104 cells in FN-coated 8-chamber slides (Nunc). Growth factors, when present, and tamoxifen (for activation of estrogen-inducible Myc [MycER]) were added at the time of plating, and cells were incubated for various times up to 22 h. To generate cell extracts, cells on FN-coated plates were lysed in a solution containing 50 mM Tris (pH 7.4), 250 mM NaCl, 2 mM EDTA, 1% NP-40, 50 mM NaF, 0.1 mM Na3VO4, 1 mM PMSF, 10 μg of leupeptin/ml, and 10 μg of aprotinin/ml. For bromodeoxyuridine (BrdU) incorporation assays, cell were incubated in chamber slides for 16 h, 10 μM BrdU was added for two to four additional hours, and then cells were fixed and stained by using a BrdU labeling kit (Boehringer Mannheim). Cells were counterstained with Hoechst 33258, and the percentage of BrdU-labeled (green) cells versus total nuclei stained (blue) was determined. Four different areas in each chamber were counted, the numbers were averaged, and the standard deviation was calculated. The t test was used to determine significance.
In vitro kinase assays.
cdk2 kinase assays were carried out as described previously (8), after cells were lysed in the cell cycle lysis buffer by using 1 μg of histone H1 as the substrate (Gibco). cdk4 kinase assays were performed as described previously (8) by using GST-Rb (amino acids 769 to 921) (Santa Cruz) as the substrate, except that cells were lysed by sonication in a solution containing 50 mM HEPES (pH 7.5), 150 mM NaCl, 0.1% Tween, 10% glycerol, 1 mM EDTA, 2.5 mM EGTA, 10 mM β-glycerolphosphate, 1 mM NaF, 1 mM dithiothreitol, 1 mM Na3VO4, 1 mM PMSF, 10 μg of aprotinin/ml, and 10 μg of leupeptin/ml. Digital images of the autoradiographs were quantified by using ScanImage software (Scion Corp.).
RESULTS
Integrin-induced activation of EGFR.
To test directly whether adhesion of cells to extracellular matrices is sufficient to activate EGFR, serum-starved Cos7 or CV1 cells were placed in suspension in the absence of serum or growth factors for 30 min. Attachment to FN-, laminin-, or collagen I-coated plates for 20 min led to induction of tyrosine phosphorylation of EGFR (Fig. 1A and B). This phosphorylation was sustained for more than 12 h after plating on FN (Fig. 1D). Adhesion to polylysine for 1 h similarly induced EGFR activation, but it was not sustained. After 6 h, EGFR levels decreased, indicating that integrin engagement is required for stable expression of EGFR (Fig. 1D). Polylysine-stimulated EGFR phosphorylation at early times was partially dependent on integrin engagement, since inhibition of integrins with blocking antibodies against α5 or β1 integrin reduced EGFR phosphorylation induced by polylysine (Fig. 1C). The ability of polylysine to stimulate integrins is most probably mediated by polylysine engagement of syndecans, as previously reported (68). Integrin-induced EGFR phosphorylation was observed in several different epithelial cells, including the prostate cell lines, DU145 and RWPE-1, telomerase-immortalized retinal epithelial cells, A431, and primary prostate epithelial cells (data not shown).
FIG. 1.
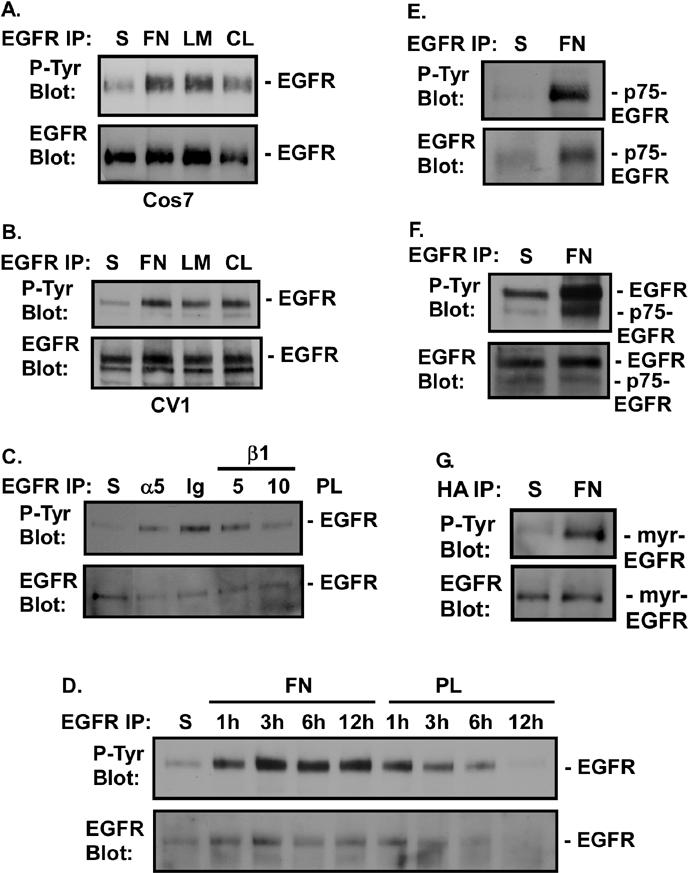

FN-induced phosphorylation of EGFR. (A and B) Cos7 (A) or CV1 (B) cells were either held in suspension (S) or plated on FN, laminin (LM), or collagen I (CL) for 20 min. (C) CV1 cells were either held in suspension or treated with 10 μg of immunoglobulin G/ml (Ig), 10 μg of an anti-α5 antibody/ml (α5), or 5 or 10 μg of an anti-β1 integrin antibody/ml (β1) for 1 h. Cells were plated on polylysine (PL) and incubated for 30 min. (D) CV1 cells were either held in suspension or plated on FN or polylysine for the indicated times. EGFR phosphorylation in EGFR immunoprecipitates was measured by immunoblotting with an anti-phosphotyrosine monoclonal antibody (P-Tyr Blot). Total levels of EGFR in the immunoprecipitates were analyzed by immunoblotting with an anti-EGFR antibody (EGFR Blot). (E through G) Rat1 cells stably expressing the p75-EGFR chimera (the p75 extracellular and transmembrane domains fused to the cytoplasmic domain of EGFR) (E) or Cos7 cells transiently transfected with either 0.5 μg of pML-p75.EGFR.F2.HA, encoding the p75-EGFR chimera (F), or 0.1 μg of pML-EGFR-F1-HA, encoding a myristoylated EGFR cytoplasmic domain (myr-EGFR) (G), were either held in suspension or plated on FN for 20 min. Levels of tyrosine phosphorylation of the chimeras were analyzed by immunoblotting of EGFR or HA immunoprecipitates with anti-phosphotyrosine antibodies. Total levels of p75-EGFR or myr-EGFR in the immunoprecipitates were measured by immunoblotting with anti-EGFR antibodies. (Note that in panel F, endogenous levels of EGFR are also shown.) (H) Control cells expressing endogenous levels of EGFR (Endog) or EGFR-overexpressing cells (EGFR1) were either placed in suspension, plated on FN for 30 min, or stimulated with 2 ng of EGF/ml for 5 min. EGFR was immunoprecipitated from cell extracts, and total levels of tyrosine phosphorylation (P-Tyr Blot) or phosphorylation of one of six different tyrosines (Y845, Y992, Y1068, Y1086, Y1148, or Y1173) was monitored by immunoblotting with anti-phospho-EGFR antibodies. Total levels of EGFR in the immunoprecipitates were analyzed by immunoblotting with an anti-EGFR antibody. Digital images were quantified by using ScanImage software (Scion Corp). The fold difference in tyrosine phosphorylation between FN- and EGF-treated cells is given below each set of panels.
Phosphorylation of EGFR is mediated intracellularly.
Integrins could induce EGFR phosphorylation through the release of EGFR ligands, through physical interactions with the EGFR extracellular domain, or through an intracellular event leading to activation of the catalytic domain. To rule out a role for EGFR ligands, we examined FN-induced activation of two different EGFR mutants lacking the extracellular binding domain. Both p75-EGFR, a chimera containing the cytoplasmic domain of EGFR and the extracellular and transmembrane domains of p75 low-affinity nerve growth factor receptor, and myr-EGFR, which contains the cytoplasmic domain of EGFR linked to a myristoylation motif for membrane association, were activated following adhesion to FN (Fig. 1E, F, and G). Additionally, conditioned medium from confluent cells failed to stimulate EGFR activation in serum-starved cells (data not shown). Furthermore, treatment of CV1 cells with the inhibitor of HB-EGF release 1,10-phenanthroline or diphtheria toxin (45) failed to block FN-induced EGFR activation (data not shown). Together these data demonstrate that the cytoplasmic domain of EGFR is sufficient for its activation through FN receptors. While these data do not rule out a role for FN in inducing the secretion of low levels of an EGFR ligand or a physical interaction between integrins and the extracellular domain of EGFR, they indicate that such events are not required for matrix-induced EGFR activation.
FN activates a subset of EGFR molecules and phosphorylation sites.
The ability of FN to activate EGFR independently of a ligand suggested that integrins might activate EGFR differently from EGF. Using phosphospecific anti-EGFR antibodies, we examined which sites on EGFR are inducibly phosphorylated following adhesion to FN (Fig. 1H). Quantitative analysis indicated that overall, EGF was 11 times more effective at inducing total EGFR tyrosine phosphorylation than FN (Fig. 1H, P-Tyr Blot). However, when individual sites were analyzed, relative to EGF, FN preferentially induced phosphorylation of Y845, Y992, Y1068, and Y1086. The relative difference between EGF and FN decreased from 11-fold overall to 5-, 2-, 4.8-, and 6.1-fold for these respective sites. FN failed to effectively phosphorylate EGF at Y1148 and Y1173. EGF was 9 and 22 times more effective at phosphorylating these sites than FN.
Overexpression of EGFR resulted in increased FN-induced tyrosine phosphorylation of EGFR overall (2.7- versus 11-fold). Phosphorylation at the sites that were poorly phosphorylated in control cells, namely, Y1148 and Y1173, was specifically increased when EGFR was overexpressed. The fold difference between FN- and EGF-induced tyrosine phosphorylation at Y1148 and Y1173 decreased from 9- and 22-fold, respectively, in control cells, to 3-fold in EGFR-overexpressing cells. There was also a 2-fold increase in phosphorylation at Y1086 (6.1- versus 3-fold). The relative phosphorylation at Y845, Y992, or Y1068 by EGF versus FN was not changed by EGFR overexpression. Together these data indicate that integrin induction of EGFR is different from that observed in response to EGF ligand. FN preferentially induces phosphorylation of EGFR on a subset of sites, and fewer EGFR molecules are activated by integrins than are activated by EGF, even when EGFR is overexpressed.
Integrin signaling is regulated by EGFR.
Stimulation of EGFR by its natural ligands leads to activation of many of the same signaling molecules that are activated by FN receptors. The evidence presented here that attachment to extracellular matrices leads to activation of EGFR raises the question of whether any of the integrin-induced signals are transduced through activated EGFR. To address this question, we examined the effects of inhibition of endogenous EGFR activity on FN-induced activation of several signaling molecules by using two different EGFR-specific inhibitors, AG1478 and PD168393.
Both EGFR inhibitors blocked FN-induced EGFR tyrosine phosphorylation (Fig. 2A), indicating that EGFR is autophosphorylated following integrin engagement. Inhibition of EGFR activation also blocked FN-induced tyrosine phosphorylation of the adaptor protein Shc and its downstream effector, the serine-threonine kinase Erk2 (Fig. 2B and C). A dominant interfering mutant of EGFR, containing a lysine-to-methionine mutation at K721, also blocked FN-induced Erk activation (Fig. 2G). Previous studies found Erk activation by integrins to be dependent on FAK (47, 49). We blocked integrin-induced FAK activation by overexpressing FRNK, an interfering mutant of FAK. Expression of FRNK blocked integrin-induced FAK phosphorylation and reduced the levels of tyrosine phosphorylation in focal adhesions (data not shown). However, expression of FRNK failed to block integrin-induced activation of Erk in CV1 (Fig. 2H) or Cos7 (data not shown) cells. Together these data indicate that the major FN-induced pathway leading to Shc and Erk2 activation in Cos7 cells involves activation of EGFR. Similar results were obtained with CV1 cells, primary prostate epithelial cells, and A431 cells (data not shown).
FIG. 2.

EGFR is required for FN-induced signaling events. Cos7 cells were placed in suspension (S), either left untreated or treated with 1 μM AG1478 or 0.5 μM PD168393, and plated on FN for 30 min. (A through F) EGFR (A), Shc (B), Erk (C), Akt (D), Cbl (E), and PLCγ (F) phosphorylation was analyzed by immunoblotting of EGFR, Shc, PLCγ, or Cbl immunoprecipitates with an anti-phosphotyrosine monoclonal antibody (P-Tyr Blot) or by immunoblotting of total-cell extracts with a phosphospecific antibody against Erk (P-Erk Blot) or Akt (P-Akt Blot). Total levels of EGFR, Shc, Erk, Akt, PLCγ, or Cbl in the immunoprecipitates or cell lysates were analyzed by immunoblotting with their respective antibodies (EGFR Blot, Shc Blot, Erk Blot, Akt Blot, PLCγ Blot, and Cbl Blot). (G) Cos7 cells were transfected with 0.2 μg of pSLV-HA-Erk2 and 1 μg of pDEST12.2-dnEGFR (Dn) or 1 μg of vector (Vec). Forty-eight hours later, cells were harvested and either placed in suspension or plated on FN. The level of Erk activation was measured by immunoblotting of HA immunoprecipitates with an anti-P-Erk antibody. Total levels of Erk2 were measured by immunoblotting with an anti-Erk antibody. The level of dnEGFR expressed (Tf Dn) compared to endogenous EGFR expression (En Wt) was measured by immunoblotting cell lysates with an anti-EGFR antibody. (H) CV1 cells were infected with a virus containing an empty pLPCX vector (Vec) or a virus expressing GFP-FRNK. Erk activation in the vector- or GFP-FRNK-expressing cells was monitored in cell lysates with a phosphospecific anti-Erk antibody. Total levels of Erk in the immunoprecipitates were analyzed by immunoblotting with anti-Erk antibodies. Levels of GFP-FRNK expression were monitored by immunoblotting with an anti-FRNK antibody. Levels of endogenous FAK are also shown.
Activation of the PI-3K/Akt pathway by FN was assayed by examining activation-specific phosphorylation of Akt/PKB by use of the Akt/PKB phosphospecific Ser473 antibody. Inhibition of EGFR kinase activity with AG1478 or PD168393 blocked FN-induced activation of Akt/PKB (Fig. 2D). Inhibition of EGFR activation also blocked FN-induced tyrosine phosphorylation of Cbl and phospholipase Cγ (PLCγ) (Fig. 2E and F). Thus, in cells where EGFR is expressed at normal levels, FN-induced activation of the Ras/Erk signaling pathway, the PI-3K/Akt pathway, and several EGFR effectors is dependent on the ability of FN to activate EGFR.
Treatment of cells with AG1478 or PD168393 had no effect on the ability of FN to activate FAK (Fig. 3A) and Src (Fig. 3B), to translocate PKCα, PKCβI, and PKCɛ to the membrane (Fig. 3C), or to induce tyrosine phosphorylation of PKCδ (Fig. 3D). Together these data indicate that FN-induced activation of FAK, Src, and several PKC isoforms is not dependent on EGFR. Similar results were obtained with CV1, A431, and primary prostate cells (data not shown). Thus, FN-induced activation of a subset of signaling pathways in epithelial cells is dependent on FN-induced activation of EGFR, and another set of signaling events is activated independently of EGFR (Fig. 3E).
FIG. 3.


EGFR is not required for all FN-induced signaling events. Cos7 cells were placed in suspension (S), either left untreated or treated with 1 μM AG1478 or 0.5 μM PD168393, and plated on FN. (A) Tyrosine phosphorylation of FAK was analyzed by immunoblotting of FAK immunoprecipitates with an anti-phosphotyrosine monoclonal antibody (P-Tyr Blot). Total levels of FAK in the immunoprecipitates were monitored by immunoblotting with an anti-FAK monoclonal antibody (FAK Blot). (B) Src activation was measured by immunoblotting of Src immunoprecipitates with an anti-phospho-Src antibody (Y-416 Blot), and total levels of Src in the immunoprecipitates were analyzed by immunoblotting with the anti-Src monoclonal antibody 327 (Src Blot). (C) Following adhesion, Cos7 cells were fractionated into soluble cytoplasmic (Su) and detergent-soluble pellet (Pe) fractions, and the level of each PKC isoform in each fraction was analyzed by immunoblotting with specific anti-PKC antibodies as indicated (PKC Blots). (D) Tyrosine phosphorylation of PKCδ was analyzed by immunoblotting of PKCδ immunoprecipitates of the cytoplasmic and detergent-soluble fractions (P-Tyr Blot). Total levels of PKCδ in the immunoprecipitates were monitored by immunoblotting with an anti-PKCδ monoclonal antibody (PKCδ Blot). Note that membrane-associated PKCδ migrates more slowly in gels due to phosphorylation. (E) Model depicting the EGFR dependence of a subset of integrin-mediated signaling events, specifically Cbl, PLCγ, Shc, Erk, and Akt. Also shown is the EGFR independence of Src, FAK, and PKC activation. Integrin activation of FAK is dependent on actin polymerization (data not shown).
EGFR does not regulate cytoskeletal events.
Both EGF and integrins can regulate actin cytoskeletal rearrangements. To determine if integrin-mediated events involving actin cytoskeletal rearrangements are controlled by EGFR, Cos7 cells were treated with AG1478 to block EGFR activation, and the effects on cell adhesion, spreading, and migration were monitored. Inhibition of EGFR activity did not alter integrin-mediated cell spreading, focal adhesion formation, or stress fiber formation of cells plated on FN (data not shown). Inhibition of EGFR did not block the ability of cells to fill in a “wound” on FN-coated plates or block their ability to migrate through transwell membranes coated with FN (data not shown).
EGFR mediates integrin-induced G1 cell cycle entry.
Both Erk and PI-3K are important regulators of the cell cycle (25), and their activation by FN is dependent on EGFR. Therefore, we explored the possibility that integrin activation of EGFR is sufficient to induce G1 cell cycle entry. Adhesion of CV1 cells to FN in the absence of exogenous growth factors induced an increase in cyclin D1 and p21 levels, Rb phosphorylation at Ser807/811, and activation of cdk4 (1.4- ± 0.1-fold) 12 h after plating on FN (Fig. 4A, C, and D). Induction of these events was similar to that observed by treatment with 40 ng of EGF/ml or was only slightly reduced in intensity. Inhibition of EGFR activity by AG1478 (Fig. 4A and B) or PD168393 (Fig. 4C and D) blocked integrin induction of these cell cycle events by FN or EGF, indicating that EGFR activation by FN is mediating G1 cell cycle entry. However, adhesion to FN failed to reduce p27 levels or induce cyclin A production and induced cdk2 activity poorly (1.74- ± 0.03-fold) (Fig. 4A, B, and D; see also Fig. 7A). In contrast, EGF induced p27 loss and cdk2 activation (2.69- ± 0.01-fold) 12 h after stimulation and an increase in cyclin A synthesis at 18 h (Fig. 4A and B). No changes in cyclin E levels were seen with FN or EGF (Fig. 4A).
FIG. 4.

FN-induced EGFR activation is required for G1 cell cycle entry. (A and B) Adherent CV1 cells were serum starved for more than 48 h (Go), placed in suspension (S), left untreated or treated with 1 μM AG1478 (AG), and plated on FN in the absence (FN) or presence (EGF) of 40 ng of EGF/ml for 12 or 18 h. The time points selected were determined to be optimal for the events monitored in time course studies (data not shown). (A) The levels of cyclin D1 (cycD1), cyclin E (cycE), p27, p21, and Rb phosphorylation at Ser807/811 (P-Rb) at 12 h and the levels of cyclin A at 18 h were monitored by immunoblotting cell lysates with their respective antibodies. Total levels of Akt were monitored by immunoblotting as a control for total protein levels in the extracts. (B) cdk2 activity was measured by using in vitro kinase assays with histone H1 as a substrate and was quantified (1.74- ± 0.03-fold induction in the presence of FN only; 2.69- ± 0.01-fold in the presence of FN and EGF; n = 3). Preimmune (PI) and no-substrate (NS) controls were included. Total levels of kinase immunoprecipitated in the assays were monitored by immunoblotting with anti-cdk2 antibodies (cdk2). (C and D) CV1 cells were prepared as described above except that cells in suspension were pretreated with either 10 μM U0126 (U0), 100 nM wortmannin (WT), or 0.5 μM PD168393 (PD) 15 min prior to plating on FN. Results obtained after plating for 12 h are shown. (C) cdk4 kinase activity was monitored by an in vitro kinase assay by measuring phosphate incorporation into GST-Rb 12 h after plating on FN and was quantified (1.4- ± 0.2-fold in the presence or absence of EGF; n = 3). Preimmune and no-substrate controls were included. Total levels of kinase immunoprecipitated in the assays were monitored by immunoblotting with anti-cdk4 antibodies (cdk4). (D) Levels of cyclin D1, p27, and Rb phosphorylation at Ser807/811 were monitored by immunoblotting cell lysates with their respective antibodies. Total levels of Akt were monitored by immunoblotting as a control for total protein levels in the extracts.
FIG. 7.


EGFR and Myc overexpression rescues S phase. (A) CV1 cells were serum starved for 48 h and either left in suspension (S) or plated on FN in the absence (FN) or presence (EGF) of 40 ng of EGF/ml for the indicated times. The levels of cyclin D1 (cycD1), p27, cyclin A (cycA), and Rb phosphorylation at Thr821 (P-Rb T821) were monitored by immunoblotting with the respective antibodies. Total levels of Akt were monitored to control for total protein levels. (B) CV1 cells (CV) were infected with retroviruses expressing EGFR (R1, R5) or cyclin D1 (D1) or with an empty vector (V). Levels of EGFR and cyclin D1 expression were monitored by immunoblotting of whole-cell extracts with their respective antibodies after plating of cells on FN for 12 h. Levels of EGFR activation in virus-infected cells held in suspension or attached to FN were monitored by immunoblotting of EGFR immunoprecipitates with anti-phosphotyrosine antibodies (P-Tyr Blot). Total levels of EGFR were monitored by immunoblotting with an anti-EGFR antibody (EGFR Blot). *, the length of film exposure to enhanced chemiluminescence was 5 times less for EGFR-overexpressing cells than for other samples. (C) Adherent CV1 cells infected with retroviral vectors encoding either EGFR, cyclin D1, or no cDNA (Vector) were serum starved for 48 h and either placed in suspension or plated on FN for the indicated times. Levels of cyclin A and p27 were monitored by immunoblotting cell lysates with their respective antibodies. Rb phosphorylation at Thr821 was monitored by immunoblotting of cell lysates with phosphospecific antibodies. Total levels of Akt were monitored by immunoblotting as a control for total protein levels in the extracts. (D) CV1 cells were serum starved for 48 h and either placed in suspension or plated on FN in the absence or presence of 40 ng of EGF/ml for the indicated times. Adherent CV1 cells infected with retroviral vectors encoding EGFR or no cDNA were serum starved for 48 h and then either placed in suspension or plated on FN for the indicated times. Levels of Myc expression were monitored by immunoblotting of cell lysates with anti-Myc antibodies. Total levels of Akt were monitored by immunoblotting as a control for total protein levels in the extracts. (E) Adherent CV1 cells infected with retroviral vectors encoding either EGFR or no cDNA were serum starved for 48 h and then either placed in suspension or plated on FN for the indicated times. Levels of cyclin D1 were monitored by immunoblotting of cell lysates with anti-cyclin D1 antibodies. Erk and Akt activation was monitored by immunoblotting of cell lysates with phosphospecific antibodies (P-Erk, P-Akt). Total levels of Erk and Akt were monitored by immunoblotting as a control for total protein levels in the extracts (Erk, Akt). (F) CV1 cells were infected with a virus containing an empty vector or expressing MycER. Levels of MycER expression were monitored by immunoblotting with anti-Myc antibodies after plating on FN in the absence (FN) or presence (Tx) of 50 nM tamoxifen. Levels of p27 and cyclin A expression in cells in suspension or at the indicated times after plating on FN in the presence or absence of 50 nM tamoxifen were monitored by immunoblotting. Total levels of Akt were monitored by immunoblotting as a control for total protein levels in the extracts.
To assess the role of Erk and PI-3K in regulating FN-mediated cell cycle entry, CV1 cells were treated with the MEK inhibitor U0126 or the PI-3K inhibitor wortmannin. Inhibition of Erk blocked Rb phosphorylation, cyclin D1 induction, and cdk4 kinase activation induced by FN, indicating that integrin activation of Erk is required for cell cycle regulation by integrins (Fig. 4C and D). Inhibition of PI-3K blocked Rb phosphorylation but had only a partial effect on cyclin D1. These data indicate that PI-3K is also required for cell cycle regulation by FN.
Integrins regulate cell cycle entry in primary cells.
Although CV1 cells are not tumorigenic and do not display properties of transformed cells in vitro, they are an immortalized cell line that has been extensively passaged in culture. To determine if integrins are capable of regulating G1 cell cycle events in primary cells, we monitored integrin-induced levels of cyclin D1, p27, cyclin A, and Rb phosphorylation in two different primary epithelial cell types, prostate epithelial cells and keratinocytes. As was observed for CV1 cells, adhesion to FN for 12 h induced increased levels of cyclin D1 and Rb phosphorylation but did not reduce p27 levels or increase cyclin A levels (Fig. 5). In these experiments, EGF did not induce a decrease in p27 levels in keratinocytes at the concentration used, consistent with a failure to induce DNA synthesis in these cells (data not shown). We failed to observe induction of any G1 cell cycle events after FN attachment of the Rat1 fibroblast cell line, which expresses very low levels of EGFR (data not shown).
FIG. 5.
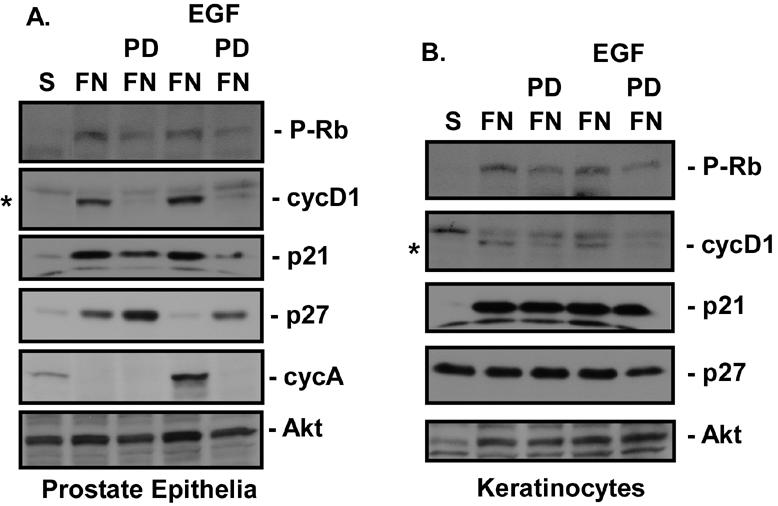
Adhesion stimulates G1 cell cycle events in primary cells. Adherent primary prostate epithelial cells (A) or primary keratinocytes (B) were serum starved for 24 h, placed in suspension (S), left untreated or treated with 0.5 μM PD168393 (PD), and plated on FN in the absence (FN) or presence (EGF) of 40 ng of EGF/ml for 18 h. Levels of cyclin D1 (cycD1), cyclin A (cycA), p27, p21, and Rb phosphorylation (P-Rb) were monitored by immunoblotting cell lysates with their respective antibodies. Note that the levels of Rb phosphorylation are less than optimal due to the late time point selected for this assay. *, cyclin D1 is the lower band. Total levels of Akt were monitored by immunoblotting as a control for total protein levels in the extracts.
Integrins are not sufficient to induce DNA synthesis.
The ability of FN to induce cyclin D1, phosphorylate Rb, and activate cdk4 raised the question whether adhesion to FN is sufficient to induce DNA synthesis. To determine if plating on FN is sufficient to stimulate entry into S phase, BrdU incorporation was monitored after plating on FN. Cells plated on FN for 18 to 20 h failed to incorporate significant amounts of BrdU; less than 4% of the cells stained positive for BrdU (Fig. 6A). Plating of cells on FN in the presence of serum, EGF, or HGF stimulated BrdU incorporation into more than 20% of the cells. Prostate epithelial cells treated with EGF or HGF were able to enter into S phase, however plating on FN alone also did not permit entry of the primary cells into S phase (Fig. 6B). These data indicate that while FN-mediated activation of EGFR is sufficient to stimulate some G1 cell cycle events, it is not sufficient for entry into S phase. Additional signals that would normally be provided by direct activation of EGFR or Met with ligands are required.
FIG. 6.

Adhesion is not sufficient for entry into S phase. (A) CV1 cells were plated onto FN-coated chamber slides in the absence (FN) or presence of 10% serum (FN + FBS), 200 ng of HGF/ml (FN + HGF), or 40 ng of EGF/ml (FN + EGF) and were allowed to adhere for 18 h. Two hours prior to fixation of cells, 10 μM BrdU was added to the medium. After fixation, cells were immunostained with anti-BrdU (green), and nuclei were counterstained with Hoechst 33258 (blue). The number of cells displaying nuclear BrdU staining was compared to the total number of cells in four fields, and the percent BrdU incorporation was calculated. A t test was used to determine significance. (B) Primary prostate epithelial cells were treated as described for panel A, except that 0.5 μM PD168393 (PD) was added to the cells 15 min prior to plating on FN. The level of BrdU incorporation was measured as described above.
Loss of p27 is not sufficient to rescue S phase.
Kinetic analysis of cell cycle markers indicated that although FN induces some early-G1 events, many late-G1 events, such as loss of p27, phosphorylation of Rb at Thr821 (a cdk2/cyclin E site [70]), and cyclin A synthesis, are not induced by FN (Fig. 7A). Treatment of FN-adherent cells with EGF was sufficient to restore induction of these late-G1 cell cycle events. Because integrin-mediated adhesion did not effectively reduce p27 levels in the absence of EGF and because p27 loss is required for subsequent G1 events (cdk2 activation and cyclin A synthesis), we proposed that down-regulation of p27 would allow cells plated on FN to enter into S phase. To address this possibility, we transfected CV1 or primary prostate epithelial cells with siRNA directed to a highly conserved region of human p27, assuming that this homology would be conserved in monkeys. Transfection of p27 siRNA resulted in a 90% loss of p27 expression as determined by immunoblotting (data not shown). However, p27 down-regulation failed to induce progression into S phase in the absence of EGF and also failed to induce activation of cdk2 (data not shown). These data indicate that down-regulation of p27 per se is not sufficient to rescue the block to DNA synthesis in cells plated on FN.
The failure to rescue cdk2 activation in the absence of p27 expression suggested that other molecules known to regulate cdk2 activity may also not be activated by integrins. Two sites of phosphorylation on cdk2, T14 and Y15, have to be dephosphorylated by cdc25A to permit cdk2 activation (23), and cdc25A levels are regulated by Myc (1). Myc also regulates p27 degradation (60). To determine if integrins are capable of activating Myc, we monitored the kinetics of Myc expression by immunoblotting of cell extracts from FN-adherent cells and EGF-treated cells. Adhesion to FN induced a small increase in Myc protein levels early in G1, around 8 h, which returned to basal levels by 12 h (Fig. 7D). Treatment with EGF resulted in a similar 8-h induction of Myc protein, but a second phase of Myc induction, which was absent in FN-adherent cells, was observed 16 to 19 h after treatment. These data suggest that the failure of FN-adherent cells to progress to S phase may be due to an inability to effectively activate Myc late in G1.
In fibroblasts, cyclin A synthesis is dependent on activation of the transcription factor E2F and of CREB, both of which bind to the promoter of cyclin A (11). Neither adhesion of CV1 cells to FN for as long as 22 h nor treatment with EGF induced a significant increase in CREB phosphorylation as measured by phosphorylation at Ser 133 (data not shown). However, we found that another CREB family member, ATF-2, was inducibly activated by EGF but not by FN (data not shown). These data raise the possibility that ATF-2, but not CREB, may be a major mediator of cyclin A synthesis in response to EGF in these cells and that adhesion to FN does not sufficiently stimulate ATF-2 activity. Together these data indicate that adhesion to FN fails to induce several steps late in G1.
Overexpression of EGFR, but not cyclin D1, is sufficient to rescue S phase.
We overexpressed EGFR in CV1 cells by retroviral transduction to determine whether increasing FN-induced EGFR activation is sufficient to induce S-phase progression. In two separate infections, we were able to overexpress EGFR 5-fold or more than 10-fold relative to expression in uninfected or vector-infected cells (Fig. 7B). As shown in Table 1, vector-infected cells failed to enter the cell cycle, as measured by BrdU incorporation, whereas as much as 20% of the cells overexpressing EGFR were labeled with BrdU, a level comparable to that observed in vector-infected cells treated with EGF. Twofold overexpression of cyclin D1 (the highest we were able to achieve), however, was not sufficient to promote S-phase entry on FN. EGFR overexpression did not result in an increase in cyclin D1 levels (Fig. 7E). These data indicate that the rescue of S-phase entry on FN driven by EGFR overexpression is not mediated by up-regulation of cyclin D1 and that cyclin D1 alone is not sufficient.
TABLE 1.
Percent BrdU incorporation in EGFR- and MycER-overexpressing cells
| Virus | % BrdU incorporation in the presence of:
|
P | |
|---|---|---|---|
| FN | FN + EGF | ||
| No virus | 5.9 ± 1.2 | 15.9 ± 1.3 | 0.0008 |
| pLPCX vector | 7.2 ± 0.7 | 21.1 ± 3.4 | 0.0016 |
| pLPCX-EGFR1 | 19.9 ± 4.5 | 20.5 ± 3.9 | 0.1999 |
| pLPCX-EGFR1 | 15.3 ± 2.8 | 15.5 ± 1.9 | 0.4666 |
| pLPCX-EGFR5 | 13.0 ± 0.2 | 15.2 ± 3.0 | 0.1810 |
| pCLXSN-MycER | 7.7 ± 2.4 | 34.4 ± 3.8 | 0.0015 |
| pCLXSN-MycER + Tx | 28.6 ± 4.0 | 40.1 ± 5.6 | 0.0321 |
| pCLXSN-MycER + Tx | 24.5 ± 1.5 | 32.9 ± 4.5 | 0.0451 |
| pCLXSN vector | 2.9 ± 0.1 | 14.3 ± 0.9 | 0.0001 |
| pCLXSN-CycD1 | 3.8 ± 0.1 | 18.5 ± 4.1 | 0.0054 |
| pCLXSN-CycD1 | 4.0 ± 1.6 | 14.1 ± 1.2 | 0.0004 |
Kinetic analysis of cell cycle markers indicated that in cells overexpressing EGFR, attachment to FN induced down-regulation of p27, increased phosphorylation of Rb at Thr821, and increased Myc and cyclin A levels (Fig. 7C and D) to an extent similar to that observed in cells treated with EGF (Fig. 7A), while cyclin D1 overexpression failed to induce this response. Furthermore, overexpression of EGFR did not significantly enhance Erk or Akt activation relative to that seen in vector-infected cells (Fig. 7E). Similar results were obtained in cells expressing a 5- or > 10-fold increase in EGFR levels.
Together these data indicate that in cells expressing endogenous levels of EGFR, adhesion to FN is capable of stimulating a subset of early-G1 cell cycle events but fails to stimulate S-phase entry. This appears to be due in part to a failure to activate Myc late in G1; a lack of efficient p27 degradation, cdk2 activation, or phosphorylation of Rb at Thr821 (cdk2 dependent [70]); and no induction of cyclin A. However, enhanced signaling through EGFR is sufficient to overcome this block and rescue cell proliferation, not by enhancing the Erk/cyclin D1 pathway but by activating other signaling pathways that lead to enhanced Myc expression, down-regulation of p27, induction of cdk2 activity, Rb Thr821 phosphorylation, and cyclin A synthesis (Fig. 7C to E).
Myc overexpression rescues S-phase entry on FN.
To determine if the failure of FN to activate Myc was responsible for the failure to enter S phase, we overexpressed MycER in CV1 cells (Fig. 7F). Activation of MycER by tamoxifen treatment resulted in a decrease in p27 levels and an increase in cyclin A synthesis (Fig. 7F) and allowed entry of CV1 cells into S phase when plated on FN (Table 1). Treatment with both EGF and tamoxifen resulted in an increase in the number of cells entering S phase only slightly greater than that with EGF or tamoxifen alone. This increase, however, was not statistically significant. Attempts to rescue DNA synthesis by stably overexpressing cyclin A in CV1 cells were unsuccessful, in keeping with previous reports that constitutive cyclin A expression can be toxic to cells (26). Together these data indicate that adhesion to FN is not sufficient to activate Myc late in G1 and that expression of Myc is sufficient to permit FN-adherent cells to progress into S phase.
DISCUSSION
The data presented here define a set of integrin-induced signaling events that are activated through a mechanism involving cross talk with EGFR. Based on these results, we propose a model where adhesion of epithelial cells to extracellular matrix proteins leads to activation of endogenous EGFRs via a ligand-independent mechanism (Fig. 3E). Integrin-induced activation of EGFR and phosphorylation of a subset of tyrosine phospho-acceptor sites on EGFR lead to recruitment and tyrosine phosphorylation of Shc, PLCγ, the p85 subunit of PI-3K, and Cbl, and to subsequent activation of the downstream targets Erk and Akt/PKB. EGFR activation is not required for all integrin signaling events, because activation of a distinct subset of signaling enzymes, including FAK, Src, and PKC's, is independent of EGFR catalytic activity. The ability of integrins to activate EGFR, and its subsequent control of a subset of specific signaling molecules, confers the ability to regulate a subset of G1 cell cycle events (Fig. 8). Integrin-mediated activation of endogenous levels of EGFR and two of its downstream effectors, Erk and PI-3K, is required for integrin-mediated induction of cyclin D1 synthesis, activation of cdk4, and Rb phosphorylation; however, these events are not sufficient for entry into S phase. Additional events, triggered by growth factor engagement of EGFR or other receptors, or by overexpression of EGFR, are required for efficient p27 down-regulation, cdk2 activation, Myc and cyclin A induction, and subsequent DNA replication. These results provide strong evidence that endogenous EGFR in epithelial cells plays an important role in mediating specific downstream functions that are activated by the engagement of integrins.
FIG. 8.

Model for integrin regulation of the cell cycle in epithelial cells. Adhesion of epithelial cells to FN is sufficient to stimulate EGFR to activate PI-3K/Akt and Erk, leading to increased synthesis of cyclin D1, subsequent activation of cdk4, and phosphorylation of a subset of sites on Rb (solid boxes). However, induction of Myc, degradation of p27, subsequent activation of cdk2, and cyclin A synthesis are poorly induced by adhesion (dashed boxes). Consequently, cells do not enter into S phase. Signals stimulated by the addition of growth factors (or by overexpression of EGFR or Myc) are required to maximally inactivate p27, stimulate cdk2 activity, and subsequently induce the synthesis of cyclin A, which is required for progression into S phase.
FN-induced activation of EGFR.
The mechanism by which integrins activate RTKs has not been firmly established. Integrins and RTKs can form complexes in cells as measured by immunoprecipitation; however, it has not been resolved whether integrins bind directly to the RTKs and whether this binding is mediated by intracellular or extracellular interactions (9, 36, 55, 69). Our results and those of others suggest that integrins activate EGFR in a ligand-independent manner (28, 36). EGFR activation could be indirect, mediated by integrin-induced signaling pathways. EGFR activation by G protein-coupled receptor (GPCR) signaling has been proposed to involve tyrosine phosphorylation of EGFR on Tyr845 in the catalytic activation loop by the Src kinase (12). Integrin-mediated EGFR activation in ECV304 and MDCK cells and Ron receptor activation in HEK293 cells have been reported to be dependent on Src (17, 37). In this study, we found that FN-induced EGFR phosphorylation in Cos7 or CV1 cells was blocked by AG1478 or PD168393 but not by the Src inhibitors PP2, PD173955, or SU6656 (C. K. Miranti, unpublished), indicating that the majority of phosphorylation on EGFR is independent of Src and is mediated by autophosphorylation. We have also observed integrin-induced activation of ErbB2 but not of ErbB3. ErbB2 activation was blocked by EGFR inhibitors, suggesting that integrins can induce EGFR-ErbB2 heterodimers (Miranti, unpublished). This finding further suggests that ErbB2 may also be contributing to integrin-mediated signaling.
Another model for GPCR-mediated EGFR activation involves induced release of the EGFR ligand HB-EGF (42); however, such a mechanism is unlikely to be responsible for the activation of EGFR by FN, since chimeric receptors lacking the extracellular domain of EGFR were activated by FN-mediated adhesion. Activation of these chimeras does not appear to involve dimerization with endogenous receptors, since no complexes were formed between the chimeras and endogenous receptors (38). Additionally, treatment of CV1 cells with the inhibitor of HB-EGF release 1,10-phenanthroline or diphtheria toxin (45) failed to block FN-induced EGFR activation or G1 cell cycle events, and conditioned medium from CV1 cells failed to stimulate EGFR activation in serum-starved cells (data not shown).
EGFR regulates integrin-dependent signaling events.
In these studies we have been able to define two sets of integrin-dependent signaling molecules: those that are dependent on EGFR and those that are not. These findings and previous studies by others (36) provide evidence for an alternative mechanism for the activation of Erk by integrins through the recruitment and activation of EGFR. We screened several different types of epithelial cells in addition to Cos7 and CV1 cells, including both primary and tumor prostate cells, as well as immortalized retinal epithelial cells and primary keratinocytes. In all cases we observed FN-induced activation of EGFR and a dependence on EGFR for Shc and Erk activation. In cells expressing very low levels of EGFR, such as Rat1 fibroblasts and A375 melanoma cells, we observed no integrin-mediated EGFR activation and no dependence on EGFR for Erk activation (Miranti, unpublished). In addition, Defilippi and coworkers have shown that FN-induced Erk activation in NIH 3T3 cells is independent of EGFR unless EGFR is overexpressed in these cells, whereupon Erk activation by attachment to FN becomes dependent on EGFR (36). Together these data indicate that either endogenous or exogenous expression of EGFR is sufficient to direct a subset of integrin signaling through EGFR.
FAK has been shown to play in important role in the activation of several pathways leading to Erk activation (47, 49). However, in Cos7 or CV1 cells, a dominant interfering mutant of FAK did not affect Erk activation by FN, in agreement with findings from other laboratories that FAK is not required for Erk activation in some cell types (29, 65). An alternate pathway whereby integrins can activate Erk is through α1, α5, or αv integrin-caveolin-1 complexes that lead to activation of Src family kinases and phosphorylation of Shc (65, 66). In reports describing these complexes, the authors did not investigate the possibility of EGFR activation in the cell lines examined (many of which were epithelial); however, we have found that EGFR is critical for Shc and Erk activation in A431 cells, one of the cell lines used in their study. Interestingly, Cos7 cells do not express caveolin (Miranti, unpublished), and thus we did not observe a dependence on caveolin for these interactions, and we saw no differences when different α integrins were engaged. This result may be due to the specific cells examined, the experimental method used to activate integrins, or an indirect requirement for caveolin because of its association with EGFR in some cell types.
In addition to Shc and Erk, we also observed that EGFR is required for signaling to other molecules, including Akt, Cbl, and PLCγ. The mechanisms by which integrins activate these three molecules have been less well characterized. Integrin signaling to PI-3K/Akt has been proposed to involve FAK or ILK (3, 13). We have not determined whether the ability of EGFR to activate Akt is also dependent on FAK or ILK. Integrin-induced Cbl phosphorylation has not been reported in cells that do not express Pyk2 and has been observed primarily in cells of lymphoid origin (48). We also observed EGFR-dependent phosphorylation of Cbl by integrins in a melanoma cell line expressing EGFR but failed to observe integrin-induced Cbl phosphorylation in melanoma cell lines lacking EGFR, despite the presence of adequate Cbl protein (Miranti, unpublished). Similarly, activation of PLCγ by integrins has been observed in only a few cell types, primarily platelets. PLCγ has been reported to be present along with RTKs in focal complexes at the sites of binding of RGD-coated beads in fibroblasts (41). Our data indicate that integrin activation of PLCγ is partially dependent on EGFR in epithelial cells.
The integrin-induced signaling molecules shown to be dependent on EGFR are molecules known to bind directly to the phosphorylated tyrosine residues of the EGFR cytoplasmic domain. Y992 is primarily a PLCγ binding site and is a secondary Shc binding site, while Y1068 and Y1086 are Grb2 binding sites which regulate Erk signaling (5, 24). Specific phosphorylation of these sites by integrins correlates with our signaling data, i.e., activation of PLCγ, Shc, and Erk. Cbl binds Y1042, a site that we have not mapped with respect to integrin activation (24). Nonetheless, we have detected Cbl in EGFR immunoprecipitates (Miranti, unpublished), indicating that there is a likely interaction between these molecules and that integrins also induce phosphorylation of EGFR at Y1042, the Cbl binding site. However, another Shc binding site, Y1148, was not phosphorylated by integrins, yet Shc was significantly activated and dependent on EGFR. Another possibility is that Erb2 (also induced by integrins), a robust activator of Shc, is cooperating with EGFR to induce Shc phosphorylation. Our findings also raise the possibility that integrin-mediated activation of other RTKs, such as Met, PDGFR, or Ron, may also contribute to integrin signaling. We have also observed adhesion-induced activation of Met in several of our epithelial cell lines (Miranti, unpublished). The roles of Met and ErbB2 in integrin-mediated signaling and cell cycle progression are currently under investigation.
Functional importance of integrin and EGFR cross talk.
The evidence that two pharmacological inhibitors of EGFR with different modes of action (reversible or irreversible) block integrin-induced cyclin D1 expression and Rb phosphorylation indicates that EGFR-dependent events are critical for this important biological response to integrin engagement. It is of interest that the epithelial cells examined in this study are able to initiate G1 cell cycle entry and progress through mid-G1 in the absence of growth factors. This response has not been observed in fibroblasts, where cooperative signals from both integrins and growth factors are required for stable Erk activation and cyclin D1 induction (2). It is likely that the ability of integrins in these epithelial cells to induce cyclin D1 is due to cross talk with EGFR, thus allowing at least a partial EGFR-initiated response. While some fibroblasts express EGFR, the number of receptors per cell is low compared to that in epithelial cells (36). Thus, the ability of epithelial cells to cross talk with EGFR is greater than in fibroblasts.
The ability of integrins to couple with EGFR or other RTKs in epithelial cells may change the spectrum of biological activities that can be induced in these cells compared to that in fibroblasts and other cell types where such interactions are weak or nonexistent. Such differences in the signaling potential in distinct cell types may be important during development and for expression of differentiated cell functions. In fibroblasts, the weaker and more transient activation of Erk through FAK or other signaling pathways may play an important role in other biological responses such as regulation of cell motility, while the longer-term integrin-mediated activation of Erk through EGFR may be important for proliferation in epithelial cells, where mid-G1 priming through integrins could allow a more efficient response to growth factor stimulation. Many types of epithelial cells proliferate and turn over as part of natural homeostasis. Thus, integration of specific receptor signaling pathways may be important for specialized cell functions. In support of our findings with CV1 cells, Kuwada and Li have shown that FN-mediated activation of EGFR is critical for cell proliferation in an EGF-dependent intestinal epithelial cell line that was engineered to overexpress α5 integrin (28). Experiments with ECV304 cells, which also express higher levels of EGFR, suggest that integrin engagement of EGFR is critical for their survival (36, 37).
Erk and PI-3K activation are required for integrin-induced progression through mid-G1, as shown by U0126 and wortmannin inhibition of cyclin D1 expression and Rb phosphorylation. These results are not surprising given previous studies demonstrating that Erk and PI-3K are required for growth factor-induced progression through mid-G1 (25). However, it is interesting that integrin-induced EGFR activation of Erk and PI-3K pathways and other integrin-induced events are not sufficient for further progression into S phase, indicating that additional signals supplied by growth factors, not triggered by FN, are required.
One pathway required for induction of DNA synthesis is an increase in cyclin A levels, mediated in part by degradation of p27 and stimulation of cdk2 activity, which in turn enhances Rb phosphorylation at specific sites and release of E2F from Rb for binding to the cyclin A promoter. Plating of cells on FN fails to effectively activate this pathway. We were unable to rescue this pathway by simply removing p27, suggesting that EGFR ligands regulate additional steps in this pathway that are not activated by integrins. Interestingly, CREB, a transcription factor previously implicated in regulating cyclin A transcription in fibroblasts independently of p27 and cdk2, does not appear to play a role in regulating the cell cycle in CV1 cells (11). However, ATF-2, a CREB family member, is activated by addition of growth factors, and its kinetics of activation parallel those of cyclin A induction (Miranti, unpublished).
Adhesion of CV1 cells to FN also failed to induce Myc expression late in G1, an event that was observed in response to EGF. In addition to its role in regulating cyclin D1 synthesis early in G1, Myc is also required later in the cell cycle to enhance p27 degradation and activate cdc25A, which is required to dephosphorylate cdk2 and permit its activation (1, 7, 23, 60). Myc is also required for entry into S phase; it is critical for inducing S-phase-specific genes such as ornithine decarboxylase (7). Thus, adhesion to FN, while capable of activating several early-G1 events, is not sufficient to activate many late-G1 events. Since Myc acts at multiple points in late G1, we predicted that ectopic expression of Myc late in G1 would rescue entry into S phase on FN. Activation of estrogen-regulated Myc was sufficient to permit S-phase entry, which was accompanied by a decrease in p27 levels and an increase in cyclin A levels.
Interestingly, we could also rescue DNA synthesis in FN-adherent cells by overexpression of EGFR. The precise mechanism by which EGFR overexpression rescues S-phase entry on FN remains unsolved; however, this report provides one potential strategy. When treated with EGF, EGFR-overexpressing cells displayed an increase in tyrosine phosphorylation of EGFR as well as increased and more prolonged activation of Erk and Akt (Miranti, unpublished). However, there was not a corresponding increase in Erk or Akt activation or overall tyrosine phosphorylation mediated by integrins, indicating that these events are not likely to be involved in regulating S-phase entry in response to matrix. Furthermore, despite prolonged activation of Erk and Akt by EGF in EGFR-overexpressing cells, EGF-stimulated EGFR-overexpressing cells did not display an increase in cell proliferation relative to that seen in control cells treated with EGF, suggesting that other events mediated by EGFR overexpression are involved. When specific sites of phosphorylation on EGFR were examined in EGFR-overexpressing cells, we similarly observed no increase in the relative levels of tyrosine phosphorylation of the sites previously shown to be regulated by integrins (namely, Y845, Y992, and Y1068) but rather observed an increase in FN-induced phosphorylation at the other sites, Y1148 and Y1173. These data suggest that overexpression of EGFR leads to an enhanced ability of integrins to activate additional signaling pathways. Indeed, EGFR overexpression did lead to FN-induced degradation of p27, activation of cdk2, Rb Thr821 phosphorylation, and increased Myc and cyclin A synthesis. These events are critical for progression from late G1 into S phase and provide the necessary signals to further cell cycle progression when cells are plated on matrix.
Many tumor cells, especially epithelial-cell-derived tumors, express elevated levels of EGFR or express mutant versions of the EGFR family (46, 67). It is possible that integrin and RTK cross talk may be an important factor in tumor progression and metastasis. In breast cancer cells, manipulations that induce the loss of either EGFR or β1 integrin result in the concomitant loss of the other receptor, suggesting a tight coupling between these two pathways (43, 62). Adhesion-dependent activation of the Met receptor is required for hepatocarcinoma formation and persistence in vivo (64), and α6β4 functions to regulate Met receptor activation during invasion and metastasis (59). Integrin-dependent, ligand-independent regulation of RTK function may be an important event during tumorigenesis. Myc is also overexpressed in many types of tumors (40). Our data indicate that elevated levels of EGFR or Myc in nontumorigenic cells permit more efficient signaling by integrins, leading to increased proliferation. Overexpression of EGFR or Myc in matrix-adherent cells may be a mechanism for increasing cell survival and cell proliferation in the absence of an EGF ligand, particularly in tumors.
It has become increasingly apparent that the functional response of a cell to any given external factor is dependent on the sum of the signals generated by that factor in cooperation with other signals generated by cross talk between different signaling pathways. Cooperation between signal transduction pathways has been best exemplified by the interaction between RTKs and integrins. Understanding the importance and the nature of integrin and RTK cross talk is vital to understanding how these are integrated to regulate cell function.
Acknowledgments
We are grateful to Brian Drucker and Tom Roberts for the monoclonal anti-phosphotyrosine antibody 4G10, to Martin Eilers for promptly providing the MycER cDNA constructs, and to Robert Kraus for the pBabe-cycD1 and pBabe-cycA constructs. C.K.M. is grateful for the technical assistance provided by Veronique Patacsil, Robert Long, and Andy Skildum.
This work was funded by grants from the National Institutes of Health (CA78773 [to J.S.B.] and CA72203 [to C.K.M.]), the Leukemia and Lymphoma Society of America (to V.R.R. and S.L.M.), the U.S. Army Medical Research and Materiel Command (to S.K.M), the Massachusetts Breast Cancer Research Program (to S.K.M.), the Fred Hutchinson Cancer Research Center (to B.K.), and the Van Andel Research Institute (to C.K.M. and H.M.B.). J.S.B. is a Research Professor of the American Cancer Society.
REFERENCES
- 1.Amati, B., K. Alevizopoulos, and J. Vlach. 1998. Myc and the cell cycle. Front. Biosci. 3:D250-D268. [DOI] [PubMed] [Google Scholar]
- 2.Assoian, R. K., and M. A. Schwartz. 2001. Coordinate signaling by integrins and receptor tyrosine kinases in the regulation of G1 phase cell-cycle progression. Curr. Opin. Genet. Dev. 11:48-53. [DOI] [PubMed] [Google Scholar]
- 3.Attwell, S., J. Mills, A. Troussard, C. Wu, and S. Dedhar. 2003. Integration of cell attachment, cytoskeletal localization, and signaling by integrin-linked kinase (ILK), CH-ILKBP, and the tumor suppressor PTEN. Mol. Biol. Cell 14:4813-4825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ausubel, F. M. (ed.). 2000. Current protocols in molecular biology. John Wiley & Sons, New York, N.Y.
- 5.Batzer, A. G., D. Rotin, J. M. Urena, E. Y. Skolnik, and J. Schlessinger. 1994. Hierarchy of binding sites for Grb2 and Shc on the epidermal growth factor receptor. Mol. Cell. Biol. 14:5192-5201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beier, R., A. Burgin, A. Kiermaier, M. Fero, H. Karsunky, R. Saffrich, T. Moroy, W. Ansorge, J. Roberts, and M. Eilers. 2000. Induction of cyclin E-cdk2 kinase activity, E2F-dependent transcription and cell growth by Myc are genetically separable events. EMBO J. 19:5813-5823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bello-Fernandez, C., G. Packham, and J. L. Cleveland. 1993. The ornithine decarboxylase gene is a transcriptional target of c-Myc. Proc. Natl. Acad. Sci. USA 90:7804-7808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Benaud, C. M., and R. B. Dickson. 2001. Adhesion-regulated G1 cell cycle arrest in epithelial cells requires downregulation of c-Myc. Oncogene 20:4554-4567. [DOI] [PubMed] [Google Scholar]
- 9.Borges, E., Y. Jan, and E. Ruoslahti. 2000. Platelet-derived growth factor receptor β and vascular endothelial growth factor receptor 2 bind to the β3 integrin through its extracellular domain. J. Biol. Chem. 275:39867-39873. [DOI] [PubMed] [Google Scholar]
- 10.Bottazzi, M. E., X. Zhu, R. M. Bohmer, and R. K. Assoian. 1999. Regulation of p21cip1 expression by growth factors and the extracellular matrix reveals a role for transient ERK activity in G1 phase. J. Cell Biol. 146:1255-1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bottazzi, M. E., M. Buzzai, X. Zhu, C. Desdouets, C. Brechot, and R. K. Assoian. 2001. Distinct effects of mitogens and the actin cytoskeleton on CREB and pocket protein phosphorylation control the extent and timing of cyclin A promoter activity. Mol. Cell. Biol. 21:7607-7616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carpenter, G. 1999. Employment of the epidermal growth factor receptor in growth factor-independent signaling pathways. J. Cell Biol. 146:697-702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen, H. C., P. A. Appeddu, H. Isoda, and J. L. Guan. 1996. Phosphorylation of tyrosine 397 in focal adhesion kinase is required for binding phosphatidylinositol 3-kinase. J. Biol. Chem. 271:26329-26334. [DOI] [PubMed] [Google Scholar]
- 14.Comoglio, P. M., C. Boccaccio, and L. Trusolino. 2003. Interactions between growth factor receptors and adhesion molecules: breaking the rules. Curr. Opin. Cell Biol. 15:565-571. [DOI] [PubMed] [Google Scholar]
- 15.Danen, E. H., R. M. Lafrenie, S. Miyamoto, and K. M. Yamada. 1998. Integrin signaling: cytoskeletal complexes, MAP kinase activation, and regulation of gene expression. Cell Adhes. Commun. 6:217-224. [DOI] [PubMed] [Google Scholar]
- 16.Danen, E. H., and K. M. Yamada. 2001. Fibronectin, integrins, and growth control. J. Cell. Physiol. 189:1-13. [DOI] [PubMed] [Google Scholar]
- 17.Danilkovitch-Miagkova, A., D. Angeloni, A. Skeel, S. Donley, M. Lerman, and E. J. Leonard. 2000. Integrin-mediated RON growth factor receptor phosphorylation requires tyrosine kinase activity of both the receptor and c-Src. J. Biol. Chem. 275:14783-14786. [DOI] [PubMed] [Google Scholar]
- 18.De Arcangelis, A., and E. Georges-Labouesse. 2000. Integrin and ECM functions: roles in vertebrate development. Trends Genet. 16:389-395. [DOI] [PubMed] [Google Scholar]
- 19.Elbashir, S. M., W. Lendeckel, and T. Tuschl. 2001. RNA interference is mediated by 21- and 22-nucleotide RNAs. Genes Dev. 15:188-200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Geiger, B., A. Bershadsky, R. Pankov, and K. M. Yamada. 2001. Transmembrane crosstalk between the extracellular matrix and the cytoskeleton. Nat. Rev. Mol. Cell Biol. 2:793-805. [DOI] [PubMed] [Google Scholar]
- 21.Giancotti, F. G., and E. Ruoslahti. 1999. Integrin signaling. Science 285:1028-1032. [DOI] [PubMed] [Google Scholar]
- 22.Gmyrek, G. A., M. Walburg, C. P. Webb, H. M. Yu, X. You, E. D. Vaughan, G. F. Vande Woude, and B. S. Knudsen. 2001. Normal and malignant prostate epithelial cells differ in their response to hepatocyte growth factor/scatter factor. Am. J. Pathol. 159:579-590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gu, Y., J. Rosenblatt, and D. O. Morgan. 1992. Cell cycle regulation of CDK2 activity by phosphorylation of Thr160 and Tyr15. EMBO J. 11:3995-4005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hynes, N. E., K. Horsch, M. A. Olayioye, and A. Badache. 2001. The ErbB receptor tyrosine family as signal integrators. Endocr. Relat. Cancer 8:151-159. [DOI] [PubMed] [Google Scholar]
- 25.Jones, S. M., and A. Kazlauskas. 2001. Growth-factor-dependent mitogenesis requires two distinct phases of signalling. Nat. Cell Biol. 3:165-172. [DOI] [PubMed] [Google Scholar]
- 26.Kang, J. S., and R. S. Krauss. 1996. Ras induces anchorage-independent growth by subverting multiple adhesion-regulated cell cycle events. Mol. Cell. Biol. 16:3370-3380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Klemke, R. L., M. Yebra, E. M. Bayna, and D. A. Cheresh. 1994. Receptor tyrosine kinase signaling required for integrin αvβ5-directed cell motility but not adhesion on vitronectin. J. Cell Biol. 127:859-866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuwada, S. K., and X. Li. 2000. Integrin α5/β1 mediates fibronectin-dependent epithelial cell proliferation through epidermal growth factor receptor activation. Mol. Biol. Cell 11:2485-2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lin, T. H., A. E. Aplin, Y. Shen, Q. Chen, M. Schaller, L. Romer, I. Aukhil, and R. L. Juliano. 1997. Integrin-mediated activation of MAP kinase is independent of FAK: evidence for dual integrin signaling pathways in fibroblasts. J. Cell Biol. 136:1385-1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lipsich, L., A. Lewis, and J. Brugge. 1983. Isolation of monoclonal antibodies that recognize the transforming proteins of avian sarcoma viruses. J. Virol. 48:352-360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Littlewood, T. D., D. C. Hancock, P. S. Danielian, M. G. Parker, and G. I. Evan. 1995. A modified oestrogen receptor ligand-binding domain as an improved switch for the regulation of heterologous proteins. Nucleic Acids Res. 23:1686-1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mainiero, F., A. Pepe, M. Yeon, Y. Ren, and F. G. Giancotti. 1996. The intracellular functions of α6β4 integrin are regulated by EGF. J. Cell Biol. 134:241-253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miranti, C. K. 2002. Application of cell adhesion to study signaling networks. Methods Cell Biol. 69:359-382. [DOI] [PubMed] [Google Scholar]
- 34.Miranti, C. K., and J. S. Brugge. 2002. Sensing the environment: a historical perspective on integrin signal transduction. Nat. Cell Biol. 4:E83-E90. [DOI] [PubMed] [Google Scholar]
- 35.Miyamoto, S., H. Teramoto, J. S. Gutkind, and K. M. Yamada. 1996. Integrins can collaborate with growth factors for phosphorylation of receptor tyrosine kinases and MAP kinase activation: roles of integrin aggregation and occupancy of receptors. J. Cell Biol. 135:1633-1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moro, L., M. Venturino, C. Bozzo, L. Silengo, F. Altruda, L. Beguinot, G. Tarone, and P. Defilippi. 1998. Integrins induce activation of EGF receptor: role in MAP kinase induction and adhesion-dependent cell survival. EMBO J. 17:6622-6632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moro, L., L. Dolce, S. Cabodi, E. Bergatto, E. B. Erba, M. Smeriglio, E. Turco, S. F. Retta, M. G. Guiffrida, M. Venturino, J. Godovac-Zimmerman, E. Schaefer, L. Beguinot, C. Tacchetti, P. Gaggini, L. Silengo, G. Tarone, and P. Defilippi. 2002. Integrin-induced epidermal growth factor (EGF) receptor activation requires c-Src and p130Cas and leads to phosphorylation of specific EGF receptor kinases. J. Biol. Chem. 277:9405-9414. [DOI] [PubMed] [Google Scholar]
- 38.Muthuswamy, S. K., M. Gilman, and J. S. Brugge. 1999. Controlled dimerization of ErbB receptors provides evidence for differential signaling by homo- and heterodimers. Mol. Cell. Biol. 19:6845-6857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ory, D. S., B. A. Neugeboren, and R. C. Mulligan. 1996. A stable human-derived packaging cell line for production of high titer retrovirus/vesicular stomatitis virus G pseudotypes. Proc. Natl. Acad. Sci. USA 93:11400-11406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pelengaris, S., M. Khan, and G. Evan. 2002. c-MYC: more than just a matter of life and death. Nat. Rev. Cancer 2:764-776. [DOI] [PubMed] [Google Scholar]
- 41.Plopper, G. E., H. P. McNamee, L. E. Dike, K. Bojanowski, and D. E. Ingber. 1995. Convergence of integrin and growth factor receptor signaling pathways within the focal adhesion complex. Mol. Biol. Cell 6:1349-1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Prenzel, N., E. Zwick, H. Daub, M. Leserer, R. Abraham, C. Wallasch, and A. Ullrich. 1999. EGF receptor transactivation by G-protein-coupled receptors requires metalloproteinase cleavage of proHB-EGF. Nature 402:884-888. [DOI] [PubMed] [Google Scholar]
- 43.Reginato, M. J., K. R. Mills, J. K. Paulus, D. K. Lynch, D. C. Sgroi, J. Debnath, S. K. Muthuswamy, and J. S. Brugge. 2003. Integrins and EGFR coordinately regulate the pro-apoptotic protein Bim to prevent anoikis. Nat. Cell Biol. 5:733-740. [DOI] [PubMed] [Google Scholar]
- 44.Roovers, K., G. Davey, X. Zhu, M. E. Bottazzi, and R. K. Assoian. 1999. α5β1 integrin controls cyclin D1 expression by sustaining mitogen-activated protein kinase activity in growth factor-treated cells. Mol. Biol. Cell 10:3197-3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roudabush, F. L., K. L. Pierce, S. Maudsley, K. D. Khan, and L. M. Luttrell. 2000. Transactivation of the EGF receptor mediates IGF-1-stimulated Shc phosphorylation and ERK1/2 activation in COS-7 cells. J. Biol. Chem. 275:22583-22589. [DOI] [PubMed] [Google Scholar]
- 46.Ruoslahti, E. 1999. Fibronectin and its integrin receptors in cancer. Adv. Cancer Res. 76:1-20. [DOI] [PubMed] [Google Scholar]
- 47.Sanders, M. A., and M. D. Basson. 2000. Collagen IV-dependent ERK activation in human Caco-2 intestinal epithelial cells requires focal adhesion kinase. J. Biol. Chem. 275:38040-38047. [DOI] [PubMed] [Google Scholar]
- 48.Sanjay, A., A. Houghton, L. Neff, E. DiDomenico, C. Bardelay, E. Antoine, J. Levy, J. Gailit, D. Bowtell, W. C. Horne, and R. Baron. 2001. Cbl associates with Pyk2 and Src to regulate Src kinase activity, αvβ3 integrin-mediated signaling, cell adhesion, and osteoclast motility. J. Cell Biol. 152:181-195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schlaepfer, D. D., and T. Hunter. 1997. Focal adhesion kinase overexpression enhances ras-dependent integrin signaling to ERK2/mitogen-activated protein kinase through interactions with and activation of c-Src. J. Biol. Chem. 272:13189-13195. [DOI] [PubMed] [Google Scholar]
- 50.Schneller, M., K. Vuori, and E. Ruoslahti. 1997. αvβ3 integrin associates with activated insulin and PDGFβ receptors and potentiates the biological activity of PDGF. EMBO J. 16:5600-5607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schoenwaelder, S. M., and K. Burridge. 1999. Bidirectional signaling between the cytoskeleton and integrins. Curr. Opin. Cell Biol. 11:274-286. [DOI] [PubMed] [Google Scholar]
- 52.Schwartz, M. A., and V. Baron. 1999. Interactions between mitogenic stimuli, or a thousand and one connections. Curr. Opin. Cell Biol. 11:197-202. [DOI] [PubMed] [Google Scholar]
- 53.Schwartz, M. A., and R. K. Assoian. 2001. Integrins and cell proliferation: regulation of cyclin-dependent kinases via cytoplasmic signaling pathways. J. Cell Sci. 114:2553-2560. [DOI] [PubMed] [Google Scholar]
- 54.Schwartz, M. A., and M. H. Ginsberg. 2002. Networks and crosstalk: integrin signalling spreads. Nat. Cell Biol. 4:E65-E68. [DOI] [PubMed] [Google Scholar]
- 55.Soldi, R., S. Mitola, M. Strasly, P. Defilippi, G. Tarone, and F. Bussolino. 1999. Role of αvβ3 integrin in the activation of vascular endothelial growth factor receptor-2. EMBO J. 18:882-892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sundberg, C., and K. Rubin. 1996. Stimulation of β1 integrins on fibroblasts induces PDGF independent tyrosine phosphorylation of PDGFβ-receptors. J. Cell Biol. 132:741-752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tanaka, M., and W. Herr. 1990. Differential transcriptional activation by Oct-1 and Oct-2: interdependent activation domains induce Oct-2 phosphorylation. Cell 60:375-386. [DOI] [PubMed] [Google Scholar]
- 58.Trusolino, L., G. Serini, G. Cecchini, C. Besati, F. S. Ambesi-Impiombato, P. C. Marchisio, and R. De Filippi. 1998. Growth factor-dependent activation of αvβ3 integrin in normal epithelial cells: implications for tumor invasion. J. Cell Biol. 142:1145-1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Trusolino, L., A. Bertotti, and P. M. Comoglio. 2001. A signaling adaptor function for α6β4 integrin in the control of HGF-dependent invasive growth. Cell 107:643-654. [DOI] [PubMed] [Google Scholar]
- 60.Vlach, J., S. Hennecke, K. Alevizopoulos, D. Conti, and B. Amati. 1996. Growth arrest by the cyclin-dependent kinase inhibitor p27Kip1 is abrogated by c-Myc. EMBO J. 15:6595-6604. [PMC free article] [PubMed] [Google Scholar]
- 61.Wadzinski, B. E., B. J. Eisfelder, L. F. Peruski, M. C. Mumby, and G. L. Johnson. 1992. NH2-terminal modification of the phosphatase 2A catalytic subunit allows functional expression in mammalian cells. J. Biol. Chem. 267:16883-16888. [PubMed] [Google Scholar]
- 62.Wang, F., V. M. Weaver, O. W. Petersen, C. A. Larabell, S. Dedhar, P. Briand, R. Lupu, and M. J. Bissell. 1998. Reciprocal interactions between β1-integrin and epidermal growth factor receptor in three-dimensional basement membrane breast cultures: a different perspective in epithelial biology. Proc. Natl. Acad. Sci. USA 95:14821-14826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang, R., R. Kobayashi, and J. M. Bishop. 1996. Cellular adherence elicits ligand-independent activation of the Met cell-surface receptor. Proc. Natl. Acad. Sci. USA 93:8425-8430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang, R., L. D. Ferrell, S. Faouzi, J. J. Maher, and J. M. Bishop. 2001. Activation of the Met receptor by cell attachment induces and sustains hepatocellular carcinomas in transgenic mice. J. Cell Biol. 153:1023-1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wary, K. K., F. Mainiero, S. J. Isakoff, E. E. Marcantonio, and F. G. Giancotti. 1996. The adaptor protein Shc couples a class of integrins to the control of cell cycle progression. Cell 87:733-743. [DOI] [PubMed] [Google Scholar]
- 66.Wary, K. K., A. Mariotti, C. Zurzolo, and F. G. Giancotti. 1998. A requirement for caveolin-1 and associated kinase Fyn in integrin signaling and anchorage-dependent cell growth. Cell 94:625-634. [DOI] [PubMed] [Google Scholar]
- 67.Wells, A. 1999. EGF receptor. Int. J. Biochem. Cell Biol. 31:637-643. [DOI] [PubMed] [Google Scholar]
- 68.Woods, A., and J. R. Couchman. 1998. Syndecans: synergistic activators of cell adhesion. Trends Cell Biol. 8:189-192. [DOI] [PubMed] [Google Scholar]
- 69.Yu, X., S. Miyamoto, and E. Mekada. 2000. Integrin α2β1-dependent EGF receptor activation at cell-cell contact sites. J. Cell Sci. 113:2139-2147. [DOI] [PubMed] [Google Scholar]
- 70.Zarkowska, T., and S. Mittnacht. 1997. Differential phosphorylation of the retinoblastoma protein by G1/S cyclin-dependent kinases. J. Biol. Chem. 272:12738-12746. [DOI] [PubMed] [Google Scholar]