

Abstract
Background
DNA adenine methyltransferase (Dam) activity is absent in many, but not all, disease isolates of Neisseria meningitidis, as a consequence of the insertion of a restriction endonuclease-encoding gene, the 'dam replacing gene' (drg) at the dam locus. Here, we report the results of a survey to assess the prevalence of drg in a globally representative panel of disease-associated meningococci.
Results
Of the known meningococcal hyper-invasive lineages investigated, drg was absent in all representatives of the ST-8 and ST-11 clonal complexes tested, but uniformly present in the representatives of the other hyper-invasive lineages present in the isolate collection (the ST-1, ST-4, ST-5, ST-32 and ST-41/44 clonal complexes). The patterns of sequence diversity observed in drg were consistent with acquisition of this gene from a source organism with a different G+C content, at some time prior to the emergence of present-day meningococcal clonal complexes, followed by spread through the meningococcal population by horizontal genetic exchange. During this spread a number of alleles have arisen by mutation and intragenic recombination.
Conclusion
These findings are consistent with the idea that possession of the drg gene may contribute to the divergence observed among meningococcal clonal complexes, but does not have a direct mechanistic involvement in virulence.
Background
Neisseria meningitidis, the causative agent of meningococcal meningitis and septicaemia, is a common inhabitant of the human nasopharynx, being asymptomatically carried by approximately 10% of the population [1,2]. There is evidence for extensive horizontal genetic exchange in populations of this antigenically and genetically variable Gram-negative bacterium [3-7] but, despite the diversity of carried meningococci, only a limited number of genotypes – the hyper-invasive lineages – are responsible for most reported disease [8]. These lineages have been identified by the techniques of multilocus enzyme electrophoresis [9] and multilocus sequence typing (MLST) [10] as clonal complexes. In recent years, the ST-1 complex (formerly subgroup I), ST-4 complex (subgroup IV), and ST-5 complex (subgroup III) have dominated meningococcal disease in Africa and Asia, while members of the ST-11 (ET-37) complex, ST-8 complex (cluster A4), ST-41/44 complex (lineage 3), and ST-32 (ET-5) complex have caused most disease in other parts of the world. Meningococci occasionally cause epidemic outbreaks of varying scale up to global pandemics, although at a given point in time disease in a given geographical locale is often dominated by a limited number of clonal complexes [8].
DNA adenine methyltransferase (Dam) is an enzyme involved in the mismatch repair system of bacteria. During DNA replication, only the parental strand is fully methylated by the enzyme, since methylation is not immediate, allowing a mismatch on the newly synthesised strand to be excised and replaced [11]. Disruption of Dam activity suggests that mismatch repair will be less effective, potentially allowing the creation of frameshift mutations in the homopolymeric or simple repeat motifs within the promoter or coding regions of surface antigen genes leading to the reversible switching of their expression state. This is an important mechanism found at bacterial loci that encode gene products that are advantageous under certain conditions but not others; such genes have been termed contingency loci [12]. There is extensive evidence for such loci in the meningococcal genome [13]. In particular, the ability to vary the expression state of surface-exposed cellular components is important for within host adaptation, for example by allowing evasion of the immune response in the nasopharynx. These mechanisms are also important in the occasional transition from harmless colonisation to invasive disease [14-16]. Examples of such loci described in the meningococcus to date include: the capsular polysaccharide [17]; the Opc outer membrane protein [18-20]; pili [21]; the PorA protein [22]; and opacity proteins [19,23].
Dam methylation has been implicated in modifying the virulence of a number of bacterial pathogens [24-26], but its role in N. meningitidis has been contentious. Bucci et al., 1999 [27] suggested that the absence of Dam activity was responsible for high rates of phase variation in the siaD capsule gene, resulting in an increased virulence of some strains. In addition, the latter work suggested that all pathogenic isolates lacked Dam activity, the dam gene inactivated by the insertion of a putative restriction endonuclease, named 'dam replacing gene' (drg), with the genotypes dam+/drg- and dam-/drg+ being mutually exclusive. However, two subsequent studies [28,29] have found no effect. The drg gene encodes a restriction enzyme, NmeBII that is similar to the Streptococcus pneumoniae DpnI restriction endonuclease which cleaves at GmeATC, but not at GATC, sequences [30,31].
In the present study the prevalence of drg and its association with disease-associated isolates of N. meningitidis was examined by a survey of its distribution in a collection of isolates chosen to represent the known diversity of pathogenic meningococci [10]. The results suggest that, while drg is present in some lineages but not others, this gene has spread in the meningococcal population by horizontal genetic exchange, possibly after the introduction of the drg gene from an exogenous source.
Results
Distribution of dam and drg among invasive meningococci
In all cases, there was an exact correlation with the presence of dam and the absence of drg, and vice-versa. Of the 84 isolates tested, 23 were indicated to be dam+/drg- and 61 to be dam-/drg+ by restriction analysis and PCR amplification (see Additional file 1). The presence of dam or drg was associated with particular clonal complexes. Of the hyper-invasive lineages included in the analysis, all ST-8 complex and ST-11 complex isolates were dam+/drg-, while all of the representatives of the ST-1, ST-4, ST-5, ST-41/44, and ST-32 complexes were dam-/drg+.
Diversity of drg gene fragment sequences
The level of nucleotide diversity, as represented by number of alleles and polymorphic sites, along with the p-distances seen within drg gene fragments was similar to that seen with the housekeeping genes used for MLST (Table 1), with a total of 19 different fragment sequences (drg-1 – drg-19) present in this collection. The ratio of non-synonymous to synonymous substitutions (dN/dS, 0.11) was also similar to that seen in the MLST gene fragments. Three drg gene fragments (drg-13, drg-14 and drg-15) exhibited polymorphisms resulting in single base frameshifts, two resulting in stop codons (Figure 1) and one fragment (drg-16) had an insertion corresponding to a 24-base direct repeat. The G+C content of drg at 42.4% was lower than that of the housekeeping genes, which were in the range 51.1–57.4%.
Table 1.
Genetic variation in MLST and drg loci
| Locus | No. of alleles (no./100 isolates) | No. (%) of polymorphic sites | dN/dS | Average p-distance | %G+C |
| abcZ | 15 (14) | 75 (17.4) | 0.05 | 0.052 | 51.1 |
| adk | 10 (9.4) | 17 (3.7) | 0.02 | 0.012 | 52.3 |
| aroE | 18 (16.8) | 166 (34) | 0.293 | 0.106 | 55.6 |
| fumC | 19 (17.8) | 38 (8.2) | 0.024 | 0.022 | 57.4 |
| gdh | 16 (15) | 28 (5.6) | 0.05 | 0.019 | 52.3 |
| pdhC | 24 (22.4) | 80 (16.7) | 0.07 | 0.054 | 55.9 |
| pgm | 21 (19.6) | 77 (17) | 0.121 | 0.050 | 54.2 |
| drg | 19 (31.1*) | 48 (7.4) † | 0.11 | 0.021 | 42.4 |
*only 61 isolates contained drg
† excluding indels – length of fragment: 650
Figure 1.

Variable sites in drg gene fragments
Split decomposition analysis of the drg fragment sequences generated a star phylogeny with some net-like phylogenetic structure involving the drg-2 allele (Figure 2). The distribution of the different drg sequences amongst the clonal complexes is also shown in Figure 2.
Figure 2.
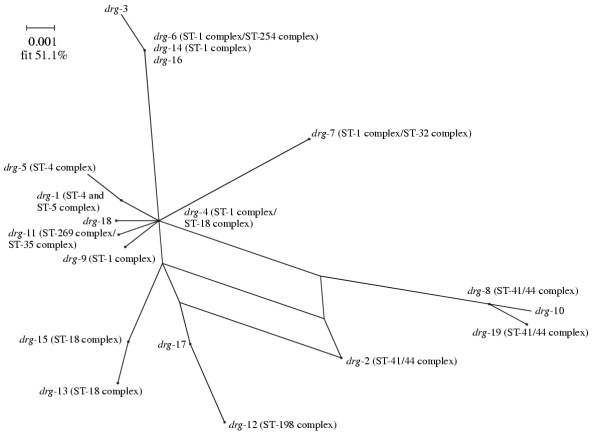
Splits graph of drg gene fragments. The drg alleles found in the major drg+ hyper-invasive lineages are annotated.
Discussion
The meningococcus is an example of an accidental pathogen, a commensal organism that rarely causes disease and which gains no evolutionary benefit from this process [32]. A complete understanding of the diseases caused by such organisms is dependent upon an appreciation of the mechanisms by which harmless carriage develops into invasive disease [33]. The observation that some meningococcal genotypes, the hyper-invasive lineages, are more likely to cause disease than others [8,10] suggests that comparative studies of disease-associated and carried meningococci may identify genetic factors responsible for disease [34]. In this context, the suggestion that the possession of a single genetic change, the insertion of the drg gene at the dam locus, was essential for virulence in meningococci [27] was attractive as it provided a single characteristic associated with the disease phenotype. Further, this proposal provided a plausible mechanistic explanation, namely the promotion of rapid switching of expression of contingency genes, for example those encoding the capsule which represents the best defined meningococcal virulence determinant [35]. This is consistent with the evidence that inactivation of Dam has been shown to affect rates of recombination and phase change in other species [25,36].
The data presented here, however, do not support the contention that the drg insertion is preferentially associated with disease-associated meningococcal isolates, even those expressing serogroup B capsular polysaccharide. There was no evidence of a difference between disease-associated and carried meningococcal isolates with 76% (45/59) of disease-associated isolates and 75% (11/15) of carrier isolates possessing the drg insertion. The isolate collection employed by the present investigation was chosen to be globally representative of meningococcal disease in the latter part of the 20th century, with multiple examples of each of the major hyper-invasive meningococci identified over this period. When analysed by clonal complex, which identifies hyper-invasive lineages, it was apparent that the drg insertion was absent from all of the isolates representing the ST-8 and ST-11 clonal complexes, both major hyper-invasive lineages but, conversely, drg was present in all representatives of the hyper invasive lineages represented by the ST-1, ST-4, ST-5, ST-41/44, and ST-32 complexes. This association was independent of the serogroup expressed by the isolates and provided a likely explanation of the earlier observations [27]. As meningococcal disease in a given locale at a given time tends to be dominated by a limited number of clonal complexes [8], it is likely from the results provided here that a given sample of disease-associated isolates collected from a given locale will be uniform at the dam locus. At a given point in time they may be dominated by drg containing meningococci, while at other times isolates without this insertion might dominate. For example, as both the ST-1 and ST 32 complexes contain the drg insertion, this insertion would be prevalent among the disease isolates recovered by the pandemic outbreaks caused by these clonal complexes [37,38]; conversely, the spread of ST-11 meningococci [39,40] would result in an increased prevalence of disease-associated meningococci without the drg insertion. This observation highlights the importance of assembling genetically defined isolate collections with a prescribed sampling frame if comparisons of meningococci with potential for virulence are to be undertaken.
It has been suggested that the drg gene has been introduced into Neisseria from an exogenous source [30]. The difference in the G+C% content of the drg gene compared to the meningococcal housekeeping genes was consistent with this idea; however, the levels of diversity among the drg alleles, which was similar to that observed in the meningococcal housekeeping genes, along with the reported occurrence of drg in other Neisseria species [30], suggested that this was a relatively old event predating the emergence of present-day clonal complexes. The ratio of non-synonymous to synonymous substitutions (dN/dS), which was also similar to that observed in meningococcal housekeeping genes, suggested that the drg gene had been subject to stabilising selection for conservation of function since its putative introduction into the meningococcal population, although the possession of frame shift mutations in some of the drg alleles demonstrated that, as with N. lactamica, not all meningococci possessing the drg insertion expressed the endonuclease [30]. In other respects the distribution of the gene among clonal complexes and the patterns of nucleotide sequence variation observed were similar to those seen in meningococcal housekeeping genes, suggesting that it is subject to similar selection pressures [41].
The presence of multiple methylation systems is a characteristic of the Neisseria and may be related to the transformable nature of these organisms [42]. Whole genome comparisons of meningococci belonging to different clonal complexes frequently yield different restriction modification systems as the principal detectable genetic differences [43,44]. It is attractive to speculate that the genetic isolation of clonal complexes may be promoted by such differences [45] and in this respect the insertion of drg in the dam locus, replacing a methylase with an endonuclease, may provide a potent barrier to genetic exchange from dam+ to drg+ meningococci; indeed, while there was evidence for the occasional horizontal genetic exchange of drg alleles among the genetically diverse meningococci that possess the insertion, there was no evidence for the transformational loss of drg from a given clonal complex among the isolates examined here. This concept of genetic isolation among lineages receives some support from the possession of porB2 alleles by members of the dam+ ST-8 and ST-11 complexes and porB3 alleles by the clonal complexes with the drg insertion [9,39], but the evidence for limitations in gene flow between these subgroups of meningococci is not at present conclusive.
Conclusions
While it is possible that the possession of the drg insertion may influence meningococcal population structure, the data presented here do not support a direct association of this genotype with meningococcal virulence.
Methods
Bacterial isolates
The 84 isolates used in this study (see Additional file 1) were a subset of a collection of 107 diverse meningococci assembled to develop and validate the MLST scheme for Neisseria meningitidis [10]. The collection included representatives of the major hyper-invasive lineages and has been characterised at many genetic loci (full details of this isolate collection are available at http://pubmlst.org/neisseria/).
Restriction digestion of chromosomal DNA
The methylation status of the chromosomal DNA of each isolate was determined by restriction digestion with the restriction endonucleases DpnI, DpnII, and Sau3AI, followed by separation of the digestion products by agarose gel electrophoresis. Each enzyme specifically recognises a cognate target site of GATC and cleaves this site depending on whether it is methylated or not: DpnI cleaves this target sequence only when N6-methyladenine is present within the recognition sequence; DpnII cleaves only unmethylated sites, and Sau3AI cleaves both methylated and unmethylated target sequences.
PCR amplification and nucleotide sequence determination
The presence of the dam gene was determined by polymerase chain reaction (PCR) performed using dam specific primers, DamF1 (5' – TAAAATGGGCAGGCGGCA – 3') and DamB2 (5' – CGTAAGGGGGATCGCAAT – 3'). These amplify a 534 bp fragment from the 5' end of the dam gene in dam+ strains but not from dam-strains. The presence of drg was determined by PCR using primers DrgF1 (5' – CATGAATTTATTTTTCGATA – 3') and DrgB2 (5' – AATTTGCAACTGTTGGCG – 3') that bind to drg internal sites and produce a 705 bp fragment in isolates containing the drg gene. PCR amplification was also performed using primer pairs Drg5F (5' – TGTCTAAAGAACTCAAAG – 3') / DrgB3 (5' – CGGTATCGAAAAATAAAT – 3') and Drg3F (5' – ATCCATCCAATTTCCCCA – 3') / DamB5 (5' – AAATGCCGTCTGAA – 3') based on dam and drg coding regions in order to confirm that dam inactivation was due to drg insertion.
Amplicons corresponding to the drg gene were purified by precipitation with poylethylene glycol and sodium chloride as described previously [46] and their nucleotide sequences determined on both strands by cycle sequencing. BigDye™ terminators (ABI, Foster city, California) and the same primers as used for amplification were employed in the extension reactions and the labelled reaction products separated on an ABI 3700 automated DNA sequencer. Each fragment was sequenced at least once on each strand and the sequences assembled with the STADEN suite of computer programs. Each unique drg sequence was assigned an arbitrary allele number in order of discovery.
Data analysis
Percentage G+C was calculated using the program START [47]. Split decomposition analysis of the drg gene fragments was performed using SPLITSTREE, version 3.1 [48]. The proportion of non-synonymous to synonymous substitutions was calculated by the method of Nei and Gojobori [49] using the program MEGA2 [50].
Authors' contributions
KAJ carried out the nucleotide sequencing and analysis of the data. LS performed the PCR screening of the isolates. ERM and MCJM conceived of the study and participated in its design and coordination.
Supplementary Material
Bacterial isolates used in the study
Acknowledgments
Acknowledegements
MCJM is a Wellcome Trust Senior Research Fellow. This work was supported by an award from the Wellcome Trust to MCJM (grant 055104).
Contributor Information
Keith A Jolley, Email: keith.jolley@medawar.ox.ac.uk.
Li Sun, Email: richard.moxon@paediatrics.ox.ac.uk.
E Richard Moxon, Email: richard.moxon@paediatrics.ox.ac.uk.
Martin CJ Maiden, Email: martin.maiden@zoo.ox.ac.uk.
References
- Broome CV. The carrier state: Neisseria meningitidis. J Antimicrob Chemother. 1986;18:25–34. doi: 10.1093/jac/18.supplement_a.25. [DOI] [PubMed] [Google Scholar]
- Cartwright K. Meningococcal carriage and disease. In: Cartwright K, editor. Meningococcal disease. Chichester: John Wiley & Sons Ltd; 1995. pp. 115–146. [Google Scholar]
- Bygraves JA, Urwin R, Fox AJ, Gray SJ, Russell JE, Feavers IM, Maiden MCJ. Population genetic and evolutionary approaches to the analysis of Neisseria meningitidis isolates belonging to the ET-5 complex. J Bacteriol. 1999;181:5551–5556. doi: 10.1128/jb.181.18.5551-5556.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feavers IM, Gray SJ, Urwin R, Russell JE, Bygraves JA, Kaczmarski EB, Maiden MCJ. Multilocus sequence typing and antigen gene sequencing in the investigation of a meningococcal disease outbreak. J Clin Microbiol. 1999;37:3883–3887. doi: 10.1128/jcm.37.12.3883-3887.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriz P, Giorgini D, Musilek M, Larribe M, Taha MK. Microevolution through DNA exchange among strains of Neisseria meningitidis isolated during an outbreak in the Czech Republic. Res Microbiol. 1999;150:273–80. doi: 10.1016/S0923-2508(99)80052-7. [DOI] [PubMed] [Google Scholar]
- Feil EJ, Enright MC, Spratt BG. Estimating the relative contributions of mutation and recombination to clonal diversification: a comparison between Neisseria meningitidis and Streptococcus pneumoniae. Res Microbiol. 2000;151:465–9. doi: 10.1016/S0923-2508(00)00168-6. [DOI] [PubMed] [Google Scholar]
- Feil EJ, Maiden MCJ, Achtman M, Spratt BG. The relative contributions of recombination and mutation to the divergence of clones of Neisseria meningitidis. Mol Biol Evol. 1999;16:1496–1502. doi: 10.1093/oxfordjournals.molbev.a026061. [DOI] [PubMed] [Google Scholar]
- Caugant DA. Population genetics and molecular epidemiology of Neisseria meningitidis. Apmis. 1998;106:505–25. [PubMed] [Google Scholar]
- Caugant DA, Mocca LF, Frasch CE, Frøholm LO, Zollinger WD, Selander RK. Genetic structure of Neisseria meningitidis populations in relation to serogroup, serotype, and outer membrane protein pattern. J Bacteriol. 1987;169:2781–2792. doi: 10.1128/jb.169.6.2781-2792.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiden MCJ, Bygraves JA, Feil E, Morelli G, Russell JE, Urwin R, Zhang Q, Zhou J, Zurth K, Caugant DA, et al. Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc Natl Acad Sci USA. 1998;95:3140–3145. doi: 10.1073/pnas.95.6.3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modrich P. Mechanisms and biological effects of mismatch repair. Annu Rev Genet. 1991;25:229–53. doi: 10.1146/annurev.ge.25.120191.001305. [DOI] [PubMed] [Google Scholar]
- Moxon ER, Rainey PB, Nowak MA, Lenski RE. Adaptive evolution of highly mutable loci in pathogenic bacteria. Curr Biol. 1994;4:24–32. doi: 10.1016/S0960-9822(00)00005-1. [DOI] [PubMed] [Google Scholar]
- Saunders NJ, Jeffries AC, Peden JF, Hood DW, Tettelin H, Rappuoli R, Moxon ER. Repeat associated phase variable genes in the complete genome sequence of Neisseria meningitidis MC58. Mol Microbiol. 2000;37:207–215. doi: 10.1046/j.1365-2958.2000.02000.x. [DOI] [PubMed] [Google Scholar]
- Hammerschmidt S, Birkholz C, Zahringer U, Robertson BD, van Putten J, Ebeling O, Frosch M. Contribution of genes from the capsule gene complex cps to lipooligosaccharide biosynthesis and serum resistance in Neisseria meningitidis. Mol Microbiol. 1994;11:885–96. doi: 10.1111/j.1365-2958.1994.tb00367.x. [DOI] [PubMed] [Google Scholar]
- Vogel U, Hammerschmidt S, Frosch M. Sialic acids of both the capsule and the sialylated lipooligosaccharide of Neisseria meningitis serogroup B are prerequisites for virulence of meningococci in the infant rat. Med Microbiol Immunol (Berl) 1996;185:81–7. doi: 10.1007/s004300050018. [DOI] [PubMed] [Google Scholar]
- Stephens DS, Spellman PA, Swartley JS. Effect of the (alpha 2–>8)-linked polysialic acid capsule on adherence of Neisseria meningitidis to human mucosal cells. J Infect Dis. 1993;167:475–479. doi: 10.1093/infdis/167.2.475. [DOI] [PubMed] [Google Scholar]
- Hammerschmidt S, Muller A, Sillmann H, Muhlenhoff M, Borrow R, Fox A, van Putten J, Zollinger WD, Gerardy-Schahn R, Frosch M. Capsule phase variation in Neisseria meningitidis serogroup B by slipped-strand mispairing in the polysialyltransferase gene siaD: correlation with bacterial invasion and the outbreak of meningococcal disease. Mol Microbiol. 1996;20:1211–1220. doi: 10.1111/j.1365-2958.1996.tb02641.x. [DOI] [PubMed] [Google Scholar]
- Virji M, Makepeace K, Ferguson DJP, Achtman M, Sarkari J, Moxon ER. Expression of the Opc protein correlates with invasion of epithelial and endothelial cells by Neisseria meningitidis. Mol Microbiol. 1992;6:2785–2795. doi: 10.1111/j.1365-2958.1992.tb01458.x. [DOI] [PubMed] [Google Scholar]
- Virji M, Makepeace K, Ferguson DJ, Achtman M, Moxon ER. Meningococcal Opa and Opc proteins: their role in colonization and invasion of humanepithelial and endothelial cells. Mol Microbiol. 1993;10:499–510. doi: 10.1111/j.1365-2958.1993.tb00922.x. [DOI] [PubMed] [Google Scholar]
- Sarkari J, Pandit N, Moxon ER, Achtman M. Variable expression of the Opc outer membrane protein in Neisseria meningitidis is caused by size variation of a promotor containing poly-cytidine. Mol Microbiol. 1994;13:207–217. doi: 10.1111/j.1365-2958.1994.tb00416.x. [DOI] [PubMed] [Google Scholar]
- Virji M, Alexandrescu C, Ferguson DJ, Saunders JR, Moxon ER. Variations in the expression of pili: the effect on adherence of Neisseria meningitidis tohuman epithelial and endothelial cells. Mol Microbiol. 1992;6:1271–1279. doi: 10.1111/j.1365-2958.1992.tb00848.x. [DOI] [PubMed] [Google Scholar]
- van der Ende A, Hopman CTP, Dankert J. Multiple mechanisms of phase variation of PorA in Neisseria meningitidis. Infect Immun. 2000;68:6685–6690. doi: 10.1128/IAI.68.12.6685-6690.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehio C, Gray-Owen SD, Meyer TF. The role of neisserial Opa proteins in interactions with host cells. Trends Microbiol. 1998;6:489–494. doi: 10.1016/S0966-842X(98)01365-1. [DOI] [PubMed] [Google Scholar]
- Garcia-Del Portillo F, Pucciarelli MG, Casadesus J. DNA adenine methylase mutants of Salmonella typhimurium show defects in protein secretion, cell invasion, and M cell cytotoxicity. Proc Natl Acad Sci USA. 1999;96:11578–83. doi: 10.1073/pnas.96.20.11578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson ME, Jr, Jarisch J, Smith AL. Inactivation of deoxyadenosine methyltransferase (dam) attenuates Haemophilus influenzae virulence. Mol Microbiol. 2004;53:651–64. doi: 10.1111/j.1365-2958.2004.04140.x. [DOI] [PubMed] [Google Scholar]
- Low DA, Weyand NJ, Mahan MJ. Roles of DNA adenine methylation in regulating bacterial gene expression and virulence. Infect Immun. 2001;69:7197–204. doi: 10.1128/IAI.69.12.7197-7204.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucci C, Lavitola A, Salvatore P, Del Giudice L, Massardo DR, Bruni CB, Alifano P. Hypermutation in pathogenic bacteria: frequent phase variation in meningococci is a phenotypic trait of a specialized mutator biotype. Mol Cell. 1999;3:435–45. doi: 10.1016/S1097-2765(00)80471-2. [DOI] [PubMed] [Google Scholar]
- Richardson AR, Stojiljkovic I. Mismatch repair and the regulation of phase variation in Neisseria meningitidis. Mol Microbiol. 2001;40:645–55. doi: 10.1046/j.1365-2958.2001.02408.x. [DOI] [PubMed] [Google Scholar]
- Martin P, Sun L, Hood DW, Moxon ER. Involvement of genes of genome maintenance in the regulation of phase variation frequencies in Neisseria meningitidis. Microbiology. 2004. [DOI] [PubMed]
- Cantalupo G, Bucci C, Salvatore P, Pagliarulo C, Roberti V, Lavitola A, Bruni CB, Alifano P. Evolution and function of the neisserial dam-replacing gene. FEBS Lett. 2001;495:178–83. doi: 10.1016/S0014-5793(01)02388-2. [DOI] [PubMed] [Google Scholar]
- Lacks S, Greenberg B. A deoxyribonuclease of Diplococcus pneumoniae specific for methylated DNA. J Biol Chem. 1975;250:4060–66. [PubMed] [Google Scholar]
- Levin BR. The evolution and maintenance of virulence in microparasites. Emerg Infect Dis. 1996;2:93–102. doi: 10.3201/eid0202.960203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiden MCJ. High-throughput sequencing in the population analysis of bacterial pathogens. Int J Med Microbiol. 2000;290:183–190. doi: 10.1016/S1438-4221(00)80089-2. [DOI] [PubMed] [Google Scholar]
- Tinsley CR, Nassif X. Analysis of the genetic differences between Neisseria meningitidis and Neisseria gonorrhoeae: two closely related bacteria expressing two different pathogenicities. Proc Natl Acad Sci U S A. 1996;93:11109–14. doi: 10.1073/pnas.93.20.11109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peltola H. Meningococcal disease: still with us. Rev Infect Dis. 1983;5:71–91. doi: 10.1093/clinids/5.1.71. [DOI] [PubMed] [Google Scholar]
- Waldron DE, Owen P, Dorman CJ. Competitive interaction of the OxyR DNA-binding protein and the Dam methylase at the antigen 43 gene regulatory region in Escherichia coli. Mol Microbiol. 2002;44:509–20. doi: 10.1046/j.1365-2958.2002.02905.x. [DOI] [PubMed] [Google Scholar]
- Caugant DA, Frøholm LO, Bovre K, Holten E, Frasch CE, Mocca LF, Zollinger WD, Selander RK. Intercontinental spread of Neisseria meningitidis clones of the ET-5 complex. Antonie Leeuwenhoek J Microbiol. 1987;53:389–394. doi: 10.1007/BF00415492. [DOI] [PubMed] [Google Scholar]
- Wang J-F, Caugant DA, Li X, Hu X, Poolman JT, Crowe BA, Achtman M. Clonal and antigenic analysis of serogroup A Neisseria meningitidis with particular reference to epidemiological features of epidemic meningitis in China. Infect Immun. 1992;60:5267–5282. doi: 10.1128/iai.60.12.5267-5282.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J-F, Caugant DA, Morelli G, Koumaré B, Achtman M. Antigenic and epidemiological properties of the ET-37 complex of Neisseria meningitidis. J Infect Dis. 1993;167:1320–1329. doi: 10.1093/infdis/167.6.1320. [DOI] [PubMed] [Google Scholar]
- Jelfs J, Munro R, Ashto FE, Caugant DA. Genetic characterization of a new variant within the ET-37 complex of Neisseria meningitidis associated with outbreaks in various parts of the world. Epidemiol Infect. 2000;125:285–98. doi: 10.1017/S0950268899004471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes EC, Urwin R, Maiden MCJ. The influence of recombination on the population structure and evolution of the human pathogen Neisseria meningitidis. Mol Biol Evol. 1999;16:741–749. doi: 10.1093/oxfordjournals.molbev.a026159. [DOI] [PubMed] [Google Scholar]
- Ritchot N, Roy PH. DNA methylation in Neisseria gonorrhoeae and other Neisseriae. Gene. 1990;86:103–6. doi: 10.1016/0378-1119(90)90120-G. [DOI] [PubMed] [Google Scholar]
- Claus H, Friedrich A, Frosch M, Vogel U. Differential distribution of novel restriction-modification systems in clonal lineages of Neisseria meningitidis. J Bacteriol. 2000;182:1296–303. doi: 10.1128/JB.182.5.1296-1303.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bart A, Dankert J, van der Ende A. Representational difference analysis of Neisseria meningitidis identifies sequences that are specific for the hyper-virulent lineage III clone. FEMS Microbiol Lett. 2000;188:111–4. doi: 10.1016/S0378-1097(00)00221-4. [DOI] [PubMed] [Google Scholar]
- Claus H, Stoevesandt J, Frosch M, Vogel U. Genetic isolation of meningococci of the electrophoretic type 37 complex. J Bacteriol. 2001;183:2570–2575. doi: 10.1128/JB.183.8.2570-2575.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Embley TM. The linear PCR reaction: a simple and robust method for sequencing amplified rRNA genes. Lett Appl Microbiol. 1991;13:171–174. doi: 10.1111/j.1472-765x.1991.tb00600.x. [DOI] [PubMed] [Google Scholar]
- Jolley KA, Feil EJ, Chan MS, Maiden MC. Sequence type analysis and recombinational tests (START) Bioinformatics. 2001;17:1230–1. doi: 10.1093/bioinformatics/17.12.1230. [DOI] [PubMed] [Google Scholar]
- Huson DH. SplitsTree: analyzing and visualizing evolutionary data. Bioinformatics. 1998;14:68–73. doi: 10.1093/bioinformatics/14.1.68. [DOI] [PubMed] [Google Scholar]
- Nei M, Gojobori T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol. 1986;3:418–26. doi: 10.1093/oxfordjournals.molbev.a040410. [DOI] [PubMed] [Google Scholar]
- Kumar S, Tamura K, Jakobsen IB, Nei M. MEGA2: molecular evolutionary genetics analysis software. Bioinformatics. 2001;17:1244–5. doi: 10.1093/bioinformatics/17.12.1244. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Bacterial isolates used in the study