

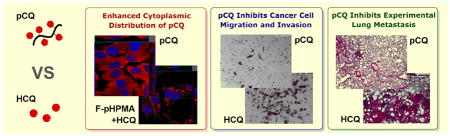
Abstract
Chloroquine (CQ) is a widely used antimalarial drug with emerging potential in anticancer therapies due to its apparent inhibitory effects on CXCR4 chemokine receptor, autophagy, and cholesterol metabolism. This study reports on polymeric CQ (pCQ) as a macromolecular drug with antimetastatic activity. The pCQ polymers were synthesized by copolymerization of methacryloylated hydroxy-CQ (HCQ) and N-(2-hydroxypropyl)methacrylamide (HPMA). The results show that pCQ is significantly more effective in inhibiting cancer cell migration and invasion when compared with the parent HCQ. The proposed mechanism of action at least partially relies on the ability of pCQ to inhibit cell migration mediated by the CXCR4/CXCL12 pathway. The pCQ also demonstrates superior inhibitory activity over HCQ when tested in a mouse model of experimental lung metastasis. Lastly, pCQ shows the ability to efficiently translocate to the cytoplasm while exhibiting lower cytotoxicity than HCQ. Overall, this study supports pCQ as a promising polymeric drug platform suitable for use in combination antimetastatic strategies and potential use in cytoplasmic drug delivery.
Keywords: Polymeric drug, Metastasis, Chloroquine, HPMA, CXCR4, Endosomal release
Graphical abstract
1. Introduction
The use of polymers has made remarkable contribution in pharmaceutical discovery and development of modern drug delivery methods. Polymer applications, whether already in clinical use or in preclinical development, range from simple functions such as providing sustained drug release to more sophisticated uses such as targeted drug delivery [1, 2]. In addition to the traditional use of polymers to improve safety and delivery efficacy of existing drugs, there has been a growing interest in the development of pharmacologically active polymers. These so-called polymeric drugs exhibit intrinsic therapeutic activity and have been employed in the treatment of various diseases [3]. Besides altered pharmacokinetics and biodistribution, polymeric drugs usually take advantage of multivalent interactions to achieve improved activity when compared with small molecule drugs [4]. This is often the result of amplified downstream signaling when compared to monovalent binding with small molecule drugs; leading to improved and/or prolonged therapeutic effects and outcomes [5, 6].
Metastasis is the leading cause of cancer-related deaths and there is a need for the development of novel antimetastatic strategies. Metastasis is a complex multistep process during which cancer cells from primary tumor migrate to secondary sites to establish new tumors. Chemokines and chemokine receptors play a prominent role in facilitating the metastatic spread and in determining the sites to which specific cancers preferentially metastasize [7]. Among the chemokine receptors, C-X-C receptor 4 (CXCR4) is most commonly overexpressed in human cancers. Primary tumor cells that overexpress CXCR4 have increased tendency to metastasize to distant organs where the levels of the CXCR4 ligand CXCL12 are elevated [8]. Mounting body of evidence shows that inhibiting the CXCR4/CXCL12 axis by CXCR4 antagonists or silencing expression of CXCR4 by siRNA, provides significant antimetastatic effect in multiple cancer models [9]. In addition to the only clinically used CXCR4 antagonist (AMD3100, Plerixafor), which has been on the market since 2008, there are multiple small molecule and peptide CXCR4 antagonists in various stages of development [10–13]. Interestingly, CXCR4 antagonism of chloroquine (CQ) and its derivatives has been recently reported [14, 15], offering a pathway to repurposing CQ for antimetastatic therapies.
CQ is a classic antimalarial drug that has been in clinical use for decades. CQ was developed from natural product quinine eighty years ago and is still widely used for the control of malaria worldwide. Besides its antimalarial properties, a broader spectrum of CQ pharmacological activities, including anti-inflammatory and anticancer activity has been discovered and explored over the years [16, 17]. CQ, and its derivatives like hydroxychloroquine (HCQ), have also been recognized as effective autophagy inhibitors that exhibit beneficial anticancer properties [18, 19]. Autophagy controls cellular homeostasis by lysosomal degradation of cytoplasmic components, including invading pathogens, cytotoxic proteins and damaged organelles. In cancer, autophagy provides a survival mechanism to allow cancer cells to support proliferation during metabolic stress [20]. Inhibition of autophagy by CQ can reverse the process and suppress the proliferation of cancer cells. Although CQ and HCQ were initially tested in cancer treatment due to their ability to inhibit autophagy, we now know that their therapeutic effect depends on other mechanisms as well [21–23]. For example, a recent report revealed that CQ-induced cancer cell death was related to its inhibitory effects on cholesterol metabolism [24]. Taking advantage of its recently uncovered CXCR4 antagonism, CQ was able to inhibit CXCR4/CXCL12-mediated pancreatic cancer cell invasion and proliferation in vitro, to eliminate established tumors, and to improve overall survival when combined with gemcitabine in vivo [15]. Taken together, CQ is a promising multi-functional agent that is well-suited for development of novel combination anticancer strategies.
In this study, we report on the properties of polymeric CQ (pCQ) as a macromolecular inhibitor of cancer metastasis and a potential carrier to improve cytoplasmic drug delivery. The pCQ polymers were synthesized by a copolymerization of N-(2-hydroxypropyl)methacrylamide (HPMA) with methacryloylated HCQ (MA-CQ). We present data evaluating pCQ as inhibitor of cancer cell migration and invasion in vitro and its antimetastatic activity in vivo in experimental lung metastasis model of breast cancer. Intracellular trafficking of pCQ was evaluated to determine the ability of the polymers to translocate to the cytoplasm.
2. Materials and Methods
2.1. Materials
Hydroxychloroquine (HCQ) sulfate (98%), trimethylamine, crystal violet, DMSO-d6 (99.8%) and chloroform-d (99.8%) were obtained from Acros Organics (Fisher Scientific, Pittsburgh, PA). Sodium acetate, dichloromethane (DCM), chloroform, methanol (MeOH), acetonitrile (HPLC grade) were from Fisher Scientific. Rhodamine isothiocyanate mixed isomers (RBITC), trifluoroacetic acid (TFA) and 2,2′-azobisisobutyronitrile (AIBN) were purchased from Sigma Aldrich (St. Louis, MO). N-(2-hydroxypropyl)methacrylamide (HPMA) and N-(3-aminopropyl)methacrylamide hydrochloride (APMA·HCl) were purchased from Polysciences (Warrington, PA). Hoechst 33342, nitrocellulose membrane, Novex 10% Tris-Glycine Midi Protein Gels and 12% Tris-Glycine Midi Protein Gels were purchased from Invitrogen (Carlsbad, CA). Phosphate buffered saline (PBS), Dulbecco's Modified Eagle Medium (DMEM), sodium pyruvate, essential amino acids and non-essential amino acids were from Hyclone (Logan, UT). Fetal bovine serum (FBS) was from Atlanta Biologicals (Flowery Branch, GA). Eagle’s Minimum Essential Medium (EMEM) was from ATCC (Manassas, VA). Gentamicin, enzyme-free cell dissociation buffer and F-12K medium were purchased from Gibco (Life Technologies, Grand Island, NY). Protease and phosphatase inhibitor cocktail, Pierce bicinchoninic acid (BCA) protein assay, RIPA buffer and Pierce ECL Western Blotting Substrate were was purchased from ThermoScientific (Waltham, MA). LC3B antibody, phospho-p44/42 MAPK (pERK) rabbit antibody, p44/42 MAPK (ERK) rabbit antibody, GAPDH rabbit antibody and anti-rabbit IgG, HRP-linked antibody were purchased from Cell Signaling Technology (Beverly, MA). Allophycocyanin (APC) mouse B anti-human CD184 and APC mouse IgG2a, κ isotype control were purchased from BD Biosciences (San Jose, CA). Human and mouse CXCL12 were purchased from Shenandoah Biotechnology (Warwick, PA). Laemmli sample buffer and 2-mercaptoethanol were purchased from Bio-rad (Hercules, CA).
2.2. Synthesis of polymers
Synthesis of pCQ
The pCQ copolymers with different CQ content were synthesized as previously reported (Scheme 1) [25]. Briefly, HCQ·HCl was converted to a base form using ammonium hydroxide and HCQ was extracted into DCM and dried. MA-CQ was prepared by dropwise addition of methacryloyl chloride in chloroform to a mixture of ice-cold HCQ and triethylamine in chloroform under vigorous stirring. The reaction mixture was washed with saturated sodium bicarbonate solution and concentrated. MA-CQ was purified by flash chromatography with DCM:MeOH (10:1). The pCQ were prepared by polymerization of different molar ratios of MA-CQ and HPMA with AIBN as initiator in MeOH at 55 °C under N2 overnight. The polymers were precipitated twice in cold diethyl ether, followed by dialysis against water for 3 days (membrane molecular weight cut-off 8,000). The final polymers were obtained by lyophilization.
Scheme 1.
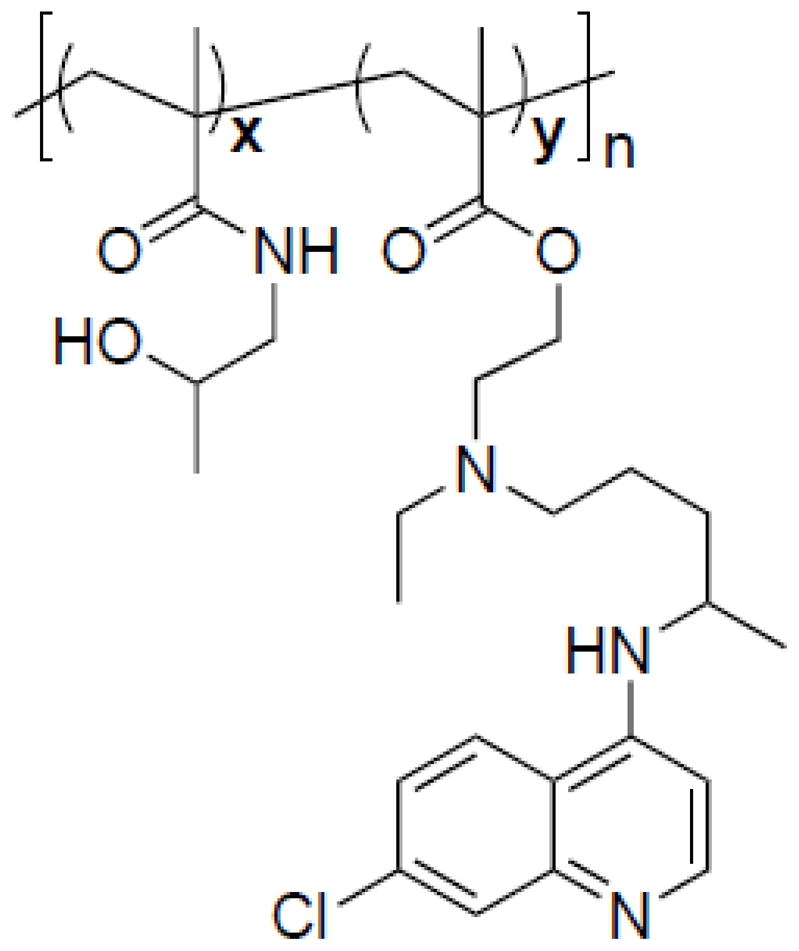
Chemical structure of pCQ.
Synthesis of fluorescently labeled poly(HPMA) (F-pHPMA)
HPMA (143 mg, 1 mmol), APMA·HCl (3.8 mg, 0.02 mmol) and AIBN (8.4 mg, 0.05 mmol) were dissolved in MeOH (1 mL), purged with N2 for 30 min, and the reaction mixture was stirred at 55 °C overnight. After double precipitation in cold diethyl ether, dialysis, and lyophilization, the polymer was obtained as white solid (pHPMA-amine, 78 mg, 53%). RBITC (10.7 mg, 0.02 mmol), pHPMA-amine (29 mg) and triethylamine (30 μL, 0.2 mmol) were dissolved in DMSO (0.5 mL) and stirred at room temperature for 48 h. The resulting solution was dialyzed against MeOH for 2 days and then against water for 5 days to remove unreacted RBITC. The F-pHPMA was obtained as dark red solid (25 mg) after lyophilization.
Synthesis of F-pCQ10.0
HPMA (123 mg, 0.86 mmol), APMA·HCl (3.6 mg, 0.02 mmol), MA-CQ (48.5 mg, 0.12 mmol) and AIBN (8.2 mg, 0.05mmol) were polymerized, purified and conjugated with RBITC following the F-pHPMA procedure above to obtain fluorescently labeled pCQ10.0 (F-pCQ10.0).
1H-NMR (Bruker Avance-III HD 500 MHz) was used to analyze the composition of all polymers and the data were analyzed by Topspin 3.5 and MestReNova 9.0 software. The molecular weights of all polymers were analyzed by gel permeation chromatography (GPC) operated in 0.1 M sodium acetate buffer (pH 5.0) using Agilent 1260 Infinity LC system equipped with a miniDAWN TREOS multi-angle light scattering (MALS) detector and a Optilab T-rEX refractive index detector (Wyatt Technology, Santa Barbara, CA). The column used was TSKgel G3000PWXL-CP (Tosoh Bioscience LLC, King of Prussia, PA) at a flow rate of 0.5 mL/min. Results were analyzed using Astra 6.1 software from Wyatt Technology. The degree of polymerization was calculated from the GPC and 1H-NMR data. High-performance liquid chromatography (HPLC) was used to confirm the complete removal of RBITC by using Agilent 1260 Infinity LC system and UV detector at 220 nm. The Agilent Eclipse Plus C18 (5 μm, 4.6 × 150 mm) column was used at a flow rate of 1 mL/min with a 5–100% gradient of acetonitrile in water (0.01% TFA).
2.3. Cell culture
Human epithelial osteosarcoma U2OS cells stably expressing functional EGFP-CXCR4 fusion protein were purchased from Fisher Scientific and cultured in DMEM supplemented with 2 mM L-glutamine, 1% Pen-Strep, 0.5 mg/mL G418 and 10% FBS. Mouse breast carcinoma 4T1 was a kind gift from Dr. Fred Miller (Wayne State University) and cultured in DMEM supplemented with 2 mM L-glutamine, 1 mM sodium pyruvate, essential amino acids, non-essential amino acids, gentamycin (0.2 mg/mL) and 10% FBS. Human hepatocellular carcinoma HepG2 cell line was purchased from ATCC (Manassas, VA) and cultured in EMEM with 10% FBS. A549 cells were from Dr. Hillman (Wayne State University) and cultured in F12-K medium with 10 % FBS and 1% Pen-Strep. All cells were maintained in an incubator at 37 °C and 5% CO2.
2.4. Cytotoxicity
Cytotoxicity was determined using CellTiter-Blue Cell Viability Assay (Promega, Madison, WI) in U2OS, 4T1 and HepG2 cells according to the manufacturer’s protocol. Cells were seeded in 96-well plates 24 h prior to treatment at a density of 6,000 cells/well for U2OS, 3,000 cells/well for 4T1, and 4,000 cells/well for HepG2. The medium was then replaced by 100 μL of serial dilutions of HCQ, pHPMA and pCQ in complete cell culture medium and the cells were incubated for 24 h. To measure cell viability, polymer/drug-containing medium was replaced with a mixture of 100 μL culture medium and 20 μL of CellTiter-Blue reagent and the cells were incubated for another 1 h. The fluorescence intensity [I] was measured using SpectraMaxM5e Multi-Mode microplate Reader (Molecular Devices, CA) at 560Ex/590Em. The relative cell viability (%) was calculated as [I]treated/[I]untreated × 100 %.
2.5. Inhibition of autophagy
Western blot was used to measure the effect of pCQ on autophagy in U2OS and 4T1 cells. Cells were treated with HCQ, pCQ10.0 or pCQ16.7 for 24 h, and then washed with cold PBS, and lysed in ice-cold lysis buffer containing protease and phosphatase inhibitors for 30 min. The lysate was centrifuged at 15,000 rpm for 10 min at 4 °C to collect the supernatant. Total protein was extracted with Laemmli lysis buffer according to the suggested protocol and the protein concentration was quantified by the BCA assay. The samples were then loaded and separated on SDS/PAGE gel, transferred to nitrocellulose membranes followed by probing with LC3B antibody and incubation with anti-rabbit IgG HRP-linked antibody. GAPDH was used as a housekeeping control. Quantification of LC3B levels was performed by ImageJ. The results are shown as mean of duplicate experiment (n=2).
2.6. Intracellular trafficking
U2OS, 4T1 and A549 cells were seeded 18 h prior to treatment at a density of 50,000 cells per confocal chamber. Then, cells were washed twice with PBS and incubated with culturing media containing Rhodamine-labeled pCQ (F-pCQ10.0) for 24 h. Rhodamine-labeled pHPMA (F-pHPMA) was used as a control either alone or co-incubated with pCQ16.7 or HCQ. Cells were washed five times with PBS and fixed with 4% paraformaldehyde at room temperature for 20 min. Fixed cells were washed four times with PBS and nuclei were stained with 1 μM Hoechst 33258 solution for 30 min before imaging. The images were obtained using Zeiss 800 Airyscan Microscope coupled with 63x oil objective and z-axis motor.
2.7. Quantification of cell surface expression of CXCR4
U2OS cells were seeded in T25 culturing flask 18 h prior to treatment. Cells were treated with different concentrations of HCQ, pHPMA and pCQ in HEPES-buffered low-serum medium (DMEM supplemented with 2 mM L-glutamine, 1% penicillin-streptomycin, 1% FBS and 10 mM HEPES) for 90 min and 24 h before flow cytometry analysis. After washing with PBS, cells were dissociated by enzyme-free cell dissociation buffer and stained with allophycocyanin (APC) Mouse B Anti-Human CD184 and APC Mouse IgG2a, κ Isotype Control according to the suggested protocol. FACSCalibur was used to analyze the cells (10,000 events per sample), and data were processed using FlowJo software V7.6.1.
2.8. CXCR4 redistribution assay
U2OS cells stably expressing EGFP-CXCR4 were seeded at a density of 50,000 cells per confocal chamber (Lab-Tek Chambered #1.0 Borosilicate Coverglass 4 chamber System) 18 h prior to treatment. Cells were washed twice with 0.5 mL PBS and incubated with HEPES-buffered low-serum medium with HCQ (300 nM) or pCQ16.7 (100 μM) for 30 min before 1 h exposure to CXCL12 (10 nM). Then cells were washed five times using PBS and fixed with 4% paraformaldehyde at room temperature for 20 min. Fixed cells were then washed four times with PBS and stained with 1 μM Hoechst 33258 solution for 30 min before imaging. The images were obtained using Zeiss 800 Airyscan Microscope coupled with 63x oil objective and z-axis motor.
2.9. CXCL12-induced cell invasion
Porous transwell inserts (pore size 8 μm, Falcon) were coated with 40 μL ice-cold diluted Matrigel (1:3 v/v with serum-free medium) and placed in 37 °C incubator for 2 h. U2OS cells were trypsinized and resuspended in serum-free medium containing HCQ, pCQ or AMD3100 for 15 min before adding to the inserts at a final concentration of 100,000 cells in 300 μL medium. Inserts were placed in a 24-well companion plate containing 20 nM CXCL12 in serum-free medium in each well. The cells were then incubated at 37 °C and allowed to invade through the Matrigel-coated insert membrane for 18 h. The non-invaded cells on the upper side of the insert membrane were removed by cotton swabs and the invaded cells attached on the bottom surface were fixed in 100% methanol and stained with 0.2% Crystal Violet solution for 15 min at room temperature. Five different areas under 20× or 40× magnification were randomly selected and imaged using EVOS xl microscope. The number of invaded cells in each area was counted and the results were expressed as average number of invaded cells/imaging area ± SD (n = 5).
2.10. Inhibition of ERK activation
Western blot was used to evaluate the effect of pCQ on inhibiting the phosphorylation of ERK induced by CXCL12 in 4T1 cells. The cells (5×106) were seeded 16 h prior to the treatment. The cells were washed with PBS, and incubated with AMD3100 (300 nM), HCQ, or pCQ in serum-free medium for 4 h before 20 min incubation with mouse CXCL12 (100 ng/mL). Total protein was extracted as above and separated by SDS-PAGE. The samples were transferred to nitrocellulose membrane, followed by probing with pERK antibody and incubation with HRP-linked secondary antibody. GAPDH and Erk were used as housekeeping controls. Quantification of the band intensities was performed by ImageJ. The results are shown as mean of duplicate experiment (n=2).
2.11. Serum-induced cell migration
4T1 cells were trypsinized, washed with PBS, and suspended in serum-free medium containing HCQ, pCQ or AMD3100 for 20 min before adding to the transwell inserts at a final concentration of 50,000 cells in 300 μL medium. Inserts were then placed in a 24-well companion plate containing medium with 10% FBS in each well. The cells were then incubated at 37 °C and allowed to migrate through the insert membrane for 8 h. The non-migrated cells on the upper side of the membrane were removed by cotton swabs and the migrated cells attached on the bottom surface were fixed, stained, imaged and counted as described above. Results were expressed as average number of migrated cells/imaging area ± SD (n = 6).
2.12. Antimetastatic activity in vivo
All animal experiments followed a protocol approved by the University of Nebraska Medical Center (UNMC) Institutional Animal Care and Use Committee. Animals were placed in a facility accredited by the Association for Assessment and Accreditation of Laboratory Animal Care upon arrival. A total of 40 female Balb/c mice (8 weeks old) were randomly assigned into five groups (n=8). Half a million 4T1 cells were treated with HCQ (20 μM), pCQ10.0 (100 μM) or pCQ16.7 (100 μM) for 4 h before intravenous injection via the tail vein (in 100 μL PBS). The animals were then intravenously administrated with HCQ (10 mg/kg or 30 mg/kg), pCQ10.0 (10 mg/kg), or pCQ16.7 (10 mg/kg) on day 3, 5, 7 and 9 for a total of four doses. The animals were sacrificed on day 11, and the lungs were inflated with 30% sucrose followed by fixation in Bouin’s solution for 18 h. The lungs were then stored in 70% ethanol before further tissue processing. Each of the five lobes was separated and all surface tumors counted using dissecting microscope. After counting, the lungs were sectioned and stained with H&E at the UNMC core facility. Major other organs, including heart, liver, spleen and kidneys were also harvested, fixed in 4% paraformaldehyde, sectioned and stained with H&E. Blinded histological analysis of the tissues was conducted by a trained pathologist at the UNMC core facility.
2.13. Statistical analysis
Results are presented as mean ± standard deviation (SD). The Student’s t-test was used to determine the statistical significance of the results obtained in all the studies of this proposal when assessing differences between two groups; ANOVA was used to determine differences among multiple groups. All statistical analysis was performed using Graphpad Prism v5. A value of p < 0.05 was considered statistically significant.
3. Results and Discussion
We recently reported synthesis of pCQ (Scheme 1) as a potential polymeric drug that takes advantage of the multivalency effect to enhance the ability of CQ to inhibit CXCR4-mediated invasion of lymphoma and leukemia cells [25]. We found that pCQ with 10 and 16.7 mol% of MA-CQ exhibited the most favorable activity profile in leukemia and lymphoma cells in vitro. In the present study, we extend our investigations to establish antimetastatic activity of pCQ in experimental lung metastasis model in vivo and to elucidate the mechanism of action of pCQ.
3.1. Synthesis and characterization of pCQ
Table 1 shows properties of all the polymers used in this study. In addition to the two pCQ (pCQ10.0 and pCQ16.7), we have synthesized pHPMA as a negative control. To enable tracking of the intracellular fate of pCQ, we synthesized a fluorescently-labeled pCQ derivative (F-pCQ10.0) and fluorescently labeled pHPMA (F-pHPMA). To fluorescently label the polymers with rhodamine, we included APMA in the polymerization mixture to introduce primary amines into the polymer structure. The primary amines were then reacted with RBITC. The rhodamine B (RBITC) content in the polymers was analyzed by measuring absorbance at 544 nm. There were 2.5 and 3.6 rhodamine B molecules present in average F-pCQ10.0 and F-pHPMA macromolecule, respectively.
Table 1.
Polymer characterization.
| mol% MA-CQ
|
Mn |
|
Pn | |||
|---|---|---|---|---|---|---|
| in feed | in polymer | |||||
| pHPMA | 0 | 0 | 33,870 | 1.6 | 235 | |
| pCQ10.0 | 9.1 | 10.0 | 18,400 | 1.5 | 109 | |
| pCQ16.7 | 20.0 | 16.7 | 18,900 | 1.8 | 101 | |
| F-pHPMA | 0 | 0 | 23,610 | 1.5 | 164 | |
| F-pCQ10.0 | 12.0 | 10.0 | 17,490 | 1.5 | 95 | |
3.2 Cytotoxicity of pCQ
Before evaluating therapeutic activity of pCQ, we first examined its cytotoxicity in three different cell lines using CellTiter-Blue Assay. In addition to the breast cancer cell line 4T1 and osteosarcoma cell line U2OS, we also included human hepatocellular carcinoma cell line HepG2, which is a well-established and frequently used in vitro toxicity model for drug screening. As shown in Figure 1, HCQ exhibited cytotoxicity in all three cell lines, with IC50 of 22, 28 and 42 μg/mL in 4T1, U2OS and HepG2 cells, respectively. The corresponding IC50 values expressed as molar concentrations were 70, 88 and 130 μM, respectively. In contrast to HCQ, both pCQ showed remarkably lowered cytotoxicity. pCQ16.7 had an estimated IC50 >2,000 μg/mL in all three cell lines and the estimated IC50 for pCQ10.0 was >3,000 ug/mL. pHPMA exhibited no toxicity in any of the cell lines within the tested concentration range. Considering the content of HPMA in pCQ, we also compared the cytotoxicity in terms of equivalent CQ concentrations. Both pCQ polymers demonstrated no toxicity at equivalent CQ concentration of 100 μM and IC50 was >1,500 μM CQ equivalent in all three cell lines. Based on the cytotoxicity findings, we selected 20 μM HCQ as a safe dose for subsequent biological studies. Concentrations up to 100 μM CQ equivalent were considered as safe for the pCQ polymers. HPMA copolymers are known to be non-toxic and non-immunogenic and have been widely applied as drug carriers for both small molecule drugs and biomacromolecules [26, 27]. Here we have shown that incorporation of CQ into HPMA copolymers greatly improves its safety in multiple cell lines.
Figure 1.

Cytotoxicity of pCQ. Cell viability of pCQ, HCQ and pHPMA was determined using CellTiter-Blue Assay. Results shown as mean cell viability ± SD (n=3).
3.3. Effect of pCQ on autophagy
With the goal of assessing possible differences in the mechanism of action between pCQ and CQ, we first evaluated the effect of pCQ on autophagy in U2OS and 4T1 cells. Autophagy is a cell survival mechanism that utilizes degradation and recycling of cellular proteins and cytoplasmic organelles. Damaged proteins or dysfunctional organelles are sequestered into autophagosomes, which then fuse with lysosomes to form autolysosomes where the contents are degraded and recycled. Autophagy is often upregulated in cancers because cancer cells use this mechanism to survive stress and starvation in the tumor microenvironment. Upregulation of autophagy promotes tumorigenesis and tumor aggressiveness [28, 29]. CQ is among several autophagy inhibitors that have been tested in combination with other anticancer drugs [18, 19, 24, 30, 31]. Although the mechanism of action is still not fully understood, CQ is believed to inhibit autophagy in cancer cells by preventing the fusion of autophagosomes and lysosomes. To investigate the effect of pCQ on autophagy, we performed Western blot to quantify the levels of autophagy marker LC3 (microtubule-associated protein 1A/1B-light chain 3). The cytosolic form of LC3 (LC3-I) is converted into LC3-II, which is bound to the autophagosomal membrane, indicating autophagic activity. Monitoring degradation of LC3-II serves as a convenient measure of autophagic activity [32]. The LC3-II degradation is blocked when cells are treated with CQ, which inhibits lysosomal acidification and leads to the accumulation of LC3-II in the cells (Figure 2A). Our results show that HCQ treatment resulted in substantial inhibitory activity indicated by the elevated levels of LC3-II in both cell lines. In contrast, pCQ showed only a modest autophagy inhibition in 4T1 cells and no inhibitory activity was observed in U2OS cells. The relative expression of LC3-II and total LC3 expression (i.e., LC3-I + LC3-II) were quantified from the Western blots (Figure 2B). HCQ treatment significantly increased LC3-II expression in both cell lines, with a 4-fold increase observed in 4T1 cells. In contrast, only 1.7- and 1.9-fold increase in LC3-II expression was observed in 4T1 cells treated with pCQ10.0 and pCQ16.7, respectively. Because of the very low LC3-II expression in untreated cells, we have quantified total LC3 levels in the U2OS cells. HCQ treatment resulted in a 5-fold increase in the total LC3 expression, with majority of the increase attributed to the LC3-II. A small non-significant increase (~1.1-fold) in LC3 expression was seen in pCQ-treated U2OS cells. These results clearly suggest that incorporation of HCQ into a polymer results in a significant loss of the underlying autophagy inhibitory activity.
Figure 2.

Effect of pCQ on autophagy in U2OS and 4T1 cells. Cells were treated with HCQ (20 μM), pCQ10.0, and pCQ16.7 (100 μM) for 24 h before Western blot analysis (A). The band intensities from two independent experiments were quantified by ImageJ (B). (* p<0.05, ** p<0.01 vs. HCQ; ANOVA with Tukey’s multiple comparison test)
3.4 Intracellular trafficking of pCQ
One of the most challenging tasks in therapeutic delivery of macromolecules and nanoparticles is the ability to traverse cell membranes and to gain access to the cytoplasm. Macromolecular drug delivery systems are typically internalized by endocytosis and subsequently sequestered by lysosomes [33–35]. The ability to avoid or escape the endo/lysosomal pathway remains a major challenge. CQ is one of the most intensively studied lysosomolytic agents that rely on endo/lysosomal buffering and osmotic imbalance to cause release of lysosomal contents into the cytoplasm [36, 37]. The ability of CQ to inhibit autophagy is directly related with its lysosomal tropism [38]. CQ is a lipophilic weak base that enters the cells and accumulates in the endosomes and lysosomes as a result of its protonation in the acidic environment. The function of CQ is highly dependent on its concentration. Low concentrations of CQ inhibit acidification of endosomes, which prevents endosome maturation. High concentrations of CQ may lead to endosomal and lysosomal swelling and rupture of the vesicles [39]. This property has been widely used to promote endosomal escape and efficient delivery of various biologicals, including proteins [37] and nucleic acids [40–42]. However, high and often toxic concentrations of CQ are needed to achieve endosomal escape which severely limits the use of CQ in the in vivo applications [43, 44].
The low activity of pCQ in inhibiting autophagy suggested altered intracellular trafficking as one of the possible reasons for the differences between HCQ and pCQ. We used confocal microscopy to investigate the intracellular fate of the pCQ. We first treated cells with F-pCQ10.0 and F-pHPMA for 24 h prior to imaging. Three different cell lines were tested, including U2OS, 4T1 and a human lung cancer cell line A549 to determine if there are any cell-dependent differences in the intracellular trafficking of the polymers. As shown in Figure 3, very low levels of accumulation of F-pHPMA were observed in the endo/lysosomes indicated by the punctate pattern of the red fluorescence. No detectable endosomal escape was seen even after 24 h. In a striking contrast, F-pCQ10.0 exhibited substantially different intracellular trafficking behavior. F-pCQ10.0 was extensively and evenly distributed in the cytoplasm, with no indication of lysosomal sequestration. The cytosolic distribution was cell line-independent and could be observed from early time points (2 h) of incubation (Figure S1).
Figure 3.

Intracellular trafficking of pCQ in U2OS, 4T1 and A549 cells. Cells were treated with Rhodamine-labeled F-pCQ10.0 (red, 100 μM) for 24 h before confocal imaging (63×). Rhodamine-labeled pHPMA (F-pHPMA) was used as a control either alone or in combination with pCQ10.0 (100 μM) or HCQ (20 μM). Cell nuclei were stained with Hoechst 33342 (blue).
In order to better understand the mechanism of the cytoplasmic translocation of pCQ, we co-incubated fluorescently labeled F-pHPMA with non-labeled pCQ (100 μM) or HCQ (20 μM). In both cases, we observed mostly punctate distribution of red fluorescence similar to F-pHPMA alone. Marginal cytosolic distribution of red fluorescence was observed in the case of F-pHPMA co-incubated with HCQ, suggesting that the local concentration of HCQ was able to slow down the maturation of endosomes but not high enough to trigger endosomal rupture and cytosolic release of the F-pHPMA [45, 46]. Interestingly, HCQ treatment increased the amount of F-pHPMA found in the cells after 24 h incubation when compared to cells treated with F-pHPMA alone. While it is possible that HCQ increased the cell uptake of F-pHPMA, we believe the observation is more likely a result of impaired endosomal recycling and an increased retention of the polymer in lysosomes of cells treated with HCQ [47–49]. The lack of cytoplasmic localization of F-pHPMA when co-incubated with pCQ suggested that pCQ most likely does not cause extensive endo/lysosomal rupture that would release the content of the vesicles into the cytoplasm. Instead, the results pointed to a pH-dependent membrane activity of pCQ that allows its translocation across endosomal membrane without significant membrane disruption. This hypothesis is supported by the results of a liposomal calcein leakage assay which showed only a modest dye release from the liposomes treated with pCQ (Figure S2). It is likely that the structure of pCQ with its hydrophobic backbone and pH-sensitive CQ side chains facilitates interaction with the endosomal membrane and cytoplasmic translocation of the copolymer. The confocal microscopy results provide evidence and possible explanation for the pCQ’s lack of inhibitory effect on autophagy and their low cytotoxicity. Unlike HCQ, pCQ does not seem to accumulate in the lysosomes, which leads to poor inhibition of autophagy. To the best of our knowledge, such intriguing intracellular behavior of synthetic polymers has not been reported previously and the results encourage further exploration of the pCQ as a cytoplasmic drug delivery carrier.
3.5. Effect of pCQ on cell surface expression of CXCR4
CQ was suggested as a CXCR4 antagonist in various types of cancers only recently [14, 15]. Traditional CXCR4 antagonists like AMD3100 exert their function by specifically binding with the CXCR4 receptors located on the cell surface, thus preventing binding of the chemokine ligand CXCL12. This inhibition of CXCL12 binding then prevents CXCR4 receptor internalization and suppresses activation of the related downstream signaling cascades. CQ and HCQ on the other hand, appear to promote internalization of the surface CXCR4 receptors and their sequestration in the lysosomes, which then makes the receptors inaccessible for binding with extracellular CXCL12 chemokine molecules [15, 25].
To investigate the effect of pCQ on CXCR4 inhibition, we used flow cytometry to quantify the changes in the surface expression of the CXCR4 receptor in U2OS cells after treatment with pCQ. The cells were treated with AMD3100, pCQ, HCQ, and pHPMA for 1.5 h and 24 h prior to incubation with anti-CXCR4 antibody. As shown in Figure 4, treatment with the CXCR4-binding compound AMD3100 resulted in a significant decrease in the amount of detectable CXCR4 receptors on the cell surface. In contrast, HCQ (20 μM) did not cause any significant change in the levels of surface CXCR4 receptors. After 1.5 h of incubation, cells treated with pCQ10.0 exhibited no decrease in the levels of CXCR4 surface expression, while pCQ16.7 resulted in a significant decrease even at this early time point. The reduction in surface CXCR4 receptor expression with both pCQ10.0 and pCQ16.7 became more pronounced after 24 h. In addition, higher concentrations of pCQ (100 μM HCQ equivalent) also resulted in a more pronounced decrease in the cell surface CXCR4 levels. As expected, pHPMA did not show any effect on the surface CXCR4 expression. All these data suggest that pCQ is considerably more effective in reducing cell surface CXCR4 than HCQ and that its effect is dependent on the concentration, HCQ content, and time of incubation.
Figure 4.

Effect of pCQ on expression of surface CXCR4 receptors in U2OS cells. Cells were treated in the absence of CXCL12 with AMD3100, HCQ, pCQ or pHPMA for 1.5 h or 24 h before flow cytometry analysis (n=2).
Further analysis of how pCQ affects the decrease of cell surface CXCR4 expression upon stimulation with CXCL12 was conducted using CXCR4 redistribution assay (Figure 5A). U2OS cells expressing EGFP-CXCR4 allow easy tracking of the CXCR4 intracellular distribution. Incubation of the cells with the CXCR4 ligand CXCL12 causes redistribution of the receptor from plasma membrane into intracellular vesicles. This process is prevented by AMD3100 as it binds CXCR4 expressed on the cell surface. The fact that AMD3100 restricts localization of the CXCR4 receptor to the cell surface suggests that the apparent decrease in cell surface expression of CXCR4 determined by flow cytometry in Figure 4 is simply a result of AMD3100 preventing binding of the staining anti-CXCR4 antibody to the receptor. In contrast, pCQ promotes CXCR4 internalization into intracellular vesicles, which makes the receptor inaccessible for binding with extracellular CXCL12. These results suggest that pCQ may inhibit CXCR4/CXCL12-mediated processes using a different mechanism of action than traditional CXCR4 antagonists like AMD3100. To further support this hypothesis, we also determined the cell surface expression of CXCR4 in the presence of CXCL12 (Figure 5B). Upon stimulation with CXCL12, a decrease of surface CXCR4 expression was observed, confirming the data in Figure 5A. Cells treated with CXCL12 and the antagonist AMD3100 showed enhanced CXCR4 surface expression when compared with CXCL12-treated cells and the levels were similar to those observed in Figure 4. In contrast, treatment with CXCL12 and pCQ16.7 resulted in further reduction in surface CXCR4 expression, confirming enhanced intracellular localization of the CXCR4 receptors.
Figure 5.

Effect of pCQ on redistribution of the CXCR4 receptors under stimulation with CXCL12. (A) U2OS cells overexpressing EGFP-CXCR4 were treated with AMD3100 (300 nM) or pCQ16.7 (100 μM) for 30 min before exposing to CXCL12 (10 nM) for 1 h. The cells were then fixed and imaged by confocal microscopy (63×). (B) Cell surface expression of CXCR4 in U2OS cells measured by flow cytometry. Cells were treated as described above in Figure 4 (n=2).
3.6. Inhibition of the CXCR4/CXCL12 axis by pCQ
The important role of CXCR4/CXCL12 axis as a therapeutic target is often highlighted by its ability to promote migration and invasion of cancer cells as an important step in metastasis. Here, we evaluated in vitro activity of pCQ in transwell cell migration and invasion assays. We first applied CXCL12 as chemoattractant in cell invasion of the CXCR4-overexpressing U2OS cells through a layer of Matrigel (Figure 6). The results showed that both pCQ10.0 and pCQ16.7 were able to completely inhibit CXCL12-induced cell invasion at 100 μM concentration. The inhibitory activity was not only considerably higher than activity achieved with safe concentrations of HCQ (~28% inhibition), but even better than the activity of the positive control AMD3100 which showed about 77% inhibition.
Figure 6.

Inhibition of CXCL12-induced cell invasion. A) U2OS cells were treated with pCQ, HCQ or AMD3100 (300 nM) and allowed to invade through a layer of Matrigel upon stimulation with CXCL12 for 18 h. B) The number of invaded U2OS cells was counted and results are shown as mean number of invaded cells/40× view ± SD (n ≥ 3). (**** p<0.0001 vs. HCQ; ANOVA with Tukey’s multiple comparison test)
The effect of pCQ on inhibiting CXCL4/CXCL12 chemokine axis was also evaluated by examining the activity on downstream signaling targets of CXCR4. ERK is one of the key downstream targets phosphorylated upon CXCR4 activation by CXCL12. Upregulation of pERK is directly associated with cancer cell migration and invasion [50]. Here, 4T1 cells were treated with AMD3100 (300 nM), HCQ (20 μM), or pCQ (100 μM) for 4 h followed by 20 min incubation with CXCL12 before Western blot analysis. As shown in Figure 7, pERK levels more than doubled after CXCL12 stimulation, and AMD3100 could inhibit the process. HCQ showed weaker inhibitory effect on pERK than AMD3100. In contrast, both pCQ10.0 and pCQ16.7 markedly decreased pERK levels, even more so than AMD3100. This finding provides supporting evidence that the mechanism of action of pCQ involves regulating the CXCR4/CXCL12 chemokine axis. The U2OS cells used in this study have impaired ERK signaling and were thus not used in this experiment.
Figure 7.
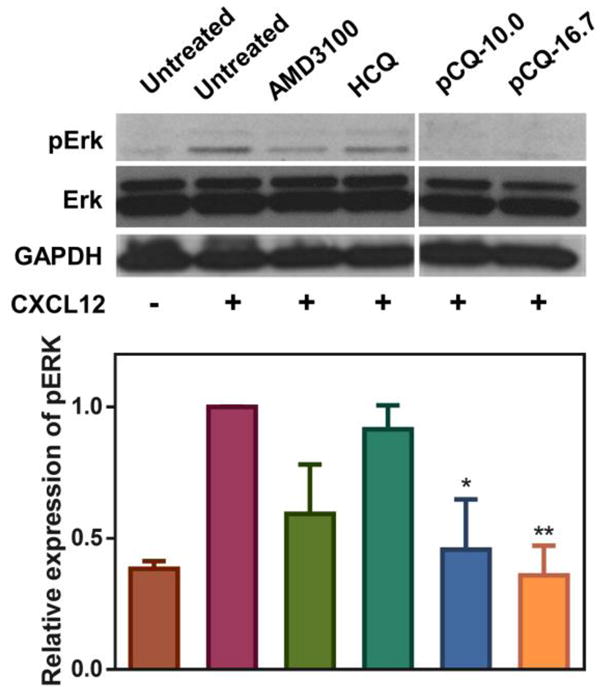
Inhibition of pERK by pCQ. 4T1 cells were treated with AMD3100 (300 nM), HCQ (20 μM), or pCQ (100 μM) for 4 h followed by 20 min incubation with CXCL12 (100 ng/mL) before lysis (n=2). (* p<0.05, ** p<0.01 vs. CXCL12+ untreated; ANOVA with Tukey’s multiple comparison test)
To further investigate if the inhibitory effect of pCQ on cancer cell motility observed in Figure 6 was specifically due to CXCR4 inhibition, we also performed a transwell migration assay using FBS as the chemoattractant. FBS contains a complex mixture of proteins that serve as chemoattractants for cancer cells. 4T1 cells were treated with pCQ, HCQ, and AMD3100 and allowed to migrate through the membrane inserts for 8 h (Figure 8). Specific CXCR4 inhibitor AMD3100 showed no inhibition of FBS-induced cell migration despite the high concentration (20 μM) used. In contrast, treatment with HCQ decreased cell migration by ~26%. Both pCQ10.0 and pCQ16.7 demonstrated even greater inhibition of the cell migration than HCQ. For example, pCQ10.0 decreased cell migration by 63% at 20 μM and by 86% at 100 μM. These results suggest that the inhibitory activity of pCQ in the cell migration and invasion studies is not CXCR4/CXCL12 specific. It appears that pCQ exerts its effect in a relatively non-specific and broad way that includes effects on other signaling pathways responsible for cancer cell motility.
Figure 8.

Inhibition of FBS-induced cell migration A) 4T1 cells were treated with pCQ, HCQ or AMD3100 (20 μM) and allowed to migrate through porous membrane upon stimulation with FBS for 8 h. B) The number of migrated 4T1 cells was counted and results are shown as mean number of invaded cells/20× view ± SD (n≥3). “Background” represents number of randomly migrating cells in the absence of any chemoattractant. “Untreated” represents the number of migrating cells in the presence of FBS. (**** p<0.0001 vs. HCQ; ANOVA with Tukey’s multiple comparison test)
3.7. Antimetastatic activity of pCQ
To investigate if the ability of pCQ to inhibit cancer cell migration and invasion in vitro translates into decreased metastasis in vivo, we used an experimental lung metastasis model of the 4T1 breast cancer. In this model, cancer cells are injected intravenously (i.v.) to colonize the lung and form lung metastasis. After the cell injection, the treatments were given via tail vein i.v. injection. The following five experimental groups were tested: (i) untreated (saline), (ii) HCQ (low dose, 10 mg/kg body weight), (iii) HCQ (high dose, 30 mg/kg), (iv) pCQ10.0 (10 mg/kg HCQ equivalent), and (v) pCQ16.7 (10 mg/kg HCQ equivalent). The lung tumors were allowed to grow for 11 days and the mice were sacrificed. The total tumor burden in the lungs was quantified by counting total number of visible surface lung metastases (regardless of their size) and further analyzed by H&E staining of lung tissue sections. As shown in Figure 9A, treatment with low dose HCQ exhibited no activity, while the high dose HCQ treatment resulted in a decreased number of surface lung metastatic lesions compared to the untreated group, however the difference was not statistically significant. The results with pCQ showed that pCQ16.7 had the highest antimetastatic activity even though only a low dose (10 mg/kg HCQ equivalent) was used. This antimetastatic inhibitory effect was higher even than the control HCQ used at the high dose. Interestingly, pCQ10.0 had no significant effect on the number of lung metastases but the size of the lung tumors in animals treated with pCQ10.0 was smaller than in the untreated animals. The low in vivo activity of pCQ10.0 was likely due to insufficient dose and less pronounced and slower effect on changes in the CXCR4 surface expression as suggested by the results in Figure 4.
Figure 9.

Antimetastatic activity of pCQ in experimental 4T1 lung metastasis model. 4T1 cells were injected i.v. in Balb/c mice, followed by 4 i.v. doses of pCQ or HCQ. L = low dose (10 mg/kg HCQ equivalent, H = high dose (30 mg/kg HCQ equivalent). (A) Total number of surface lung metastases. Results shown as average of total number of surface lung mets ± SD (n = 8) (** p<0.01; *** p<0.001). (B) Representative images of the whole lung and H&E staining of the lung tissue sections (4× and 10×).
In addition to surface lung metastases, we also evaluated the tumor burden in the lungs by histopathological analysis (Figure 9B). H&E staining of the lung sections revealed that the number of metastases in the lungs correlated well with the number of surface lesions. Treatment with pCQ16.7 resulted not only in a decreased number of metastases in the lung, but also smaller sizes of the metastases. Untreated animals typically exhibit signs of mortality related to the tumor burden in the lungs around 14–18 days after injection of the 4T1 cells. We therefore expect the pCQ treatment may improve animal survival since we observed significantly reduced tumor burden in the lungs as late as day 11. These in vivo results provide important evidence for antimetastatic activity of pCQ16.7 in 4T1 lung metastatic model, which is one of the most aggressive cancer models in mice. The significant enhancement of antimetastatic activity of pCQ16.7 compared with HCQ was confirmed not only by the overall decrease in the lung metastases, but also by the ability to achieve such effect at a much lower dose than HCQ.
3.8. Toxicity of pCQ in vivo
Although exhibiting significantly lowered toxicity in vitro compared with HCQ, it was important to investigate the possible toxicity of pCQ in vivo. As illustrated in Figure 10A, no apparent loss of body weight was observed in any of the treatment groups until the end of the experiment, indicating that pCQ is well tolerated when given systemically. To further explore the effect on major organs including heart, liver, spleen and kidney, a blinded histopathological examination on H&E stained tissue sections was performed by a pathologist (Figure 10B). No significant morphological differences or tissue damage were observed in any of the treatment groups when compared with the untreated controls. These results confirm safety of pCQ16.7 after multiple administered doses in vivo.
Figure 10.
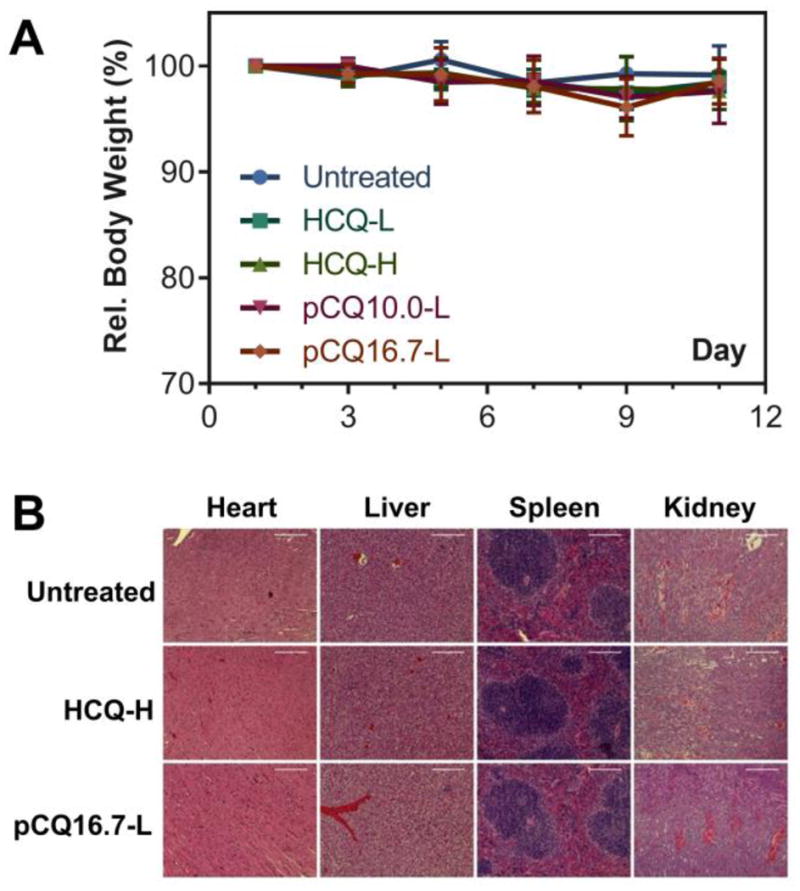
Toxicity evaluation of pCQ in vivo. (A) Relative body weight. Results shown as % body weight relative to the body weight on day 0. (B) H&E staining of major organs in different treatment groups (heart 20×, liver 20×, spleen 20×, kidney 10×).
4. Conclusions
We developed CQ-based polymeric drugs with antimetastatic activity and ability to effectively translocate to the cytoplasm. The pCQ exhibited lowered cytotoxicity, enhanced inhibition of cancer cell migration and invasion, and improved antimetastatic activity in vivo when compared with parent HCQ. Although not fully understood yet, our results revealed that pCQ mechanism of action involves, in part, inhibition of the CXCR4/CXCL12 chemokine axis. More importantly, pCQ shows the ability to efficiently translocate to the cytoplasm, suggesting a promise as a potential delivery platform for combination anticancer therapy to achieve simultaneous antimetastatic effect and cytoplasmic drug delivery.
Supplementary Material
Acknowledgments
Financial support from the University of Nebraska Medical Center, National Institutes of Health (EB015216, EB020308, EB019175), and Changjiang Scholar Program is gratefully acknowledged. We thank Dr. Geoffrey Talmon for histophathological analysis of the tissues and Janice Taylor and James Talaska for help with confocal microscopy. Support for the UNMC Advanced Microscopy Core Facility was provided by the Nebraska Research Initiative, the Fred and Pamela Buffett Cancer Center Support Grant (P30CA036727), and an Institutional Development Award (IDeA) from the NIGMS (P30GM106397).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Liechty WB, Kryscio DR, Slaughter BV, Peppas NA. Polymers for drug delivery systems. Annu Rev Chem Biomol Eng. 2010;1:149–173. doi: 10.1146/annurev-chembioeng-073009-100847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Duncan R, Vicent MJ. Polymer therapeutics-prospects for 21st century: the end of the beginning. Adv Drug Deliv Rev. 2013;65:60–70. doi: 10.1016/j.addr.2012.08.012. [DOI] [PubMed] [Google Scholar]
- 3.Dhal PK, Polomoscanik SC, Avila LZ, Holmes-Farley SR, Miller RJ. Functional polymers as therapeutic agents: concept to market place. Adv Drug Deliv Rev. 2009;61:1121–1130. doi: 10.1016/j.addr.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 4.Fasting C, Schalley CA, Weber M, Seitz O, Hecht S, Koksch B, Dernedde J, Graf C, Knapp EW, Haag R. Multivalency as a chemical organization and action principle. Angew Chem Int Ed Engl. 2012;51:10472–10498. doi: 10.1002/anie.201201114. [DOI] [PubMed] [Google Scholar]
- 5.Kiessling LL, Gestwicki JE, Strong LE. Synthetic multivalent ligands as probes of signal transduction. Angew Chem Int Ed Engl. 2006;45:2348–2368. doi: 10.1002/anie.200502794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li J, Yu F, Chen Y, Oupicky D. Polymeric drugs: Advances in the development of pharmacologically active polymers. J Controlled Rel. 2015;219:369–382. doi: 10.1016/j.jconrel.2015.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Joyce JA, Pollard JW. Microenvironmental regulation of metastasis. Nat Rev Cancer. 2009;9:239–252. doi: 10.1038/nrc2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burger JA, Kipps TJ. CXCR4: a key receptor in the crosstalk between tumor cells and their microenvironment. Blood. 2006;107:1761–1767. doi: 10.1182/blood-2005-08-3182. [DOI] [PubMed] [Google Scholar]
- 9.Teicher BA, Fricker SP. CXCL12 (SDF-1)/CXCR4 pathway in cancer. Clin Cancer Res. 2010;16:2927–2931. doi: 10.1158/1078-0432.CCR-09-2329. [DOI] [PubMed] [Google Scholar]
- 10.Fahham D, Weiss ID, Abraham M, Beider K, Hanna W, Shlomai Z, Eizenberg O, Zamir G, Izhar U, Shapira OM, Peled A, Wald O. In vitro and in vivo therapeutic efficacy of CXCR4 antagonist BKT140 against human non-small cell lung cancer. J Thorac Cardiovasc Surg. 2012;144:1167–1175e1161. doi: 10.1016/j.jtcvs.2012.07.031. [DOI] [PubMed] [Google Scholar]
- 11.Wong D, Korz W. Translating an Antagonist of Chemokine Receptor CXCR4: from bench to bedside. Clin Cancer Res. 2008;14:7975–7980. doi: 10.1158/1078-0432.CCR-07-4846. [DOI] [PubMed] [Google Scholar]
- 12.Gerlach LO, Skerlj RT, Bridger GJ, Schwartz TW. Molecular interactions of cyclam and bicyclam non-peptide antagonists with the CXCR4 chemokine receptor. J Biol Chem. 2001;276:14153–14160. doi: 10.1074/jbc.M010429200. [DOI] [PubMed] [Google Scholar]
- 13.Scala S. Molecular Pathways: Targeting the CXCR4–CXCL12 Axis—Untapped Potential in the Tumor Microenvironment. Clin Cancer Res. 2015;21:4278–4285. doi: 10.1158/1078-0432.CCR-14-0914. [DOI] [PubMed] [Google Scholar]
- 14.Kim J, Yip MR, Shen X, Li H, Hsin LYC, Labarge S, Heinrich EL, Lee W, Lu J, Vaidehi N. Identification of anti-malarial compounds as novel antagonists to chemokine receptor CXCR4 in pancreatic cancer cells. PLoS One. 2012;7:e31004. doi: 10.1371/journal.pone.0031004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Balic A, Sorensen MD, Trabulo SM, Sainz B, Jr, Cioffi M, Vieira CR, Miranda-Lorenzo I, Hidalgo M, Kleeff J, Erkan M, Heeschen C. Chloroquine targets pancreatic cancer stem cells via inhibition of CXCR4 and hedgehog signaling. Mol Cancer Ther. 2014;13:1758–1771. doi: 10.1158/1535-7163.MCT-13-0948. [DOI] [PubMed] [Google Scholar]
- 16.Solomon VR, Lee H. Chloroquine and its analogs: A new promise of an old drug for effective and safe cancer therapies. Eur J Pharmacol. 2009;625:220–233. doi: 10.1016/j.ejphar.2009.06.063. [DOI] [PubMed] [Google Scholar]
- 17.Srivastava V, Lee H. Chloroquine-based hybrid molecules as promising novel chemotherapeutic agents. Eur J Pharmacol. 2015;762:472–486. doi: 10.1016/j.ejphar.2015.04.048. [DOI] [PubMed] [Google Scholar]
- 18.Janku F, McConkey DJ, Hong DS, Kurzrock R. Autophagy as a target for anticancer therapy. Nature reviews Clin Oncol. 2011;8:528–539. doi: 10.1038/nrclinonc.2011.71. [DOI] [PubMed] [Google Scholar]
- 19.Zhang X, Zeng X, Liang X, Yang Y, Li X, Chen H, Huang L, Mei L, Feng SS. The chemotherapeutic potential of PEG-b-PLGA copolymer micelles that combine chloroquine as autophagy inhibitor and docetaxel as an anti-cancer drug. Biomaterials. 2014;35:9144–9154. doi: 10.1016/j.biomaterials.2014.07.028. [DOI] [PubMed] [Google Scholar]
- 20.Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, Mukherjee C, Shi Y, Gelinas C, Fan Y, Nelson DA, Jin S, White E. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell. 2006;10:51–64. doi: 10.1016/j.ccr.2006.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boya P, Gonzalez-Polo RA, Poncet D, Andreau K, Vieira HL, Roumier T, Perfettini JL, Kroemer G. Mitochondrial membrane permeabilization is a critical step of lysosome-initiated apoptosis induced by hydroxychloroquine. Oncogene. 2003;22:3927–3936. doi: 10.1038/sj.onc.1206622. [DOI] [PubMed] [Google Scholar]
- 22.Maycotte P, Aryal S, Cummings CT, Thorburn J, Morgan MJ, Thorburn A. Chloroquine sensitizes breast cancer cells to chemotherapy independent of autophagy. Autophagy. 2012;8:200–212. doi: 10.4161/auto.8.2.18554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eng CH, Wang Z, Tkach D, Toral-Barza L, Ugwonali S, Liu S, Fitzgerald SL, George E, Frias E, Cochran N, De Jesus R, McAllister G, Hoffman GR, Bray K, Lemon L, Lucas J, Fantin VR, Abraham RT, Murphy LO, Nyfeler B. Macroautophagy is dispensable for growth of KRAS mutant tumors and chloroquine efficacy. Proc Natl Acad Sci U S A. 2016;113:182–187. doi: 10.1073/pnas.1515617113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.King MA, Ganley IG, Flemington V. Inhibition of cholesterol metabolism underlies synergy between mTOR pathway inhibition and chloroquine in bladder cancer cells. Oncogene. 2016 doi: 10.1038/onc.2015.511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yu F, Xie Y, Wang Y, Peng Z-H, Li J, Oupický D. Chloroquine-Containing HPMA Copolymers as Polymeric Inhibitors of Cancer Cell Migration Mediated by the CXCR4/SDF-1 Chemokine Axis. ACS Macro Lett. 2016:342–345. doi: 10.1021/acsmacrolett.5b00857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kopeček J. Polymer–drug conjugates: Origins, progress to date and future directions. Adv Drug Delivery Rev. 2013;65:49–59. doi: 10.1016/j.addr.2012.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Duncan R. Polymer therapeutics: Top 10 selling pharmaceuticals - what next? J Controlled Rel. 2014;190:371–380. doi: 10.1016/j.jconrel.2014.05.001. [DOI] [PubMed] [Google Scholar]
- 28.White E. The role for autophagy in cancer. J Clin Invest. 2015;125:42–46. doi: 10.1172/JCI73941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kondo Y, Kanzawa T, Sawaya R, Kondo S. The role of autophagy in cancer development and response to therapy. Nat Rev Cancer. 2005;5:726–734. doi: 10.1038/nrc1692. [DOI] [PubMed] [Google Scholar]
- 30.Kimura T, Takabatake Y, Takahashi A, Isaka Y. Chloroquine in cancer therapy: a double-edged sword of autophagy. Cancer Res. 2013;73:3–7. doi: 10.1158/0008-5472.CAN-12-2464. [DOI] [PubMed] [Google Scholar]
- 31.Yang ZJ, Chee CE, Huang S, Sinicrope FA. The Role of Autophagy in Cancer: Therapeutic Implications. Mol Cancer Ther. 2011;10:1533–1541. doi: 10.1158/1535-7163.MCT-11-0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mizushima N, Yoshimori T, Levine B. Methods in Mammalian Autophagy Research. Cell. 2010;140:313–326. doi: 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Duncan R, Richardson SCW. Endocytosis and Intracellular Trafficking as Gateways for Nanomedicine Delivery: Opportunities and Challenges. Mol Pharm. 2012;9:2380–2402. doi: 10.1021/mp300293n. [DOI] [PubMed] [Google Scholar]
- 34.Varkouhi AK, Scholte M, Storm G, Haisma HJ. Endosomal escape pathways for delivery of biologicals. J Controlled Rel. 2011;151:220–228. doi: 10.1016/j.jconrel.2010.11.004. [DOI] [PubMed] [Google Scholar]
- 35.Sahay G, Alakhova DY, Kabanov AV. Endocytosis of Nanomedicines. J Controlled Rel. 2010;145:182–195. doi: 10.1016/j.jconrel.2010.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mezzaroba N, Zorzet S, Secco E, Biffi S, Tripodo C, Calvaruso M, Mendoza-Maldonado R, Capolla S, Granzotto M, Spretz R, Larsen G, Noriega S, Lucafo M, Mansilla E, Garrovo C, Marin GH, Baj G, Gattei V, Pozzato G, Nunez L, Macor P. New potential therapeutic approach for the treatment of B-Cell malignancies using chlorambucil/hydroxychloroquine-loaded anti-CD20 nanoparticles. PloS one. 2013;8:e74216. doi: 10.1371/journal.pone.0074216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Caron NJ, Quenneville SP, Tremblay JP. Endosome disruption enhances the functional nuclear delivery of Tat-fusion proteins. Biochem Biophys Res Commun. 2004;319:12–20. doi: 10.1016/j.bbrc.2004.04.180. [DOI] [PubMed] [Google Scholar]
- 38.Mellman I, Fuchs R, Helenius A. Acidification of the endocytic and exocytic pathways. Annu Rev Biochem. 1986;55:663–700. doi: 10.1146/annurev.bi.55.070186.003311. [DOI] [PubMed] [Google Scholar]
- 39.El-Sayed A, Futaki S, Harashima H. Delivery of macromolecules using arginine-rich cell-penetrating peptides: ways to overcome endosomal entrapment. AAPS J. 2009;11:13–22. doi: 10.1208/s12248-008-9071-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cheng J, Zeidan R, Mishra S, Liu A, Pun SH, Kulkarni RP, Jensen GS, Bellocq NC, Davis ME. Structure-function correlation of chloroquine and analogues as transgene expression enhancers in nonviral gene delivery. J Med Chem. 2006;49:6522–6531. doi: 10.1021/jm060736s. [DOI] [PubMed] [Google Scholar]
- 41.Kim TI, Rothmund T, Kissel T, Kim SW. Bioreducible polymers with cell penetrating and endosome buffering functionality for gene delivery systems. J Controlled Rel. 2011;152:110–119. doi: 10.1016/j.jconrel.2011.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hyndman L, Lemoine JL, Huang L, Porteous DJ, Boyd AC, Nan X. HIV-1 Tat protein transduction domain peptide facilitates gene transfer in combination with cationic liposomes. J Controlled Rel. 2004;99:435–444. doi: 10.1016/j.jconrel.2004.07.023. [DOI] [PubMed] [Google Scholar]
- 43.Abes S, Williams D, Prevot P, Thierry A, Gait MJ, Lebleu B. Endosome trapping limits the efficiency of splicing correction by PNA-oligolysine conjugates. J Controlled Rel. 2006;110:595–604. doi: 10.1016/j.jconrel.2005.10.026. [DOI] [PubMed] [Google Scholar]
- 44.Choi YH, Liu F, Kim J-S, Choi YK, Jong Sang P, Kim SW. Polyethylene glycol-grafted poly-l-lysine as polymeric gene carrier. J Controlled Rel. 1998;54:39–48. doi: 10.1016/s0168-3659(97)00174-0. [DOI] [PubMed] [Google Scholar]
- 45.Kadlecova Z, Rajendra Y, Matasci M, Baldi L, Hacker DL, Wurm FM, Klok HA. DNA delivery with hyperbranched polylysine: a comparative study with linear and dendritic polylysine. J Controlled Rel. 2013;169:276–288. doi: 10.1016/j.jconrel.2013.01.019. [DOI] [PubMed] [Google Scholar]
- 46.Katav T, Liu L, Traitel T, Goldbart R, Wolfson M, Kost J. Modified pectin-based carrier for gene delivery: cellular barriers in gene delivery course. J Controlled Rel. 2008;130:183–191. doi: 10.1016/j.jconrel.2008.06.002. [DOI] [PubMed] [Google Scholar]
- 47.Smith RM, Jarett L. Ultrastructural basis for chloroquine-induced increase in intracellular insulin in adipocytes: alteration of lysosomal function. Proc Natl Acad Sci U S A. 1982;79:7302–7306. doi: 10.1073/pnas.79.23.7302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhou Q, Yang X, Xiong M, Xu X, Zhen L, Chen W, Wang Y, Shen J, Zhao P, Liu QH. Chloroquine Increases Glucose Uptake via Enhancing GLUT4 Translocation and Fusion with the Plasma Membrane in L6 Cells. Cell Phys Biochem. 2016;38:2030–2040. doi: 10.1159/000445562. [DOI] [PubMed] [Google Scholar]
- 49.Lee CM, Tannock IF. Inhibition of endosomal sequestration of basic anticancer drugs: influence on cytotoxicity and tissue penetration. Brit J Cancer. 2006;94:863–869. doi: 10.1038/sj.bjc.6603010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Alsayed Y, Ngo H, Runnels J, Leleu X, Singha UK, Pitsillides CM, Spencer JA, Kimlinger T, Ghobrial JM, Jia X, Lu G, Timm M, Kumar A, Cote D, Veilleux I, Hedin KE, Roodman GD, Witzig TE, Kung AL, Hideshima T, Anderson KC, Lin CP, Ghobrial IM. Mechanisms of regulation of CXCR4/SDF-1 (CXCL12)-dependent migration and homing in multiple myeloma. Blood. 2007;109:2708–2717. doi: 10.1182/blood-2006-07-035857. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.