

Abstract
A high-resolution structure of the histidine-containing phosphocarrier protein (HPr) from Staphylococcus aureus was obtained by heteronuclear multidimensional nuclear magnetic resonance (NMR) spectroscopy on the basis of 1,766 structural restraints. Twenty-three hydrogen bonds in HPr could be directly detected by polarization transfer from the amide nitrogen to the carbonyl carbon involved in the hydrogen bond. Differential line broadening was used to characterize the interaction of HPr with the HPr kinase/phosphorylase (HPrK/P) of Staphylococcus xylosus, which is responsible for phosphorylation-dephosphorylation of the hydroxyl group of the regulatory serine residue at position 46. The dissociation constant Kd was determined to be 0.10 ± 0.02 mM at 303 K from the NMR data, assuming independent binding. The data are consistent with a stoichiometry of 1 HPr molecule per HPrK/P monomer in solution. Using transversal relaxation optimized spectroscopy-heteronuclear single quantum correlation, we mapped the interaction site of the two proteins in the 330-kDa complex. As expected, it covers the region around Ser46 and the small helix b following this residue. In addition, HPrK/P also binds to the second phosphorylation site of HPr at position 15. This interaction may be essential for the recognition of the phosphorylation state of His15 and the phosphorylation-dependent regulation of the kinase/phosphorylase activity. In accordance with this observation, the recently published X-ray structure of the HPr/HPrK core protein complex from Lactobacillus casei shows interactions with the two phosphorylation sites. However, the NMR data also suggest differences for the full-length protein from S. xylosus: there are no indications for an interaction with the residues preceding the regulatory Ser46 residue (Thr41 to Lys45) in the protein of S. xylosus. In contrast, it seems to interact with the C-terminal helix of HPr in solution, an interaction which is not observed for the complex of HPr with the core of HPrK/P of L. casei in crystals.
The histidine-containing phosphocarrier protein (HPr) plays a central role in the uptake of carbohydrates by the phosphoenolpyruvate-dependent phosphotransferase system (PTS) and in the regulation of carbohydrate metabolism in bacteria (for a review, see reference 54). In the transport system, it is part of a phosphate shuttle, which transfers a phosphate group from phosphoenolpyruvate to the carbohydrate transported through the cell membrane. As a second function, HPr is involved in gene regulation of the PTS carbon catabolite repression system. In that system, it acts as an activator of gene repression. Both of these mechanistically very different processes are controlled by HPr through phosphorylation-dephosphorylation reactions. In the phosphate shuttle, the amino acid that participates in phosphorylation-dephosphorylation reactions in HPr is a histidine residue at position 15. It is phosphorylated by enzyme I (EI) at Nδ1 and transfers this group to Nɛ2 of a histidine residue of the enzyme IIA domain of the enzyme II (EII) complex. In most gram-positive and some pathogenic gram-negative bacteria, the second phosphorylation site in HPr is Ser46, which can be phosphorylated by the ATP-dependent HPr kinase/phosphorylase (HPrK/P), the product of the hprK gene (7, 8, 13, 34, 50, 48). Phosphoserine-HPr functions in a regulatory fashion, down regulating catabolic activity by its interaction with catabolite control protein A (CcpA) (25, 52). Simultaneously, the phosphorylation of Ser46 inhibits phosphocarrier activity by perturbing the interaction with phosphorylated EI (3, 48). Furthermore, in some bacteria P-Ser-HPr seems to be involved in additional regulatory mechanisms, called inducer expulsion and inducer exclusion (9, 54, 61, 62).
HPr proteins from different microorganisms have been structurally characterized by X-ray crystallography and nuclear magnetic resonance (NMR) spectroscopy (10, 21, 23, 24, 29, 32, 41, 46, 53, 60). Although they differ largely in primary structure, their general folding structure is well conserved. It consists of a four-stranded antiparallel β-pleated sheet and three α-helices arranged in a βαββαβα open β-sandwich topology.
The X-ray structure of the catalytic domain of HPrK/P (amino acids 128 to 319) from Lactobacillus casei (11) and the structures of the full-length HPrK proteins from Staphylococcus xylosus (40) and Mycoplasma pneumoniae (1) have been solved, and data for the complex of HPrK from L. casei with its substrate HPr from Bacillus subtilis are available (12). The catalytic mechanism of the bifunctional protein kinase and its precise interaction with its substrate protein were explained on the basis of the complex data (37, 42).
With the capability to interact with numerous other proteins, HPr is an ideal system for the study of protein-protein interactions. These complexes are far larger than the 40-kDa size which up to now was considered the limit for studies using NMR. With the transversal relaxation optimized spectroscopy (TROSY) technique first described by Pervushin et al. (44) making possible the investigation of proteins and complexes with molecular masses of far more than 40 kDa, the interaction of HPr with HPrK/P, with the focus on HPr, is a system that is now suited for NMR investigation.
For HPr from Staphylococcus aureus, only a low-resolution NMR structure, which was solved exclusively with homonuclear methods under a low magnetic field, has been published (29). In this paper, we present a high-resolution structure of HPr from S. aureus as the basis for a study of its interaction with HPrK/P from S. xylosus. HPr from S. aureus is closely related to HPr from S. xylosus (with five amino acid differences) and is thus a suitable binding partner for HPrK/P from S. xylosus.
MATERIALS AND METHODS
Protein expression and purification.
Unlabeled wild-type HPr from S. aureus (molecular mass, 9.496 kDa) was expressed and purified as described previously (27). For the preparation of uniformly 13C- and 15N-enriched HPr, the plasmid pT7-5 ptsH, coding for HPr from S. aureus, was transformed into Escherichia coli BL21(DE3), which was initially grown in TBY medium (1% casein hydrolysate, 0.5% yeast extract, 0.5% NaCl). For protein expression, cells were inoculated into 200 ml of M9 minimal medium containing uniformly 13C-enriched (99%) glucose-1-hydrate (25 g/liter), MgSO4 · 7H2O (0.26 g/liter), CaCl2 (0.02 g/liter), Na2HPO4 · 2H2O (7.2 g/liter), KH2PO4 (3.1 g/liter), NaCl (0.52 g/liter), 15NH4Cl (99% isotope enrichment, 1.25 g/liter), thiamine (10 g/liter), and 50 mg of ampicillin/liter and were grown overnight at 310 K. This culture was diluted 1:10 in 2 liters of minimal medium and grown until the cells reached an A578 of 0.8. Protein expression was induced by the addition of 0.3 mM isopropyl-β-d-thiogalactopyranoside (IPTG) to the medium. The cells were harvested by centrifugation after 3 h. From 2 liters of culture, 3.5 g of cells (wet weight) could be obtained. The enriched protein was purified by the same procedure as that used for unlabeled HPr. The yield of isotope-enriched HPr was 12 mg/g of cells.
HPrK/P from S. xylosus was prepared as described previously (40). The molecular mass of a monomer of the most probably hexameric protein is 35,324 Da. It was shown earlier that freeze-drying does not influence the activity of HPrK/P, and therefore freeze-dried protein was used for NMR spectroscopy (as for X-ray crystallography).
Sample preparation.
For homonuclear measurements, 3 mM unlabeled HPr protein was dissolved in 500 μl of 99.75% D2O or 90% H2O-10% D2O at pH 7.0. For heteronuclear experiments, 3 mM uniformly 13C- and 15N-labeled HPr protein was dissolved in 500 μl of 99.75% D2O or 90% H2O-10% D2O. The pH was adjusted to 7.0 by the addition of appropriate quantities of 0.4% perdeuterated KOH to the sample. For the study of the interaction of HPr with HPrK/P, 0.5 mM uniformly 13C- and 15N-enriched HPr from S. aureus was dissolved in 500 μl of 99.75% D2O or 90% H2O-10% D2O. Unlabeled freeze-dried HPrK/P from S. xylosus was added in well-defined quantities to the sample. 4,4-Dimethyl-4-silapentane-sulfonic acid (DSS) (0.1 mM) was added as an internal reference.
NMR spectroscopy.
1H NMR spectra were recorded on Bruker DMX-600 and -800 spectrometers operating at proton resonance frequencies of 600 and 800 MHz, respectively. All two-dimensional (2D) homonuclear spectra were collected in the phase-sensitive mode by use of the time proportional phase incrementation method (39), with 8,192 data points in the direct dimension and 512 data points in the indirect dimension. Nuclear Overhauser enhancement spectroscopy (NOESY) spectra (22) were recorded with a mixing time of 100 ms, and total correlation spectroscopy (TOCSY) spectra (4) were recorded with a spin-lock time of 60 ms, using the DIPSI-2 (49) sequence for isotropic mixing. All 3D spectra were acquired with 1,024 or 2,048 data points in the direct proton dimension; 128 data points in the 13C dimension, using constant time evolution and States-time proportional phase incrementation acquisition; and 64 data points in the 15N dimension, using echo-antiecho type selection (51). Forward linear prediction in the indirect dimensions resulted in a spectral resolution of 5 Hz/data point for 1H, 23 Hz/data point for 13C, and 36 Hz/data point for 15N. The data were referenced indirectly by using the 1H chemical shift of the methyl group in DSS and multiplying this value by 0.25144953 for 13C and 0.101329118 for 15N (59). All data were recorded at 303 K.
Assignment strategy.
The complete assignment of the backbone of HPr from S. aureus was accomplished by using the standard 3D NMR experiments HNCA (17), HN(CO)CA (16), CBCANH (17), CBCA(CO)NH (18), and HNCO (16) on 13C- and 15N-labeled protein. The side chain atoms were assigned by using HCCH-TOCSY (31) and 2D 13C-heteronuclear single quantum correlation (HSQC) (55) experiments. Aromatic chemical shifts were assigned by using data from 1H-,13C-HSQC and HCAN (57) experiments. An exhaustive analysis led to the identification of the chemical shifts of all but approximately 5% of the NMR active nuclei present in HPr.
Structure calculations.
Utilizing resonance assignments, we extracted distance information from both 2D NOESY and 13C- and 15N-edited NOESY spectra (30). NOESY cross peaks were integrated by using the integration routine of the AURELIA software (43) and were calibrated to the geminal Hα-Hα cross peaks of glycine residues set to 0.179 nm in the homonuclear 2D NOESY spectrum and the 3D 13C-edited NOESY spectrum. Calibration of the 15N-separated NOESY spectra was accomplished by calibration of the HN-Hα cross peaks of atom pairs whose mutual distances were previously calculated from the 2D NOESY data. The calculated distances were corrected by factors implemented in DYANA software to take into account pseudo-atom effects arising from nonstereospecific assignments (19). All non-pseudo-atom constraints were converted into upper and lower bounds by assuming an error of 30%. 3JHNHα coupling constants were determined from cross-peak-to-diagonal-peak ratios in the HNHA (35) spectrum and were corrected for relaxation time effects (56). The generation of additional φ and ψ restraints from Cα chemical shifts was done as implemented by DYANA (38). Hydrogen bonds were identified by a 2D version of HNCO as proposed by Cordier and Grzesiek (5) and were transformed to additional distance restraints. The obtained distance and angle restraints were used as input for the high-temperature angular simulated annealing and dynamics protocol as implemented in DYANA. The protocol consists of the generation of 440 random torsion angle structures by use of a random number seed. Each starting structure was subjected to restrained simulated annealing, with a total of 4,000 steps. Of these, 800 were high-temperature steps followed by 3,200 steps with slow cooling. The temperature is defined in DYANA as the units of the target function per degree of freedom. Finally, 1,000 steps of conjugate gradient minimization were performed. An ensemble of 16 structures was selected with regard to minimal penalty functions, correct backbone dihedral angles in the Ramachandran plot, and low NOE violations. The quality of the structures was checked with the program PROCHECK (36). The NMR structures were deposited in the protein database under the accession number 1KA5.
Random coil chemical shifts.
Random coil chemical shifts for the side chain nitrogens of Arg were determined in a solution of 3 mg of the tetrapeptide Ac-Gly-Gly-Arg-Ala dissolved in 500 μl of 95% H2O-5%D2O at 283 K and pH 7.0. The nitrogen Nη1,2 resonances could only be detected after lowering the pH to 3.9 but are most probably also true for pH 7.0 since the corresponding proton shifts were not influenced by the pH shift from 7.0 to 3.9. 15N chemical shifts were obtained by natural abundance HSQC spectroscopy without isotope enrichment of the peptide. The arginine chemical shifts were 7.25 and 84.61 ppm for Hɛ and Nɛ, respectively. The Hη1,2 and Nη1,2 resonances were found at 6.64, 70.23, 6.92, and 71.93 ppm.
NMR binding studies.
Differential line-broadening experiments were performed by the titration of 13C- and 15N-labeled HPr from S. aureus with HPrK/P from S. xylosus and by the recording for each titration step of a 1D homonuclear spectrum, a 2D 1H-,15N-HSQC TROSY spectrum, and a 2D version of the 3D HNCO-TROSY experiment. For these experiments, suitable quantities of freeze-dried unlabeled HPrK/P were added to the sample, thus leading to only a small dilution of the sample. In addition, an HPrK/P sample from S. xylosus in D2O-50 mM Tris-HCl, pH 7.5, was titrated with unlabeled HPr. 1D 1H spectra were recorded at 303 K at HPr-to-HPrK/P ratios of 0, 0.25, 0.5, 0.75, 1, 1.25, 1.5, 1.75, 2, 2.5, 3, 3.5, and 4. After normalization of the intensities of the spectra to the intensity of the Tris peak, the intensity of the single HPr methyl peak of Leu81 at −0.18 ppm was used to determine the concentration of free HPr. The absolute signal intensity was calibrated with well-resolved methyl peaks of HPrK/P with known concentrations in the initial spectrum in the absence of HPr.
The peak volume changes observed in the HSQC spectra when HPrK/P was added to HPr were fitted, assuming independent binding to N independent binding sites of molecule B (HPrK/P), with an individual dissociation constant, Kd. If PA and PB are the probabilities that molecules A and B are not complexed and PAB is the probability that molecule A (HPr) is bound to molecule B, then PA is given by the equation
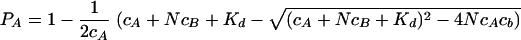 |
(1) |
with cA and cB representing the total concentrations of A and B in the solution. Different limiting cases must be considered concerning the polarization transfer time as well as the evolution and detection time. The exchange correlation time τe may be much smaller or much larger than time 2τ in the n polarization transfer periods, which means that signal losses due to T2 relaxation may be either averaged or not averaged during this period. With T2A and T2AB representing the transversal relaxation time for molecule A in the free state and the bound state, respectively, and with J representing the relevant coupling constant for insensitive nuclei enhanced by polarization transfer (INEPT), the magnetization value MA is given in the first order by the equation
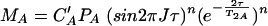 |
(2) |
If T2A = aT2AB, MAB is given by the equation
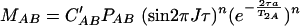 |
(3) |
C′A and C′AB are constants which are not necessarily equal and correct for factors such as saturation and hydrogen exchange. When no averaging occurs during the mixing time (τe ≫ τ) and detection time (τe ≫ 1/ΔωA,AB) with the frequency separation of the resonances in the free and bound states, the cross-peak volume VA of A is given by the equation
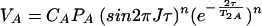 |
(4) |
When averaging occurs during time τ but not during the evolution time, t1, and the acquisition time, t2 (τe ≪ τ and τe ≫ 1/ΔωA,AB), VA is given by the equation
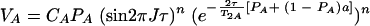 |
(5) |
When averaging occurs during all periods (τe ≪ τ and τe ≪ 1/ΔωA,AB), the observed cross-peak volume V is given by the equation
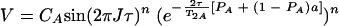 |
(6) |
The situation becomes more complicated when intermediate exchange correlation times cause additional line broadening, which further reduces the INEPT efficiency. In a first approximation, the concentration-dependent effective transverse relaxation time would be a factor in equations 4 to 6.
Equations 4 to 6 can be simplified somewhat if the peak volumes are normalized to the volume V0 in the absence of the ligand (PA = 1). One then obtains the following equation for τe ≫ τ and τe ≫ 1/ΔωA,AB:
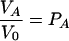 |
(7) |
For τe ≪ τ and τe ≫ 1/ΔωA,AB, the equation is as follows:
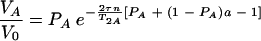 |
(8) |
For τe ≪ τ and τe ≪ 1/ΔωA,AB, the equation is as follows:
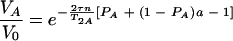 |
(9) |
RESULTS
Resonance assignments and high-resolution structure of HPr from S. aureus.
With the strategy described in Materials and Methods, all backbone nitrogens except the nitrogen of the N-terminal amino group and the imino group of Pro18, all Cα resonances, and all carbonyl carbons except that of Arg17 and the C-terminal amino acid could be sequentially assigned. For the backbone proton resonances, a similar result was obtained: only the N-terminal amino protons could not be observed since they exchange too quickly with the bulk water. The Hα resonances were completely assigned. From the side chain carbon resonances, 92% were assigned only a few resonances, as the Cγ resonances of aromatic residues could not be detected by the experiments used for this study. The resonances of the nonexchangeable protons of the side chains were all observable. Interestingly, the hydroxyl proton of Ser31 was directly observable in TOCSY spectra at 5.60 ppm. Arg17 is a conserved amino acid in the active center of HPr. The Nɛ and Nη1,2 resonances could also be detected, at 84.77 and 71.92 ppm, respectively. These resonances can be critical indicators of protein-protein interactions involving the active center of HPr.
The analysis of the Hα, Cα, Cβ, and C′ chemical shifts led to a reliable prediction of the secondary structure elements (Fig. 1). It predicted β-strands from amino acids 2 to 9 (strand A), 33 to 37 (strand B), 40 to 43 (strand C), and 60 to 66 (strand D). Analogously, α-helices were predicted from amino acids 16 to 27 (helix a), 48 to 51 (helix b), and 70 to 83 (helix c) from the secondary structure-dependent chemical shifts.
FIG. 1.

Secondary structure elements predicted from chemical shifts by the program CSI-PLOT (58) on the basis of Hα, Cα, Cβ, and C′ chemical shifts.
The 3D structure of HPr protein was determined by a simulated annealing approach in torsional angle space (19) as described in Materials and Methods. In total, 1,562 NOEs, 78 J couplings, 23 hydrogen bond distance restraints derived from an H(N)CO experiment, and 178 φ,ψ restraints obtained from TALOS (6) were used for the structure calculation. The calculated structures were well-defined, with an overall root mean square deviation (RMSD) of 0.016 nm for the backbone atoms and 0.054 nm for all heavy atoms (Table 1). The experimental restraints were well fulfilled and the DYANA target functions were in the expected range. Figure 2a shows the superposition of the backbone of the 16 lowest energy structures. The obtained structures are obviously rather well-defined. The 3D arrangement of the secondary structure elements in HPr from S. aureus is depicted in Fig. 2c together with the hydrogen bonds determined directly by 2D HNCO. HPr from S. aureus consists of a four-stranded antiparallel β-pleated sheet (strands A, B, C, and D) and three helices located on one side of the sheet (helices a, b, and c). This arrangement has been found in all HPr structures solved so far. The lengths of the canonical secondary elements in HPr vary somewhat with the microorganism and are also dependent on the actual method used for their determination. Identification of the secondary structure elements in HPr from S. aureus was achieved by using an algorithm described by Kabsch and Sander (26) as implemented in the program MOLMOL (33). Applied to the 16 lowest energy structures, it recognized essentially the same secondary structure elements predicted from the chemical shift analysis, but the lengths and positions differed sometimes: helix a is two residues longer and helix c is one residue longer at its C terminus than predicted and β-strand A is two residues shorter at its C terminus than predicted from chemical shifts.
TABLE 1.
Structural statistics
| Constraint | Value or no. of occurrences |
|---|---|
| NOEs | 1,562 |
| Intraresidual (i, i) | 673 |
| Sequential (i, i+1) | 255 |
| Intermediate range | |
| (i, i+2) | 68 |
| (i, i+3) | 116 |
| (i, i+4) | 41 |
| Long range (i,j with j > i+4) | 409 |
| Dihedral angle restraints from J couplings | |
| ϕ angles from 3JHN-Hα | 78 |
| H bonds from H(N)CO main chain | 23 |
| ϕ/ψ restraints from Cα chemical shifts | 103 |
| Structural statistics for the 16 lowest energy structures (from 440 calculated structures) | |
| NOE violations of >0.05 nm | 43 |
| Maximum NOE violation (nm) | 0.062 |
| Violation of dihedral angle restraints of >2.5° | 0 |
| Maximum violation of dihedral angle restraints (°) | 1.9 |
| Minimal target function (structure 1) | 24.4 |
| Maximal target function (structure 16) | 26.5 |
| RMSDs relative to the mean structure | |
| Amino acids 1 to 88 (backbone heavy atomsa) (nm) | 0.0156 ± 0.008 |
| Amino acids 1 to 88 (all nonhydrogen atoms) (nm) | 0.0535 ± 0.013 |
NH, Cα, and C′ atoms of the backbone.
FIG. 2.

3D structure of HPr from S. aureus. (a) Superposition of the 16 lowest energy structures. The active center histidine at position 15 is represented in red, the regulatory serine at position 46 is shown in yellow, and helix a is shown in orange. (b) Representation of structural precision as a tube, where higher precision corresponds to a smaller diameter of the tube. (c) 3D structure of HPr and the network of directly detected hydrogen bonds. The hydrogen bonds detected by H(N)CO are indicated by red bars. (d) NOEs defining the position of His15. (e) Space-filling model of the environment of His15.
The backbone φ and ψ angles of the lowest energy structures obtained are all located in the energetically allowed region of the Ramachandran plot, as expected for well-resolved structures. Seventy-seven percent of all angles are located in the most favored range and only one residue is located in the least favored but allowed region of the plot. Deviations from ideal Ramachandran plot geometry in the active site around residue His15 have been observed before for HPr from E. coli (53) and might arise from conformational averaging on the microsecond-millisecond timescale.
Usually, the hydroxyl protons of serine or threonine residues are exchanged too quickly with the solvent to be observable in COSY-like spectra. As described earlier (28), for the HPr protein of S. aureus there is an interesting exception: the cross peaks between the Hγ proton of the hydroxyl group and the Hα and Hβ protons of Ser31 can be observed directly in the TOCSY spectra at 303 K. This is a typical spectroscopic feature of HPr proteins which has been described for HPr from Staphylococcus carnosus (14) as well as HPr from E. coli (20), which both contain a conserved serine residue at this position. The hydroxyl proton resonances of Ser31 could be observed at 5.58 ppm in HPr from S. carnosus and at 5.77 ppm in HPr from E. coli, very close to the resonance position of 5.56 ppm obtained for HPr from S. aureus. In the NOESY spectra, strong contacts between the Hγ of Ser31 and the amide resonances of Asp69 (0.23 nm) and Glu70 (0.27 nm) were observed. Especially strong NOEs to the backbone amide protons of Asp69 and Glu70 were also observed earlier for HPr from S. carnosus, although for this study no unique, strong hydrogen bond to these amides was obtained. However, for the new structures of HPr from S. aureus, the program MOLMOL detected a clear hydrogen bond to the amide of Asp69. Structurally, this hydrogen bond fixes the position of L2 to the loop L6 and the N terminus of helix c.
The most important part of the structure is the active center around His15 which is transiently phosphorylated during phosphotransfer between EI and EIIA. The imidazole ring caps the N terminus of helix a (Fig. 2). Figures 2d and e show the position of the imidazole ring together with the NOEs defining this position. The phosphoryl group acceptor Nδ1 of His15 is directed to the solvent and is freely accessible for water (or the phosphohistidyl residue of EI during the phosphotransfer reaction). All exchangeable protons of the conserved Arg17 in the active center could be observed in the 1H,15N HSQC spectra of isotope-enriched protein. Whereas the observation of the Hɛ resonances of arginine residues is usually possible in proteins, the Hη resonances are usually not observable at a high temperature (303 K). We found cross peaks for the ɛ-NH group at 7.68 and 84.77 ppm. The proton chemical shift value differs significantly from the random coil value of 7.25 ppm; however, the nitrogen shift is close to the random coil value of 84.61 ppm. For the protons and nitrogens of the guanidino group, cross peaks at 6.89 and 71.92 ppm were observed, which were again close to the random coil values of 6.92 and 71.93 ppm found in random coil peptides. However, the second set of Hη1,2 and Nη1,2 resonances found in random coil peptides at 6.46 and 70.23 ppm could not be identified in HPr. The nitrogen resonances may be degenerated by a fast motional averaging which is supported by the chemical shifts not far away from the random coil values (2; also data from this study). The side chain protons Hβ and Hγ of Arg17 show NOE contacts to the Hd of Pro18. Therefore, its position is not completely undefined, although the missing NOEs to the Hδ and Hɛ would allow some side chain mobility, which is in line with the exposure of the positively charged side chain to the solvent.
Characterization of the complex of HPr with HPrK/P.
Differential line-broadening experiments were performed by the titration of 13C- and 15N-labeled HPr from S. aureus with HPrK/P from S. xylosus and by the recording for each titration step of a 1D homonuclear spectrum, a 2D 1H,15N-HSQC TROSY spectrum, and a 2D version of the 3D HNCO-TROSY experiment. For these experiments, suitable quantities of freeze-dried unlabeled HPrK/P were added to the sample, thus leading to only a small dilution of the sample. Possible pH shifts were excluded by the direct measurement of the pH after the last titration step and an analysis of resonances which are known to be pH sensitive in the pH range.
A selected region of the 2D HNCO-TROSY spectra of HPr in the absence and presence of HPrK/P is shown in Fig. 3. The HNCO-TROSY signals at a ratio of 0.5 mM HPr to 0.25 mM monomers of HPr kinase were much reduced, with the magnitude of this reduction of intensity varying from cross peak to cross peak. At higher HPrK/P concentrations, almost all signals disappeared. No significant chemical shift changes or new cross peaks were observed after the addition of HPrK/P. The same was true for the 1D 1H NMR spectrum and the 2D 1H,15N-HSQC TROSY spectrum recorded under identical conditions. However, the high sensitivity of the 1H,15N-HSQC TROSY spectrum still allows the detection of several cross peaks at saturating concentrations of HPrK/P.
FIG. 3.

Binding of HPrK/P to HPr detected in a 2D TROSY-H(N)CO experiment. An overlay plot of 2D TROSY-H(N)CO spectra of HPr in the presence or absence of HPr-K/P recorded at an 800-MHz proton frequency at 303 K is shown. Only a part of the spectrum is shown. 1H and 13C′ cross peaks of free HPr and those of HPr in the presence of HPrK/P are shown. The residues that are strongly broadened in the presence of HPr-K/P are denoted by arrows. The sample initially contained 320 μl of 0.5 mM uniformly 13C- and 15N-enriched HPr from S. aureus in 90% H2O-10% D2O, pH 7.0 (gray cross peaks). Appropriate amounts of freeze-dried HPrK/P were added to obtain a solution containing 0.25 mM (monomer concentration) HPrK/P (black cross peaks).
Using the previously assigned chemical shift data for free HPr, we could identify the lines from HPr in the presence of HPrK/P. In Fig. 4, the volume changes of the HPr cross peaks at half saturation with HPrK/P are depicted as a function of the HPr sequence. A few cross peaks in the 15N TROSY spectra show a significantly higher reduction in intensity (larger than the mean value <ΔVi> plus the standard deviation s). The corresponding cross peaks are summarized in Table 2. As expected, most of them are located close to Ser46 in the regulatory phosphorylation site. However, some of the signals are sequentially and structurally close to the second phosphorylation site at His15. A similar picture was obtained from the shifts in the H(N)CO spectrum (Table 2), in which some of the signals were also significantly influenced by the presence of HPrK/P.
FIG. 4.

Volume changes of amide and carbonyl cross peaks after addition of HPrK/P. The same set of samples was used as that described for Fig. 3. The graphs show the relative volume decrease ΔVi {ΔVi = [Vi(HPrK/P = 0)] − [Vi (HPrK/P = 0.25 mM)]/Vi(HPrK/P = 0)} of the 1H,15N-HSQC TROSY (A) and 2D TROSY-H(N)CO (B) cross peaks at half saturation of HPr with HPrK/P. Data were recorded at an 800-MHz proton frequency and 303 K. The mean values <ΔVi > (broken lines) and the range defined by the mean value ± the standard deviation s are depicted as black boxes.
TABLE 2.
Intermolecular contacts in the HPr-HPrK/P complexa
| HPr residueb | Interaction visible in X-ray structure
|
Interaction visible by NMRe
|
||||
|---|---|---|---|---|---|---|
| Atomc | HPrK/P residued | Atomc | HNCO | 15N HSQC | 1H NMR | |
| Thr12(Ser) | O | Ile301 | sc | × | × | |
| Gly13 | Hα | Ile301 | sc | × | × | |
| Ile14 | × | × | ||||
| His15 | Hα | Leu297 | sc | × | × | |
| Hα | Ile301 | sc | ||||
| sc | Leu297 | sc | ||||
| sc | Ile301 | sc | ||||
| Ala16 | HN | Leu297 | sc | × | × | |
| sc | Asn294(Glu) | sc | ||||
| Arg17 | × | × | ||||
| Ala19 | × | |||||
| Gln24 | × | |||||
| Tyr37 | sc | Asn308 | sc | × | ||
| Lys40 | sc | Thr132(Arg) | Hα | |||
| sc | Thr132(Arg) | C′ | ||||
| sc | Thr132(Arg) | O | ||||
| sc | Thr132(Arg) | sc | ||||
| sc | Ser134 | sc | ||||
| sc | Asn176(Arg) | sc | ||||
| Lys41(Thr) | O | Ser134 | sc | |||
| Val42 | sc | Asn176(Arg) | sc | |||
| Asn43 | sc | His136 | sc | |||
| Lys45 | C′ | Ser153 | sc | |||
| O | Ser153 | sc | ||||
| sc | Ser153 | sc | ||||
| Ser46 | sc | His136 | sc | × | ||
| sc | Asp175 | sc | ||||
| Ile47 | sc | Glu200 | sc | × | ||
| Met48 | HN | Asp175 | sc | × | ||
| O | Ile195(Leu) | sc | ||||
| sc | Asp175 | sc | ||||
| sc | Ile195(Leu) | sc | ||||
| sc | Glu200 | sc | ||||
| Gly49 | HN | Asp175 | sc | |||
| Hα | Asn176(Arg) | sc | ||||
| Val50 | × | |||||
| Met51 | O | Leu297 | sc | × | × | × |
| sc | Phe293 | sc | ||||
| sc | Leu297 | sc | ||||
| Ser52 | Cα | Leu194(Ile) | sc | × | ||
| Hα | Leu194(Ile) | sc | ||||
| O | Asn176(Arg) | sc | ||||
| O | Leu194(Ile) | sc | ||||
| O | Glu300(Leu) | sc | ||||
| sc | Asn176(Arg) | sc | ||||
| sc | Leu194(Ile) | sc | ||||
| sc | Ile195(Leu) | sc | ||||
| Leu53 | Hα | Asn176(Arg) | sc | × | ||
| Gly54 | Cα | Ile301 | sc | × | ||
| Hα | Leu297 | sc | ||||
| Hα | Glu300(Leu) | sc | ||||
| Hα | Ile301 | sc | ||||
| Hα | Asn304 | sc | ||||
| C′ | Ile301 | sc | ||||
| C′ | Asn304 | sc | ||||
| O | Ile301 | Hα | ||||
| O | Ile301 | sc | ||||
| O | Arg303(His) | sc | ||||
| O | Asn304 | sc | ||||
| Val55(Ile) | N | Ile301 | sc | × | ||
| C′ | Ile301 | sc | ||||
| O | Ile301 | sc | ||||
| Gly56(Ala) | Cα | Ile301 | sc | × | ||
| Hα1 | Asn304 | sc | ||||
| Hα1 | Ile301 | sc | ||||
| sc | Leu297 | Hα | ||||
| sc | Ile301 | O | ||||
| sc | Ile305 | sc | ||||
| sc | Arg303(His) | HN | ||||
| sc | Arg303(His) | sc | ||||
| sc | Asn304 | sc | ||||
| sc | Gly305(Glu) | sc | ||||
| Lys57 | O | Gly305(Glu) | sc | |||
| sc | Gly305(Glu) | sc | ||||
| Ser78(Glu) | × | |||||
| Leu81(Met) | × | |||||
The X-ray structure of the complex of unphosphorylated HPr from B. subtilis with HPrK/P (1KKL.pdb) from L: casei (12) was used for the analysis. Hydrogen atoms were added with the program Molmol and intermolecular contacts were identified. Contacts were defined as having an interatomic distance of <0.29 nm.
Residue types and numbering are for HPr from S. aureus. The corresponding residues in HPr from B. subtilis are given in parentheses, and residues that are strictly conserved in all HPr proteins are given in bold. Compared to HPr from S. aureus, HPr from S. xylosus has five mutated residues, namely Asn4-Lys, Ser71-Thr, Gln75-Glu, Ser78-Thr, and Val80-Ile.
sc, side chain atoms.
Residue types and numbering are for HPrK/P from S. xylosus. Note that the alignment of the two sequences leads to a shift of four residues. For residues that are not conserved, the corresponding amino acid HPrK/P from L. casei is given in parentheses.
×, an interaction was visible.
For a more quantitative evaluation of the data, the dissociation constant Kd of HPr from HPrK/P and the number of binding sites N of HPrK/P were determined, assuming as a first approximation an independent binding of HPr to the kinase (see Materials and Methods). The interpretation of homonuclear 1D spectra is much more direct than that of 2D HSQC spectra, for which possible variations in the polarization transfer must be taken into account. As an example, the resonances of the methionine methyl groups are shown in Fig. 5 at different HPrK/P concentrations. Generally, no line shifts were observed, but two different cases could be observed; either the line width did not change significantly but the intensity decreased with the addition of HPrK/P or the line was broadened extremely with increasing HPrK/P concentrations. The first case would be typical for slow exchange conditions for interacting groups, and the second case would be typical for noninteracting residues. Only residues of the first group are useful for the calculation of Kd and N since they allow measurements of the free concentration of HPr in the presence of HPrK/P. A series of NMR spectra with unlabeled proteins with well-defined concentrations were created, the well-resolved 1D signal from free HPr (the methyl group of Leu81) was selected (Fig. 6), and its intensity changes upon the addition of HPrK/P were fitted to equation 1. From the NMR data, the dissociation constant Kd and the number of binding sites N per HPrK/P monomer were determined to be 0.10 ± 0.02 mM and 1.02 ± 0.05, respectively, at 303 K.
FIG. 5.

Spectral changes observed in 1D NMR spectra of HPr induced by the interaction with HPrK/P. A small part of an 800-MHz 1H NMR spectrum of HPr from S. aureus in the presence and absence of HPrK/P is shown. Note that HPr is 13C and 5N enriched. The doublet resonances of the methyl protons of the four methionine residues of HPr are labeled with 1, 2, and 3, with 1 corresponding to Met21, 2 corresponding to Met1 and Met51, and 3 corresponding to Met48. Identical experimental conditions and samples were used as those described for Fig. 3. The molar ratios of monomers of HPrK/P to HPr were 0, 0.5, 1.0, and 2.0. The quantity of HPr in the active volume of the probe head was held constant.
FIG. 6.

Binding constant of HPr to HPrK/P and fit of volume changes induced by HPr-HPrK/P interaction with different models. (Top) HprK/P (0.2 mM) from S. xylosus in D2O in 50 mM Tris-HCl, pH 7.5, was titrated with a 4 mM HPr S. aureus solution in the same buffer. 1D 1H spectra were recorded at 303 K at different HPr-to-HPrK/P ratios. The concentration of free HPr was determined from the intensity of the Hδ resonance of Leu81. The concentration c(HPrfree) of free HPr was fitted as function of the total concentration c(HPrtotal) of HPr (and corrected for changes of the HPrK/P concentration) with equation 1, calculated by using c(HPrfree) = c(HPrtotal) × PA. A dissociation constant Kd and a number N of binding sites per HPrK/P monomer were obtained as 0.10 ± 0.02 mM and 1.02 ± 0.05. (Bottom) The same set of samples was used as that described for Fig. 3. The volume dependence of the cross peaks of a few selected residues in the 1H,15N-HSQC TROSY spectra on the HPrK/P concentration was calculated with an N of 1.02 and a Kd of 0.10 mM with either equation 7 (broken line), 8 (solid bold line), or 9 (solid thin line). The data for Leu53 (+), Gly54 (×), and Val55 (▵) which are most probably involved in the protein-protein interaction are shown. Note that the signal of Val55 was too weak to be observed at higher concentrations.
In the 1D spectra, the ring resonances of His15 and Tyr37, one of the Hδ resonances of Leu81, and the Hɛ resonance of Met48 show a behavior that is typical for slow exchange, and they are thus candidates for protein-protein interactions. The resonances of Met1 and Met51 are superposed, but at least one of the lines again shows a dependence on the HPrK/P concentration, which is typical for slow exchange. From the 3D structure, Met51 is a reasonable candidate for interaction with HPrK/P.
With the above parameters, the concentration dependence of the cross-peak volumes in the 1H,15N-HSQC spectra can be predicted on the basis of equations 7 to 9, describing different interaction models. The factor a, defining the ratio of transverse relaxation times in free and bound HPr, was fixed to 28.3, that is, to a value calculated from the molecular masses of free HPr and the hexameric HPrK/P decorated with six HPr molecules. It is clear that this is only an approximation since a must be concentration dependent because a distribution of HPr-HPrK/P complexes with different numbers of HPrs (and hence different masses) must exist in solution. In addition, exchange broadening is not taken into account. Simulations show that the value of a has a negligible influence on the fit of the data for a rather wide range of values.
The volume changes of the peaks in the 1H,15N-HSQC TROSY spectra were fitted as a function of the HPrK/P concentration with the three different models (equations 7 to 9). Residues which would show a large chemical shift change induced by the binding of HPrK/P (i.e., described by slow exchange conditions) should be best fitted by equation 7 or 8, and residues not involved in the protein-protein interaction should be best fitted by equation 9. It turned out that equation 7 was in no case the optimal solution, which means it cannot describe the system sufficiently well. This is reasonable since it would require a very small off-rate of HPr, which is not consistent with the rather weak binding of the protein. As an example, Fig. 6 shows the calculations using the three different equations for three residues assumed to be involved in protein-protein interaction. Clearly, equation 7 gives a wrong prediction and equation 8 gives a somewhat better fit of the data than equation 9, although the differences are not very large. Therefore, the complete information from all spectra recorded [1H,15N-HSQC TROSY, 2D TROSY-H(N)CO, and 1D spectra] must be used.
DISCUSSION
Assignments and properties of the refined structure.
The application of heteronuclear experiments to 15N- and 13C-labeled HPr protein allowed an almost complete assignment with the high reliability of heteronuclear methods. Compared to the structure of HPr from S. aureus determined earlier solely by homonuclear methods at 500 MHz (28), the sequential assignment of some resonances had to be corrected (see the BioMagRes database). However, these corrections do not influence the general fold of the molecule. Compared to the already published structure, a much larger number of restraints could be obtained by including the data for the direct identification of hydrogen bonds via the weak J coupling through the hydrogen bonds. The low-resolution structure had been calculated from 6 NOEs per residue; with 17.8 NOEs per residue, a substantially higher level of precision for the structure could be obtained, as the RMSD value of the main chain dropped from 0.087 to 0.016 nm. The R-factor (R-factor R5 according to Gronwald et al. [15]) calculated for an 800-MHz spectrum of HPr in H2O is substantially smaller than that obtained for the low-resolution structure. The regions with still larger structural variabilities encompass the regulatory helix (helix b), the active center loop, and the C terminus. Since a similar picture was also obtained for the low-resolution structures published earlier and also for HPr from other microorganisms, this seems to be a typical feature of HPrs and probably describes the lack of a unique, rigid structure in these regions. This is also reasonable since it comprises the sites of the proteins that are most probably involved in protein-protein interactions.
The function of the strictly conserved residue Arg17 of HPr is largely discussed in the literature, and its position relative to the active center histidine (His15) varies from structure to structure. In contrast to the case for the structure of HPr from S. aureus determined earlier, Arg17 does not seem to be in close contact with the histidine ring system. This could be due to differences in pH since the structure presented here was determined at pH 7.0, whereas the older structure (28) was solved at a pH of 7.8, at which the histidine ring is completely uncharged. At pH 7.0, the interaction between the partial charge and the positive charge of the arginine side chain will probably disfavor any close contact between the two side chains. This is also in line with the observation that the resonances of the guanidino groups are averaged here but were clearly not equivalent at pH 7.8. In general, the position of the side chain of Arg17 may be strongly dependent on the functional state of HPr; in particular, the phosphorylation of the histidine ring and the interaction with other proteins during the phosphotransfer may require different conformations. Analogous to the arginine finger of the Ras-RasGAP system, it may facilitate the transfer of the phosphoryl group bound to the active center histidine of EI to HPr.
Interaction of HPr with HPrK/P.
The crystal structures of the isolated catalytic domain of HPrK/P from L. casei (11) and of full-length HPrK/P from S. xylosus (40) and M. pneumoniae (1) have been reported previously. A crystal structure of the complex of HPr from B. subtilis with the nucleotide binding domain (amino acids 128 to 319) of HPrK/P from L. casei was published recently by Fieulaine et al. (12). The HPr protein from B. subtilis has only 64% identity with HPr from L. casei, which means that some of the features observed in the crystal structures of HPrK/P from L. casei and HPr from B. subtilis could be due to the use of a heterologous system. The system used in the solution NMR studies is much more closely related since the HPr proteins from S. xylosus and S. aureus used here exhibit a sequence identity of 94%.
In the crystals the nucleotide binding domain of HPrK/P from L. casei forms a hexamer with six HPr proteins bound to two adjacent subunits of the kinase. The structure shows that HPr mainly interacts via helix b (with Ser46 located in the loop L4 at its N terminus) and with the preceding β-strand C. A second interaction site of HPr with a different subunit of HPrK/P involves helix b and the N terminus of helix a, which is capped by the active center histidine His15. The residues with atoms whose interatomic distances are smaller than 0.29 nm are listed in Table 2. For a better comparison with our data, the numbering and residue types for our complex are given (HPr from S. aureus and HPrK/P from S. xylosus). The sequences of HPr from B. subtilis and HPrK/P from L. casei were replaced with the corresponding residues in HPr from S. aureus and HPrK/P from S. xylosus. The residues assumed to interact with HPrK/P according to our NMR data are depicted in Fig. 7.
FIG. 7.

Interaction site of HPr with HPrK/P. (Left) Topological model of HPr. (Right) Same view as in the left panel of a surface model of HPr in which all residues which show signs of being involved in the interaction with HPrK/P are depicted in red. Residues were assumed to be potential interaction partners when one of the markers summarized in Table 2 was applied.
Since the kinase phosphorylates HPr at position 46, which is located at the loop preceding helix b, an interaction with HPrK/P is required. In the X-ray structure of the HPr-HPrK/P complex, all residues in the region between Lys40 and Lys57 of HPr are in contact with residues of HPrK/P, with the only exception being Val50. In the NMR data, the first part of the putative interaction site of HPr (Lys40 to Lys45) shows no signs of a contact with HPrK/P. The subsequent region (Ser46 to Lys57) is clearly involved in the interaction; the only difference is that Gly49 and Lys57 do not show a significant response upon HPrK/P binding. However, in contrast to the case for the X-ray structure, Val50 shows a response upon HPrK/P binding (Table 2). As shown in Table 2, most of the HN and CO chemical shift changes are not caused by direct contacts but are transmitted via changes of the side chain environment. Therefore, it is not surprising that small differences between the X-ray and NMR data exist. However, the interaction with β-strand C and loop L4 (Fig. 2c) seems not to exist in solution.
The interaction with the region around the active center His15 encompasses in the X-ray structure the residues of loop L1 (Ser12 to Ala16); the same residues are identified in HPr from S. aureus. In addition, Arg17 and two residues of the N-terminal part of helix a (Ala19 and Gln24) are influenced by the binding of HPrK/P from S. xylosus. Note that in the X-ray structure of the complex between phosphorylated HPr and HPrK/P, additional contacts were observed for Thr20 and Gln24 of HPr.
The ring of Tyr37 is located between β-strand B and helices b and c and is part of the interaction surface in the X-ray structure, and a probable interaction is also predicted in solution (Table 2). However, the chemical shift changes of Ser78 and Leu81 located in the C-terminal part of helix c were not expected from the X-ray data. Ser78 is replaced by a threonine residue in HPr from S. xylosus, whereas Leu81 is conserved in the two HPr proteins. Although the observed chemical shift changes could be due to changes induced indirectly by conformational changes after binding, they could also represent additional interaction sites of HPrK/P, maybe involving the N-terminal domain of the molecule, which is not present in the published X-ray structure.
Conclusions.
The NMR data for HPr in the presence of HPrK/P from S. xylosus indicate the formation of a complex with a stoichiometry of one HPr molecule per monomer of HPrK/P. The 1H NMR data can be interpreted as indicating a polymer in solution whose size would be in agreement with the hexameric structure observed for the crystals of HPrK/P from L. casei. The data show that the second binding site of HPr for HPrK/P is not a crystal artifact but also exists in solution. In principle, the kinase and phosphorylase activities of HPrK/P should depend on the metabolic situation in the cell, which is also reflected in the phosphorylation state of His15. An interaction with this region can be used for the recognition of the phosphorylation state of the active center of HPr and thus can regulate the kinase and phosphorylase activities. The data also explain the fact that HPr lacks the capability to be phosphorylated at the His15 residue when in complex with HPrK/P and while phosphorylated at the regulatory Ser46 residue (47). A possible additional interacting site could be the C-terminal part of helix c in solution, maybe involving the N-terminal domain of HPrK/P.
Acknowledgments
We thank J. Scheiber for modeling of the HPr-HPrK/P complex, T. Graf for providing random coil shifts, and C. Cabrele for synthesizing the corresponding model peptide.
This work was supported by the EU (SPINE QL62-CT-2002-00988) and the Deutsche Forschungsgemeinschaft (SFB 521).
REFERENCES
- 1.Allen, G. S., K. Steinhauer, W. Hillen, J. Stülke, and R. G. Brennan. 2003. Crystal structure of HPr kinase/phosphatase from Mycoplasma pneumoniae. J. Mol. Biol. 326:1203-1217. [DOI] [PubMed] [Google Scholar]
- 2.Arnold, M. R., W. Kremer, H.-D. Luedemann, and H. R. Kalbitzer. 2002. 1H NMR parameters of common amino acid residues measured in aqueous solutions of the linear tetrapeptides Gly-Gly-X-Ala at pressures between 0.1 and 200 MPa. Biophys. Chem. 96:129-140. [DOI] [PubMed] [Google Scholar]
- 3.Audette, G., R. Engelmann, W. Hengstenberg, J. Deutscher, K. Hayakawa, J. W. Quail, and L. Delbaere. 2000. The 1.9 Å resolution structure of phospho-serine 46 HPr from Enterococcus faecalis. J. Mol. Biol. 303:545-553. [DOI] [PubMed] [Google Scholar]
- 4.Braunschweiler, L., and R. R. Ernst. 1983. Coherence transfer by isotropic mixing: application to proton correlation spectroscopy. J. Magn. Reson. 61:306-320. [Google Scholar]
- 5.Cordier, F., and S. Grzesiek. 1999. Direct observation of hydrogen bonds in proteins by scalar couplings. J. Am. Chem. Soc. 121:1601-1602. [DOI] [PubMed] [Google Scholar]
- 6.Cornilescu, G., F. Delaglio, and A. Bax. 1999. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J. Biomol. NMR 13:289-302. [DOI] [PubMed] [Google Scholar]
- 7.Deutscher, J., and M. H. Saier. 1983. ATP-dependent protein kinase-catalyzed phosphorylation of a seryl residue in HPr, a phosphate carrier protein of the phosphotransferase system in Streptococcus pyogenes. Proc. Natl. Acad. Sci. USA 80:6790-6794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deutscher, J., B. Pevec, K. Beyreuther, H. H. Kiltz, and W. Hengstenberg. 1986. Streptococcal phosphoenolpyruvate-sugar phosphotransferase system: amino acid sequence and site of ATP-dependent phosphorylation of HPr. Biochemistry 25:6543-6551. [DOI] [PubMed] [Google Scholar]
- 9.Dossonnet, V., V. Monedero, M. Zagorec, A. Galinier, G. Pérez-Martinez, and J. Deutscher. 2000. J. Bacteriol. 182:2582-2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.El-Kabbani, O. A. L., E. B. Waygood, and L. T. J. Delbaere. 1987. Tertiary structure of histidine-containing protein of the phosphoenolpyruvate:sugar phosphotransferase system of Escherichia coli. J. Biol. Chem. 262:12926-12929. [PubMed] [Google Scholar]
- 11.Fieulaine, S., S. Morera, S. Poncet, V. Monedero, V. Gueguen-Chaignon, A. Galinier, J. Janin, J. Deutscher, and S. Nessler. 2001. X-ray structure of HPr kinase: a bacterial protein kinase with a P-loop nucleotide-binding domain. EMBO J. 20:3917-3927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fieulaine, S., S. Morera, S. Poncet, L. Mijakovic, A. Galinier, J. Janin, J. Deutscher, and S. Nessler. 2002. X-ray structure of a bifunctional protein kinase in complex with its protein substrate HPr. Proc. Natl. Acad. Sci. USA 99:13437-13441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Galinier, A., M. Kravanja, R. Engelmann, W. Hengstenberg, M.-C. Kilhoffer, J. Deutscher, and J. Haiech. 1998. New protein kinase and protein phosphatase families mediate signal transduction in bacterial catabolite repression. Proc. Natl. Acad. Sci. USA 95:1823-1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Görler, A., W. Hengstenberg, M. Kravanja, W. Beneicke, T. Maurer, and H. R. Kalbitzer. 1999. Solution structure of histidine containing phosphocarrier protein (HPr) from Staphylococcus carnosus. Appl. Magn. Reson. 17:465-480. [Google Scholar]
- 15.Gronwald, W., R. Kirchhöfer, A. Görler, W. Kremer, B. Ganslmeier, K.-P. Neidig, and H. R. Kalbitzer. 2000. RFAC, a program for automated NMR-R-factor estimation. J. Biomol. NMR 17:137-151. [DOI] [PubMed] [Google Scholar]
- 16.Grzesiek, S., and A. Bax. 1992. Improved 3D triple-resonance NMR techniques applied to a 31-kDa protein. J. Magn. Reson. 96:432-440. [Google Scholar]
- 17.Grzesiek, S., and A. Bax. 1992. An efficient experiment for sequential backbone assignment of medium-sized isotopically enriched proteins. J. Magn. Reson. 99:201-207. [Google Scholar]
- 18.Grzesiek, S., and A. Bax. 1993. Amino acid type determination in the sequential assignment procedure of uniformly 13C/15N enriched proteins. J. Biomol. NMR 3:185-204. [DOI] [PubMed] [Google Scholar]
- 19.Güntert, P., C. Mumenthaler, and K. Wüthrich. 1997. Torsion angle dynamics for NMR structure calculation with the new program DYANA. J. Mol. Biol. 273:283-298. [DOI] [PubMed] [Google Scholar]
- 20.Hammen, P. K., E. B. Waygood, and R. E. Klevit. 1991. Reexamination of the secondary and tertiary structure of histidine-containing protein from Escherichia coli by homonuclear and heteronuclear NMR-spectroscopy. Biochemistry 30:11842-11850. [DOI] [PubMed] [Google Scholar]
- 21.Herzberg, O., P. Reddy, S. Sutrina, M. H. Saier, J. Reizer, and G. Kapadia. 1992. Structure of the histidine-containing phosphocarrier protein HPr from Bacillus subtilis at 2.0 Å resolution. Proc. Natl. Acad. Sci. USA 89:2499-2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jeener, J., B. H. Meier, P. Bachmann, and R. R. Ernst. 1979. Investigation of exchange processes by two-dimensional NMR spectroscopy. J. Chem. Phys. 71:4546-4553. [Google Scholar]
- 23.Jia, Z., J. W. Quail, E. B. Waygood, and L. T. J. Delbaere. 1993. The 2.0-Å resolution structure of Escherichia coli histidine-containing phosphocarrier protein HPr. A redetermination. J. Biol. Chem. 268:22490-22501. [DOI] [PubMed] [Google Scholar]
- 24.Jia, Z., M. Vandonselaar, W. Hengstenberg, J. W. Quail, and L. T. J. Delbaere. 1994. The 1.6 Å structure of the histidine-containing phosphocarrier protein HPr from Streptococcus faecalis. J. Mol. Biol. 236:1341-1355. [DOI] [PubMed] [Google Scholar]
- 25.Jones, B. E., V. Dossonnet, E. Kuester, W. Hillen, J. Deutscher, N. Schnell, and R. E. Klevit. 1997. Binding of the catabolite repressor protein CcpA to its DNA target is regulated by phosphorylation of its corepressor HPr. J. Biol. Chem. 272:26530-26535. [DOI] [PubMed] [Google Scholar]
- 26.Kabsch, W., and C. Sander. 1983. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22:2577-2637. [DOI] [PubMed] [Google Scholar]
- 27.Kalbitzer, H. R., W. Hengstenberg, P. Rösch, P. Muss, P. Bernsmann, R. Engelmann, M. Dörschug, and J. Deutscher. 1982. HPr proteins of different microorganisms studied by hydrogen-1 high resolution nuclear magnetic resonance: similarities of structures and mechanisms. Biochemistry 21:2879-2885. [DOI] [PubMed] [Google Scholar]
- 28.Kalbitzer, H. R., K.-P. Neidig, and W. Hengstenberg. 1991. Two-dimensional 1H-NMR studies on HPr protein from Staphylococcus aureus: complete sequential assignments and secondary structure. Biochemistry 30:11186-11192. [DOI] [PubMed] [Google Scholar]
- 29.Kalbitzer, H. R., and W. Hengstenberg. 1993. The solution structure of the histidine-containing protein (HPr) from Staphylococcus aureus as determined by two-dimensional 1H-NMR spectroscopy. Eur. J. Biochem. 216:205-214. [DOI] [PubMed] [Google Scholar]
- 30.Kay, L. E., P. Keifer, and T. Saarinen. 1992. Pure absorption gradient enhanced heteronuclear single quantum correlation spectroscopy with improved sensitivity. J. Am. Chem. Soc. 114:10663-10665. [Google Scholar]
- 31.Kay, L. E., G. Y. Xu, A. U. Singer, D. R. Muhandiram, and J. D. Forman-Kay. 1993. A gradient-enhanced Hcch Tocsy experiment for recording side-chain H-1 and C-13 correlations in H2O samples of proteins. J. Magn. Reson. B. 101:333-337. [Google Scholar]
- 32.Klevit, R. E., and E. B. Waygood. 1986. Two-dimensional 1H NMR studies of histidine-containing protein from Escherichia coli. Secondary and tertiary structure as determined by NMR. Biochemistry 25:7774-7781. [DOI] [PubMed] [Google Scholar]
- 33.Koradi, R., M. Billeter, and K. Wüthrich. 1996. MOLMOL: a program for display and analysis of macromolecular structures. J. Mol. Graphics 14:51-55. [DOI] [PubMed] [Google Scholar]
- 34.Kravanja, M., R. Engelmann, V. Dossonnet, M. Bluggel, H. E. Meyer, R. Frank, A. Galinier, J. Deutscher, N. Schnell, and W. Hengstenberg. 1999. The hprK gene of Enterococcus faecalis encodes a novel bifunctional enzyme: the HPr kinase/phosphatase. Mol. Microbiol. 31:59-66. [DOI] [PubMed] [Google Scholar]
- 35.Kuboniwa, H., S. Grzesiek, F. Delaglio, and A. Bax. 1994. Measurement of HN-H alpha J couplings in calcium-free calmodulin using new 2D and 3D water-flip-back methods. J. Biomol. NMR 4:871-878. [DOI] [PubMed] [Google Scholar]
- 36.Laskowski, R. A., M. W. MacArthur, D. S. Moss, and J. M. Thornton. 1993. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Cryst. 26:283-291. [Google Scholar]
- 37.Lavergne, J.-P., J.-M. Jault, and A. Galinier. 2002. Insights into the functioning of Bacillus subtilis HPr kinase/phosphatase: affinity for its protein substrates and role of cations and phosphate. Biochemistry 41:6218-6225. [DOI] [PubMed] [Google Scholar]
- 38.Luginbühl, P., T. Szyperski, and K. Wüthrich. 1995. Statistical basis for the use of 13C chemical shifts in protein structure determination. J. Magn. Reson. B 109:229-233. [Google Scholar]
- 39.Marion, D., and K. Wüthrich. 1983. Application of phase-sensitive two-dimensional correlated spectroscopy (COSY) for measurements of 1H-1H spin-spin coupling constants in proteins. Biochem. Biophys. Res. Commun. 113:967-974. [DOI] [PubMed] [Google Scholar]
- 40.Marquez, J. A., S. Hasenbein, B. Koch, S. Fieulaine, S. Nessler, R. B. Russell, W. Hengstenberg, and K. Scheffzek. 2002. Structure of the full length HPr kinase/phosphatase from Staphylococcus xylosus at 1.95 Å resolution: mimicking the product/substrate of the phosphotransfer reactions. Proc. Natl. Acad. Sci. USA 99:3458-3463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maurer, T., R. Döker, A. Görler, W. Hengstenberg, and H. R. Kalbitzer. 2001. Three-dimensional structure of the histidine containing phosphocarrier protein (HPr) from Enterococcus faecalis in solution. Eur. J. Biochem. 268:635-644. [DOI] [PubMed] [Google Scholar]
- 42.Mijakovic, I., S. Poncet, A. Galinier, V. Monedero, S. Fieulaine, J. Janin, S. Nessler, J. A. Marquez, K. Scheffzek, S. Hasenbein, W. Hengstenberg, and J. Deutscher. 2002. Pyrophosphate-producing protein dephosphorylation by HPr kinase/phosphorylase: a relic of early life? Proc. Natl. Acad. Sci. USA 99:13442-13447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Neidig, K. P., M. Geyer, A. Görler, C. Antz, R. Saffrich, W. Beneicke, and H. R. Kalbitzer. 1995. AURELIA, a program for computer-aided analysis of multidimensional spectra. J. Biomol. NMR 6:255-270. [DOI] [PubMed] [Google Scholar]
- 44.Pervushin, K., R. Riek, G. Wider, and K. Wüthrich. 1997. Attenuated T2 relaxation by mutual cancellation of dipole-dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules. Proc. Natl. Acad. Sci. USA 94:12366-12371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Postma, P. W., J. W. Lengeler, and G. R. Jacobson. 1993. Phosphoenolpyruvate-carbohydrate phosphotransferase systems of bacteria. Microbiol. Rev. 57:543-549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pullen, K., P. Rajagopal, B. R. Branchini, M. E. Huffine, J. Reizer, M. H. Saier, J. M. Scholtz, and R. E. Klevit. 1995. Phosphorylation of serine-46 in HPr, a key regulatory protein in bacteria, results in stabilization of its solution structure. Protein Sci. 4:2478-2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Reizer, J., M. J. Novotny, W. Hengstenberg, and M. H. Saier, Jr. 1984. Properties of ATP-dependent protein kinase from Streptococcus pyogenes that phosphorylates a seryl residue in HPr, a phosphocarrier protein of the phosphotransferase system. J. Bacteriol. 160:333-340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Reizer, J., C. Hoischen, F. Titgemeyer, C. Rivolta, R. Rabus, J. Stülke, D. Karamata, M. H. Saier, Jr., and W. Hillen. 1998. A novel protein kinase that controls carbon catabolite repression in bacteria. Mol. Microbiol. 27:1157-1169. [DOI] [PubMed] [Google Scholar]
- 49.Rucker, S. P., and A. J. Shaka. 1989. Broadband homonuclear cross polarization in 2D N.M.R. using DIPSI-2. Mol. Phys. 68:509-517. [Google Scholar]
- 50.Russell, R. B., J. A. Marquez, W. Hengstenberg, and K. Scheffzek. 2002. Evolutionary relationship between the bacterial HPr kinase and the ubiquitous PEP-carboxykinase: expanding the P-loop nucleotidyl transferase superfamily. FEBS Lett. 517:1-6. [DOI] [PubMed] [Google Scholar]
- 51.Schleucher, J., M. Schwendinger, M. Sattler, P. Schmidt, O. Schledletzky, S. J. Glaser, O. W. Sorensen, and C. Griesinger. 1994. A general enhancement scheme in heteronuclear multidimensional NMR employing pulsed field gradients. J. Biomol. NMR 4:301-306. [DOI] [PubMed] [Google Scholar]
- 52.Stülke, J., and W. Hillen. 1999. Carbon catabolite repression in bacteria. Curr. Opin. Microbiol. 2:195-201. [DOI] [PubMed] [Google Scholar]
- 53.Van Nuland, N. A. J., R. Boelens, R. M. Scheek, and G. T. Robillard. 1995. High-resolution structure of the phosphorylated form of the histidine-containing phosphocarrier protein HPr from Escherichia coli determined by restrained molecular dynamics from NMR-NOE data. J. Mol. Biol. 246:180-193. [DOI] [PubMed] [Google Scholar]
- 54.Viana, R., V. Monedero, V. Dossonnet, C. Vadeboncoeur, G. Pérez-Martinez, and J. Deutscher. 2000. Enzyme I and HPr from Lactobacillus casei: their role in sugar transport, carbon catabolite repression and inducer exclusion. Mol. Microbiol. 36:570-584. [DOI] [PubMed] [Google Scholar]
- 55.Vuister, G. W., and A. Bax. 1992. Resolution enhancement and spectral editing of uniformly C-13-enriched proteins by homonuclear broad-band C-13 decoupling. J. Magn. Reson. 98:428-435. [Google Scholar]
- 56.Vuister, G. W., and A. Bax. 1993. Quantitative J correlation—a new approach for measuring homonuclear 3-bond J(H(N)H(alpha) coupling-constants in N-15-enriched proteins. J. Am. Chem. Soc. 115:7772-7777. [Google Scholar]
- 57.Wang, A. C., S. Grzesiek, R. R. Tschudin, P. J. Lodi, and A. Bax. 1995. Sequential backbone assignment of isotopically enriched proteins in D2O by deuterium-decoupled Ha(CA)N and Ha(CACO)N. J. Biomol. NMR 5:376-382. [DOI] [PubMed] [Google Scholar]
- 58.Wishart, D. S., and B. D. Sykes. 1994. The C-13 chemical-shift index—a simple method for the identification of protein secondary structure using C-13 chemical-shift data. J. Biomol. NMR 4:171-180. [DOI] [PubMed] [Google Scholar]
- 59.Wishart, D. S., C. G. Bigam, J. Yao, F. Abildgaard, H. J. Dyson, E. Oldfield, J. L. Markley, and B. D. Sykes. 1995. 1H, 13C and 15N chemical-shift referencing in biomolecular NMR. J. Biomol. NMR 6:135-140. [DOI] [PubMed] [Google Scholar]
- 60.Wittekind, M., P. Rajagopal, B. R. Branchini, J. Reizer, M. H. Saier, and R. E. Klevit. 1992. Solution structure of the phosphocarrier protein HPr from Bacillus subtilis by two-dimensional NMR spectroscopy. Protein Sci. 1:1363-1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ye, J. J., J. Reizer, X. Cui, and M. H. Saier, Jr. 1994. Inhibition of the phosphoenolpyruvate:lactose phosphotransferase system and activation of a cytoplasmic sugar-phosphate phosphatase in Lactococcus lactis by ATP-dependent metabolite-activated phosphorylation of serine-46 in the phosphocarrier protein HPr. J. Biol. Chem. 269:11837-11844. [PubMed] [Google Scholar]
- 62.Ye, J. J., and M. H. Saier, Jr. 1995. Allosteric regulation of the glucose:H+ symporter of Lactobacillus brevis: cooperative binding of glucose and HPr(ser-P). J. Bacteriol. 177:1900-1902. [DOI] [PMC free article] [PubMed] [Google Scholar]