

Abstract
The virulence of the intracellular pathogen Rhodococcus equi in foals is dependent on the presence of an 81-kb virulence plasmid encoding the virulence protein VapA. Expression of this protein is induced by exposure to oxidative stress, high temperatures, and low pHs, which reflect the conditions encountered by R. equi when it enters the host environment. The aim of this study was to determine whether the LysR-type transcriptional regulator VirR, which is encoded by the virulence plasmid, is required for the expression of vapA. It was shown that the virR gene is cotranscribed with four downstream genes, one of which encodes a two-component response regulator. The expression of VapA, as monitored by Western blotting, was completely dependent on the presence of virR. Maximal expression was observed when vapA was present together with the complete virR operon, suggesting that at least one of the virR operon genes, in addition to virR, is required for the expression of vapA to wild-type levels. The transcriptional start site of vapA was determined to be a cytidine located 226 bp upstream from the vapA initiation codon. His-tagged VirR protein was expressed in Escherichia coli and purified by nickel affinity chromatography. DNA binding studies showed that purified VirR binds to a DNA fragment containing the vapA promoter. We therefore conclude that VirR is required for the activation of vapA transcription.
The gram-positive bacterium Rhodococcus equi is a facultative intracellular pathogen of alveolar macrophages. Although young foals are the primary host of this pathogen, the incidence of R. equi infection in immunocompromised humans has increased markedly over the past 15 years (9, 23, 46). Infection with R. equi leads to life-threatening pyogranulomatous pneumonia accompanied by gross lesions such as macroabscesses and cavitation (32). The virulence of R. equi in foals is dependent on an indigenous plasmid, which varies in size between 80 and 85 kb (40, 42). Plasmid-cured strains are unable to proliferate in macrophages (12, 17). A recent analysis of the nucleotide sequences of two virulence plasmids revealed the presence of a 27.5-kb DNA fragment characterized by a significantly lower G+C content than the remainder of the virulence plasmid (39). The expression of genes located within this region of the virulence plasmid is upregulated following the internalization of R. equi by macrophages, suggesting that this part of the plasmid is a pathogenicity island (33). One of the proteins encoded within the pathogenicity island is VapA, a highly immunogenic, lipid-modified, surface-expressed protein (39, 41). A deletion of vapA results in the attenuation and rapid host clearance of an R. equi vapA mutant strain in mice, showing that VapA is a virulence factor (19). The pathogenicity island encodes six VapA homologues, one of which (VapF) is a pseudogene (39). VapC, -D, and -E are secreted (4); VapG and -H contain a signal sequence and are therefore likely to be secreted.
The expression of vapA is controlled by environmental parameters such as temperature, pH, oxidative stress, and the concentrations of calcium, iron, and magnesium, which reflect the conditions encountered by R. equi when it enters the host environment (2, 33, 38). To date, it remains unclear how these environmental signals are transduced to the transcriptional apparatus. The pathogenicity island contains two open reading frames (ORF4 and ORF8) that display a high degree of similarity to genes encoding transcriptional regulators. ORF4 encodes a protein belonging to the family of LysR-type transcriptional regulators (LTTR) and ORF8 encodes a response regulator which is part of a two-component regulatory system. LTTRs are present in a wide range of bacterial species and represent the largest family of prokaryotic transcriptional regulators (47). These proteins are involved in regulating a diverse range of cellular processes, including CO2 fixation (43), the oxidative stress response (6), and virulence (8, 10). The first crystal structure of a full-length LTTR was recently reported (28). The N-terminal DNA binding domains of LTTRs contain a helix-turn-helix motif that is required for binding to inverted repeats containing a thymidine and an adenine separated by 11 nucleotides (T-N11-A) (13, 35). The expression of LysR-encoding genes is often autoregulated, and they are divergently transcribed from the gene(s) that they control.
Since ORF4 is located within the pathogenicity island, it is likely that it is required for the expression of one or more genes located within this region of the virulence plasmid. The aim of this study was to determine whether the LTTR encoded by ORF4 is required for the expression of vapA. The transcriptional organization of vapA and the gene cluster containing ORF4 and ORF8 was determined, followed by mapping of the transcriptional start site of vapA. It was subsequently shown that the expression of vapA is dependent on the presence of the protein encoded by ORF4 (VirR) and that this protein binds adjacent to the vapA promoter.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
R. equi ATCC 33701 was obtained from the American Type Culture Collection. An avirulent, plasmid-cured strain of R. equi ATCC 33701 (P−) was obtained from S. Takai, Kitasato University, Towada, Japan. Escherichia coli DH5α (Bethesda Research Laboratories) and E. coli BL21(DE3) pLysE (Novagen) were used for general cloning procedures and for expression of the protein encoded by ORF4, respectively, (Table 1). Bacterial strains were grown in Luria-Bertani (LB) broth (34). When appropriate, the following supplements were added: kanamycin, 50 μg ml−1 (E. coli) or 200 μg ml−1 (R. equi); ampicillin, 50 μg ml−1; chloramphenicol, 30 μg ml−1; 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal), 20 μg ml−1; isopropyl-β-d-thiogalactopyranoside (IPTG), 0.1 mM. For solid medium, agar was added to 1.5% (wt/vol).
TABLE 1.
Bacteria and plasmids used for this study
| Strain or plasmid | Genotype or characteristics | Source or reference |
|---|---|---|
| Strain E. coli | ||
| DH5α | supE44 lacU169 (φ80lacZ M15) hsdR17 recA1 endA1 gyrA96 thi-1 relA1 | Bethesda Research Laboratories |
| BL21 (DE3) pLysE | F−ompT hsdSB(rB− mB−) dcm gal λ(DE3) pLysE Cmr | Novagen |
| R. equi | ||
| ATCC 33701 | Virulent strain, 81-kb virulence plasmid p33701 | American Type Culture Collection |
| ATCC 33701 (P−) | Avirulent strain, virulence plasmid cured | 40 |
| Plasmids | ||
| p33701 | Virulence plasmid of R. equi ATCC 33701 | |
| pET3b | Apr | Novagen |
| pBluescript II KS | Apr | Stratagene |
| pRE7 | Kanr, pMB1 oriR R. equi oriR | 48 |
| pRTOsm | Kanr, pMB1 oriR R. equi oriR vapA, omega transcriptional terminator, Smr | This study |
| pR6VT | pRE7, 1,529-bp fragment containing vapA and 679 bp upstream of its translational start site | This study |
| pBlyFR2 | pBluescript KS, 1,830-bp fragment containing virR and 826 bp upstream of its translational start site | This study |
| PRvip10 | pRE7, 1,529-bp fragment containing vapA and 679 bp upstream of its translational start site, 1,867-bp fragment containing virR and 877 bp upstream of its translational start site | This study |
| pBlueRegP1 | pBluescript KS, 6,680-bp fragment containing ORF3 to ORF8 from the R. equi ATCC 33701 virulence plasmid | This study |
| pForlan21 | pRE7, 1,529-bp fragment containing vapA and 679 bp upstream of its translational start site, 6,761-bp fragment containing ORF3 to ORF8 from pBlueRegP1 | This study |
| pEDAR1012 | pBluescript KS, 322-bp fragment containing the vapA promoter region | This study |
| pBluevirRhis | pBluescript KS, virR with a six-His tag at the 3′ end | This study |
| pET3bvirRhis | pET3b, 931-bp fragment containing virR with a six-His tag at the 3′ end | This study |
| pPCR-RR | pBluescript KS, 736-bp fragment containing 396-bp section of ORF8 | This study |
DNA manipulations.
Chromosomal DNA was isolated as described previously (29). Plasmid DNAs were isolated via the alkaline lysis method of Birnboim and Doly (3) or by the use of a Wizard Plus SV miniprep kit as described by the manufacturer (Promega). DNA fragments were isolated from agarose gels by the use of a Genelute DNA purification kit as described by the manufacturer (Sigma-Aldrich). PCRs were performed with Taq DNA polymerase (Promega) or Deep Vent DNA polymerase (New England Biolabs) as described by the manufacturer. Other DNA manipulations were done in accordance with standard protocols (34).
Plasmid construction.
vapA and its promoter region were amplified with Deep Vent DNA polymerase and oligonucleotides PR600F and Takrev (Table 2). The product was ligated into the EcoRV site of pRE7 (48), yielding pR6VT.
TABLE 2.
Oligonucleotide primers used for this study
| Primer name | Sequence (5′ to 3′) | Strand | Location (position or ORF)a |
|---|---|---|---|
| PR400F | GCGCCATGGGCTGGTCCCTGATGA | + | 12072 |
| PR600F | GCGCCATGGAGACCAACATCGTTCG | + | 11867 |
| 103R | CGATGTCCTGACCACAAGAA | − | 12384b |
| LYF | GCGGCTAGCAGCCTTATCGTCGCAACTGT | + | ORF3 |
| LYR | GCGTTCGAAGGTTGGGCCTAGTGTCCAATTC | − | 5927b |
| CY5VAPA200R | [Cy5]TTGAGAATCGCACTGCTGC | − | vapA |
| VAPA200R | TTGAGAATCGCACTGCTGC | − | vapA |
| Takrev | GCGGGATCCCTGCAGTCAGCGCGATTG | − | ORF13 |
| 102R | TTTCCACATGGACGAGTGAG | − | 9645b |
| 008F | GAACAACTGGGAATGGTGGT | + | ORF8 |
| LYSR-FORW | GCGCATATGAATGTTGACGAACTCCG | + | virR |
| VIR-HIS | TCAATGATGATGATGATGATGGGCGCCCTCTTGTCCGCGGTTTGC | − | virR |
| Vap1 | GCGCATATGACCGTTCTTGATTCCGGT | + | vapA |
| Vap2 | CGCCTCGAGGGCGTTGTGCCAGCTACC | − | vapA |
| 003R | AGCCTTATCGTCGCAACTGT | + | ORF3 |
| 004R | CAAAGACGATTTGGGGTACG | − | virR |
| 004F | CGGACGAGTTCGACTGGTAT | + | virR |
| 005R | GAGTCGCAGACGAGGTAAGC | − | ORF5 |
| 005F | CTCTTCCTGATCGGAGTTGC | + | ORF5 |
| 006R | TACCGATTACGGAGCTCACC | − | vapH |
| 006F | AGGGTTATGCAGGTGGATTG | + | vapH |
| 007NR | GGTGGGCTGGATTGACGCGCA | − | ORF7 |
| 007NF | ATGCACTCCCTGAAAACTATC | + | ORF7 |
| 008R | GTTCGCCGTTTCTAGACGAA | − | ORF8 |
| 008F | GAACAACTGGGAATGGTGGT | + | ORF8 |
| 009R | TACAACGCAACGGCGTAAAG | − | ORF9 |
When the oligonucleotide does not bind to an ORF, the 3′ binding location is given.
Position on complementary strand.
pForlan21 was constructed as follows. ORF8 was amplified with Deep Vent DNA polymerase and oligonucleotides 008F and 102R (Table 2). The resulting PCR product was digested with EcoRI and the 736-bp fragment was ligated into SmaI-EcoRI-digested pBluescript II KS (Stratagene), yielding pPCR-RR. A 5,988-bp KpnI-EcoRI fragment of p33701 was ligated into KpnI-EcoRI-digested pPCR-RR, yielding pBlueRegP1. pBlueRegPI was digested with NotI, end filled with Klenow DNA polymerase, and further digested with KpnI. The resulting 6,761-bp fragment was ligated into pR6VT which had been digested with NcoI, end filled with Klenow DNA polymerase, and further digested with KpnI.
pRvip10 was constructed as follows. ORF4 and its promoter region were amplified with Deep Vent DNA polymerase and oligonucleotides LYF and LYR (Table 2). The product was ligated into the EcoRV site of pBlueScript II KS, yielding pBlyFR2. The 1,867-bp NotI-NheI fragment from pBlyFR2 was ligated into NotI-NheI-digested pR6VT (Table 1).
pEDAR1012 was constructed by amplifying the promoter region of vapA with Deep Vent DNA polymerase and the oligonucleotides PR400F and 103R (Table 2). This product was ligated into EcoRV-digested pBluescript II KS.
pET3bvirRhis was constructed by amplifying the coding region of ORF4 with Deep Vent DNA polymerase and oligonucleotides LYSR-FORW and VIR-HIS (Table 2). This product was ligated into EcoRV-digested pBluescript II KS, yielding pBluevirRhis. The 931-bp NdeI-BamHI fragment from pBluevirRhis was ligated into NdeI-BamHI-digested pET3b (Novagen) (Table 1).
Electroporation of R. equi.
R. equi was made electrocompetent by use of the method described by Zheng et al. (48); the pR6VT, pRvip10, and pForlan21 constructs were introduced into R. equi ATCC 33701 (P−) by electroporation as previously described (27).
Overexpression and purification of VirR-his.
E. coli BL21(DE3) harboring pLysE and pET3bvirRhis was grown at 28°C and 200 rpm in 400 ml of LB medium containing ampicillin and chloramphenicol. At an optical density at 650 nm of 0.5, IPTG was added to a final concentration of 1 mM and growth was continued for another 4 h. The cells were harvested by centrifugation at 4,100 × g and were resuspended in 16 ml of binding buffer (5 mM imidazole, 0.5 M NaCl, 20 mM Tris-HCl [pH -pH pH 7.9]) supplemented with 1 mM phenylmethylsulfonyl fluoride. The cells were broken by three passages through a French press at 1,000 lb/in2 (Aminco), and the cell debris was subsequently removed by centrifugation (25 min, 20,000 × g, 4°C). His-tagged VirR (VirR-his) was purified by the use of a His-Bind purification kit as described by the manufacturer (Novagen). Purified VirR was desalted with a HiTrap desalting column (Amersham Biosciences) equilibrated with electrophoretic mobility shift assay (EMSA) binding buffer (25 mM Tris-HCl [pH 7.5], 1 mM EDTA, 0.1 mM dithiothreitol [DTT], 50 mM KCl, 10% glycerol).
Preparation and labeling of DNA fragment used for EMSAs.
To obtain a radiolabeled DNA fragment containing the vapA promoter region, we digested pEDAR1012 with BglII and HindIII and labeled the resulting 262-bp DNA fragment with [α-32P]dATP (Perkin-Elmer) in a mixture containing 50 ng of DNA, 100 μM dCTP, 100 μM dGTP, 100 μM dTTP, 5 μCi of [α-32P]dATP, 2 U of Klenow DNA polymerase (Promega), 50 mM Tris-HCl (pH 7.2), 10 mM MgSO4, and 0.1 mM DTT which was then incubated at 30°C for 30 min. The reaction was stopped by the addition of 25 mM EDTA. The labeled fragment was purified by use of a Qiaquick PCR purification kit according to the manufacturer's instructions (Qiagen).
EMSA.
Radiolabeled DNA fragments (2 ng) were incubated with purified VirR-his at 30°C for 30 min in EMSA binding buffer, 20 μg of bovine serum albumin, and 1 μg of poly(dI-dC) DNA (Amersham Biosciences) in a volume of 20 μl. The samples were separated by electrophoresis in a prerun 5% nondenaturing polyacrylamide gel containing TBE (45 mM Tris base, 45 mM boric acid, 1 mM EDTA) and run at 4°C and 10 V cm−1. After drying, the gel was analyzed by autoradiography.
RNA isolation.
Total bacterial RNA was isolated from a 4-ml culture grown to mid-logarithmic phase (optical density at 610 nm, 0.25) and harvested by centrifugation for 45 s at 20,000 × g at 4°C. The cells were resuspended in 1 ml of RLT buffer (RNeasy mini kit; Qiagen) and added to 0.5 ml of DEPC-treated 0.1-mm-diameter zirconia-silica beads (BioSpec). The samples were lysed for 2 min with a Ribolyser (Hybaid) at a speed setting of 6.5. Total RNA was isolated by the use of an RNeasy RNA mini kit (Qiagen) according to the manufacturer's instructions, except that a 60-min on-column DNA digestion with 60 U of RNase-free DNase I (Qiagen) was performed. After elution, 4 U of DNA-free DNase was incubated with 5 μg of total RNA according to the manufacturer's instructions (Ambion).
Fluorescent primer extension and DNA sequencing.
A Cy5-labeled oligonucleotide, CY5VAPA200R (Table 2), complementary to the sequence 113 to 131 bp downstream from the initiation codon of the vapA gene was used in primer extension reactions. Total RNA (5 μg) and 1 μM CY5VAPA200R (Table 2) were incubated at 70°C for 5 min, followed by reverse transcription (RT) at 42°C for 60 min with 5 U of Improm-II reverse transcriptase in a volume of 100 μl as recommended by the manufacturer (Promega). After treatment of the sample with 20 μg of RNase A at 37°C for 30 min, the cDNA was precipitated and dissolved in 12 μl of nuclease-free water. The primer extension product (0.5 ng) was combined with 0.5 μl of the DNA size standard kit-600 (Beckman Coulter) and 40 μl of CEQ sample loading solution (Beckman Coulter) and analyzed with the CEQ 8000 fragment analysis system on a CEQ 8000 DNA sequencer (Beckman Coulter). In addition, a dideoxy sequencing reaction containing VAPA200R (Table 2) and 60 ng of EcoRV-digested pRvip10 (Table 1) was performed by the use of a CEQ DCTS kit as described by the manufacturer (Beckman Coulter). The Cy5-labeled primer extension product (50 pg) was added to the sample prior to analysis of the sequence with a CEQ 8000 DNA sequencer.
RT-PCR.
Total RNA (1 μg) was used for the synthesis of cDNA with 0.5 μg of random hexamers (Promega) and 1 U of ImProm-II reverse transcriptase (Promega) used according to the manufacturer's instructions. One-tenth of the reaction mixture was used as a template for PCR amplification with Taq DNA polymerase (Promega) as described by the manufacturer. The oligonucleotides utilized for PCR amplification are listed in Table 2.
Extraction of cellular proteins.
Cells were harvested in late-logarithmic phase by centrifugation (10 min, 4,000 × g, 4°C) and resuspended in 10 mM Tris-HCl (pH 8.0) and 1 mM EDTA. The cells were broken by three passages through a French pressure cell (Aminco) at 1,000 lb/in2, followed by centrifugation (10 min, 14,000 × g, 4°C) to remove cell debris.
Western blot analysis.
Cell extracts were boiled for 5 min in a sodium dodecyl sulfate (SDS) solution (62.5 mM Tris-HCl [pH 6.8], 10% [vol/vol] glycerol, 2% [wt/vol] SDS, 5% [vol/vol] 2-mercaptoethanol, 0.02% [wt/vol] bromophenol blue). SDS-polyacrylamide gel electrophoresis was performed on a 15% polyacrylamide gel by the method of Laemmli (26). After electrophoresis, the proteins were transferred to a polyvinylidene difluoride membrane (Immobilon-P; Millipore) according to the manufacturer's instructions. Immunoblot analysis was performed with a chemiluminescence Western blot analysis system (Lumi-Light Western blotting substrate; Roche). A monoclonal antibody against VapA (Mab103, provided by S. Takai) was used for immunoblotting procedures.
Northern hybridization.
After electrophoresis in a denaturing formaldehyde gel (34), RNAs were transferred to a positively charged membrane according to the manufacturer's instructions (Roche). The vapA probe used for Northern blot analysis was synthesized with the oligonucleotides Vap1 and Vap2 (Table 2) and Taq DNA polymerase (Promega) in the presence of a 0.2 mM concentration (each) of dATP, dCTP, and dGTP; 0.13 mM dTTP; and 0.07 mM digoxigenin 11-dUTP. The reaction mixture was incubated at 94°C for 2 min and was subsequently subjected to 30 cycles of 94°C for 30 s, 50°C for 45 s, and 74°C for 1 min, followed by an incubation at 74°C for 7 min. Prehybridization, hybridization, and chemiluminescent detection of the labeled probe with DIG Easy Hyb and CDP-Star kits (Roche) were done according to the manufacturer's recommendations.
RESULTS
Nucleotide sequence analysis of ORF4 gene cluster.
In the initial annotation of the pathogenicity island of the virulence plasmid of R. equi (accession number NC_004854), the start codon of ORF4, encoding an LTTR, is an ATG located at position 4993 (39). A characteristic of LTTRs is the presence of a helix-turn-helix motif at the amino-terminal end of the protein which facilitates DNA binding. An inspection of the protein encoded by ORF4 reveals that half of this DNA binding domain is missing. This strongly suggests that the initiation codon is more likely a GTG at position 4942, located 51 bp upstream of the annotated ATG initiation codon, because the protein encoded by this revised ORF4 contains a complete helix-turn-helix motif.
The spacing between ORF4 and ORF5 is 493 bp in the original annotation of the virulence plasmid, with the initiation codon of ORF5 being an ATG at nucleotide 6322. However, a careful inspection of ORF5 indicated that the true initiation codon of this ORF is more likely to be a GTG at position 5947. The protein encoded by the revised ORF5 displays a high degree of similarity with transport proteins belonging to the major facilitator superfamily, in particular to proteins belonging to the arabinose efflux permease family (COG2814).
Two base pairs downstream of ORF8 is a region (nucleotides 9295 to 9692) that displays similarity to genes encoding the VapA protein family. This ORF starts at position 9295 of the virulence plasmid but is interrupted by a stop codon at position 9544 and contains a −1 frame shift at position 9630. It is therefore a pseudogene, similar to vapF, which contains two frameshift mutations. Furthermore, as is the case for vapF, the vap pseudogene downstream of ORF8 lacks a signal sequence. When corrected for the presence of the stop codon and the frame shift, the hypothetical protein encoded by this pseudogene is most similar to vapE.
The ORF4-8 cluster is cotranscribed.
An analysis of the nucleotide sequence of the pathogenicity island indicated that ORF4 to ORF8 may form a polycistronic operon. These five ORFs are transcribed in the same direction and are separated from each other by a maximum of 136 bp (ORF5 and ORF6) and a minimum of 57 bp (ORF6 and ORF7). To determine the transcriptional organization of this region, we performed RT-PCRs with oligonucleotide pairs (Table 2) complementary to adjacent ORFs to span each gene junction (Fig. 1A). Reverse transcriptase-dependent amplification generated products of the predicted sizes from oligonucleotide pairs complementary to the following genes: ORF4-ORF5, ORF5-ORF6, ORF6-ORF7, and ORF7-ORF8 (Fig. 1B). These results showed that ORF4 to ORF8 are transcribed as a single message, showing that this cluster is an operon which is transcribed from a promoter located between ORF3 and ORF4. As expected, RT-PCR products were not observed when we used oligonucleotide pairs complementary to ORF3 and ORF4, nor were any observed when we used oligonucleotides complementary to ORF8 and ORF9. RT-PCR products were not observed when reverse transcriptase was omitted from the reaction mixture.
FIG. 1.

Transcriptional organization of virR gene cluster. (A) Genetic organization of DNA region harboring ORF3 to ORF9. The arrow above the map denotes the direction of transcription of the polycistronic message. Arrows below the illustration indicate oligonucleotide primers used for RT-PCR. (B) Results of RT-PCR analyses. Each oligonucleotide pair was used in three amplification reactions, with 2 μl of the reverse transcriptase-containing reaction (cDNA), without reverse transcriptase (−RT), and with R. equi ATCC 33701 genomic DNA (DNA). The oligonucleotide pairs used were 003R and 004R (i), 004F and 005R (ii), 005F and 006R (iii), 006F and 007NR (iv), 007NF and 008R (v), and 008F and 009R (vi). The size of each band is indicated.
Mapping of vapA transcriptional start site.
It was shown previously that the vapA transcript is monocistronic in R. equi 85F, a strain which contains an 85-kb virulence plasmid (2). To determine the transcriptional organization of vapA in R. equi ATCC 33701, we isolated mRNAs from R. equi grown under conditions that induce the expression of vapA (pH 6.5, 37°C) and analyzed them by Northern hybridization, using vapA as a probe. A single transcript of approximately 700 nucleotides was observed, indicating that vapA is transcribed as a monocistronic transcript, as is the case for R. equi 85F (data not shown). The transcriptional start site of vapA was mapped for this strain by rapid amplification of cDNA ends (RACE) following induction with H2O2 (2). To determine whether both strains employ the same transcriptional start site, we performed a primer extension reaction using a fluorescently labeled oligonucleotide complementary to vapA and mRNAs isolated from R. equi grown under inducing conditions. A single 357-bp DNA fragment was observed (Fig. 2A), indicating that vapA is transcribed from a single promoter under these growth conditions. Interestingly, the transcriptional start site of vapA in R. equi ATCC 33701 is not the same as that in R. equi 85F. In the former strain, transcription starts at a cytidine (Fig. 2B) 226 bp upstream of the translational start site of vapA, whereas the transcriptional start of vapA in R. equi 85F was determined to be 69 bp upstream of vapA (2). The vapA transcriptional start site of R. equi 85F was determined after the induction of vapA expression by exposure to H2O2, whereas a low pH and high temperatures were used for R. equi ATCC 33701. To determine whether vapA is transcribed from different promoters under these two growth conditions, we determined the transcriptional start of vapA in R. equi ATCC 33701 after exposure to H2O2. Transcription initiated at the same nucleotide (226 bp upstream of vapA), indicating that R. equi ATCC 33701 employs the same promoter under high-temperature, low-pH growth conditions as it does after exposure to H2O2 (data not shown). Interestingly, the −10 and −35 regions (Fig. 2C) of the vapA promoter are similar to the consensus σhrdB promoter (−10, TAGART; −35, TTGaCA) of Streptomyces coelicolor (21, 36).
FIG. 2.

Determination of vapA transcriptional start site in R. equi ATCC 33701. Fluorescent primer extension was performed with the Cy5-labeled primer CY5VAPA200R and 5 μg of total cellular RNA extracted from R. equi grown under vapA-inducing conditions (37°C, pH 6.5). CY5VAPA200R is complementary to a sequence 131 bp downstream from the vapA initiation codon. (A) Cy5-labeled primer extension product combined with DNA size standards and analyzed with the CEQ 8000 fragment analysis system. (B) Nucleotide sequence obtained by using VAPA200R. A dideoxy sequencing reaction mix was spiked with the Cy5-labeled primer extension product. The arrow indicates the transcriptional start site where the Cy5-labeled cDNA and the sequencing product overlapped. (C) Sequence of vapA promoter region. The transcriptional start site (+1) and putative −10 and −35 regions are boxed, and putative LysR motifs (T-N11-A) are indicated with brackets.
ORF4 is required for expression of vapA.
The expression of vapA is controlled by a range of environmental parameters, including temperature and pH (38). In order to determine whether regulatory proteins encoded on the virulence plasmid are required for the expression of vapA, we introduced a plasmid (pRTOsm) containing vapA and the vapA promoter into a virulence plasmid-free strain of R. equi. The inserted plasmid contained a transcriptional terminator between the vector and the insertion site of the DNA fragment containing vapA, preventing readthrough from vector-borne promoters into the vapA gene. Expression of vapA was not observed under inducing conditions (pH 6.5, 37°C) (data not shown). This shows that expression is at least partly dependent on virulence plasmid-encoded transcriptional regulators. The pathogenicity island contains two genes encoding proteins similar to transcriptional regulators, suggesting that these may be required for vapA expression. To determine whether vapA expression is dependent on the LTTR encoded by ORF4, we introduced a plasmid containing ORF4 and vapA (pRvip10) into R. equi ATCC 33701 (P−). In contrast to a strain harboring only vapA, the presence of ORF4 in addition to vapA resulted in the expression of the latter, as detected by Western blotting (Fig. 3A). This shows that ORF4 is required for the expression of vapA and represents a functional gene (virR). Interestingly, the expression of vapA was increased under inducing conditions (pH 6.5, 37°C) relative to noninducing conditions (pH 8.0, 30°C). However, the expression level of vapA was lower than that of the wild-type strain (Fig. 3A), suggesting the involvement of additional transcriptional regulators. A plasmid (pForlan21) harboring the virR-ORF8 operon in addition to vapA was introduced into R. equi ATCC 33701 (P−), followed by incubation under inducing and noninducing conditions. A Western blot analysis of the proteins extracted from this strain showed that wild-type vapA expression levels were restored under inducing conditions (Fig. 3B). Furthermore, the regulation of vapA expression under inducing and noninducing conditions was comparable to that in the wild-type strain, indicating that the virR-ORF8 operon harbors all of the virulence plasmid genes that are required for the normal expression and regulation of vapA.
FIG. 3.

Effect of virR and the virR operon on expression of vapA. The expression of VapA was determined by Western blotting with VapA monoclonal antibodies. (A) Lanes 1 and 2, R. equi wild-type strain grown at 37°C and pH 6.5 (lane 1) and at 30°C and pH 8.0 (lane 2); lanes 3 and 4, R. equi (P−) harboring pRvip10 (virR vapA) grown at 37°C and pH 6.5 (lane 3) and at 30°C and pH 8.0 (lane 4); lane 5, R. equi (P−) grown at 37°C and pH 6.5. Each lane was loaded with 2 μg of protein. (B) Lanes 1 and 2, R. equi wild-type strain grown at 37°C and pH 6.5 (lane 1) and at 30°C and pH 8.0 (lane 2); lanes 4 and 5, R. equi (P−) harboring pForlan21 (virR operon and vapA) grown at 37°C and pH 6.5 (lane 4) and at 30°C and pH 8.0 (lane 5); lane 3, R. equi (P−) grown at 37°C and pH 6.5. Each lane was loaded with 2 μg of protein.
Purification of VirR-His.
To facilitate the purification of VirR, we constructed an efficient expression system by replacing the GTG initiation codon of virR with ATG and by placing the virR gene downstream of the T7 promoter carried on pET3b. In addition, six histidine codons were added to the 3′ end of virR to create a histidine tag. After the induction of the T7 RNA polymerase in E. coli BL21(pET3bvirRhis), an abundant protein representing VirR-His which was absent from E. coli BL21 grown under the same conditions was observed in the cell extract. The molecular mass of this protein (31 kDa) corresponds to the predicted molecular mass of VirR (32,667 Da). VirR-His was subsequently purified by Ni2+ affinity chromatography; the resulting preparation was shown to be homogeneous by SDS-polyacrylamide gel electrophoresis (Fig. 4).
FIG. 4.
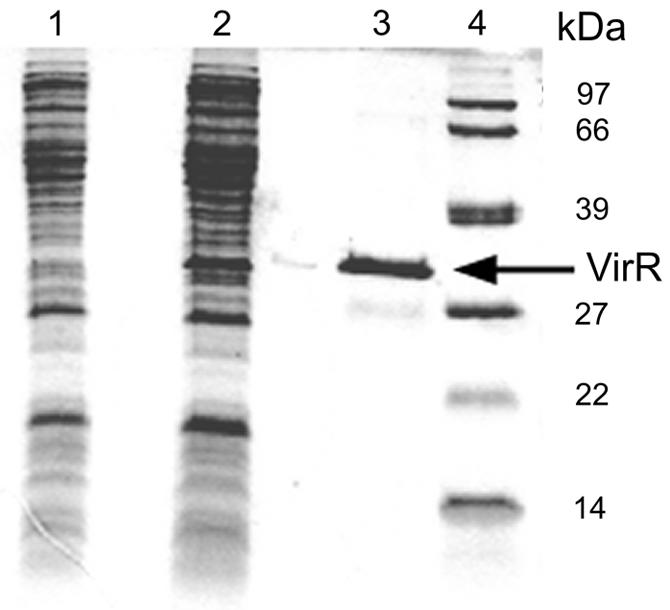
Coomassie brilliant blue-stained denaturing polyacrylamide gel showing cell extracts of E. coli BL21 harboring pET3b (lane 1) or pET3bvirRhis (lane 2). VirR-His was purified by Ni2+ affinity chromatography (lane 3). The sizes of the molecular mass standards are shown in kilodaltons (lane 4).
VirR binds to the vapA promoter region.
Although VirR was required for the expression of vapA, it was not clear whether this regulator interacts with the vapA promoter or whether the stimulatory effect of VirR on vapA expression was mediated through another transcriptional regulator. To determine whether VirR binds to a 262-bp DNA fragment containing the vapA promoter, we performed a band shift experiment. After being radiolabeled, the DNA fragment harboring the vapA promoter was incubated with increasing concentrations of purified VirR-His. With increasing concentrations of VirR-His, two protein-DNA complexes were observed (Fig. 5). The high-mobility protein-DNA complex was present at all concentrations of VirR-His, whereas the second, low-mobility complex was only observed at high concentrations.
FIG. 5.

EMSA analysis of VirR binding to the vapA promoter region. Various concentrations of VirR were incubated with 2 ng of radiolabeled DNA (262 bp) containing the vapA promoter region. The amount of protein added to each lane was as follows: lane 1, radiolabeled DNA fragment only; lane 2, 50 ng; lane 3, 100 ng; lane 4, 200 ng; lane 5, 300 ng; and lane 6, 400 ng. Protein-DNA complexes are indicated with black arrows. Nonbound DNA is indicated with a gray arrow.
To rule out the possibility that the observed band shifts were due to the presence of contaminating E. coli proteins in the purified VirR-His preparation, we performed a band shift assay with 20 μg of soluble protein from E. coli BL21 harboring either pET3bvirRhis or pET3b. A band shift was only observed for the cell extract of E. coli expressing VirR-His (pET3bvirRhis), not for E. coli BL21(pET3b), showing that the observed band shifts are due to an interaction of VirR-His with DNA (results not shown).
LTTRs generally bind to degenerate inverted repeats that have a T-N11-A motif at their core (35). An inspection of the sequence upstream of the vapA transcriptional start site revealed six T-N11-A motifs. Two of these which are on the same side of the DNA helix and are centered at −43 and −64 occur adjacent to the −35 region (Fig. 2C).
DISCUSSION
It is well documented that the expression of the virulence gene vapA is regulated by environmental parameters that signal that R. equi has entered the host environment. These include high temperature, low pH, low iron concentrations, and oxidative stress. However, a transcriptional regulator that is required for the expression of vapA and that may be involved in transducing some of these environmental signals to the transcriptional apparatus has not yet been described. This study shows that the LTTR VirR, encoded by ORF4 of the virulence plasmid, is required for the transcription of vapA. LTTRs are frequently transcriptional activators, although there are some that control gene expression by repression (15, 22). LTTRs control a wide range of biological processes, including virulence. The SpvR protein in Salmonella enterica serovar Dublin induces the expression of the spvABCD operon during the stationary phase of growth (14). The plasmid-encoded spv locus is essential for growth in the liver and the spleen (16). The AphB protein of Vibrio cholerae is required for the activation of the ToxR virulence cascade by transcriptionally activating the tcpPH operon (24).
The majority of LTTR-encoding genes are monocistronic and are transcribed divergently from the genes under their control (18, 25, 30). In contrast, the virR gene is cotranscribed with four other genes located downstream from virR. Although this is an unusual configuration, it is not unprecedented. CatR, an LTTR of the actinomycete Streptomyces setonii, was shown to be translationally coupled to two downstream genes required for the metabolism of aromatic compounds (31). The Mycobacterium tuberculosis genome contains five LTTR genes; one of these, Rv3678c, is transcribed in the same direction as the downstream Rv3677c gene (7). Since the spacing between these genes is only six nucleotides, it is extremely likely that these too are cotranscribed. In both instances, the first gene of these putative operons is the LTTR gene, as is the case for virR. The significance of this, if any, remains to be established.
The transcriptional start site of vapA in R. equi ATCC 33701 was determined to be 226 bp upstream of the initiation codon of vapA. The induction of vapA transcription either by incubation at a high temperature and a low pH or by exposure to H2O2 gave rise to the same transcriptional start site, indicating that vapA is transcribed from a single identical promoter under these conditions. The long 5′ untranslated region (5′-UTR) of vapA may serve several functions. 5′-UTRs are frequently involved in stabilization of the downstream mRNA, as is the case for the cryIIIA toxin gene of Bacillus thuringiensis (1) and ompA of E. coli (5). The prfA gene of Listeria monocytogenes encodes a transcriptional regulator that activates the transcription of virulence genes. Similar to the case for vapA, the expression of virulence genes in L. monocytogenes is controlled by temperature, with high expression levels at 37°C, not at 30°C. It was recently shown that this temperature-dependent expression of virulence genes is controlled by the 5′-UTR of prfA. At 37°C, the structure of the 5′-UTR of prfA unfolds, exposing the ribosome binding site of this gene and allowing translation to initiate (20). The function of the 5′-UTR of vapA remains to be established.
The fact that purified VirR binds to a DNA fragment containing the vapA promoter strongly suggests that this protein activates vapA transcription by a direct interaction with RNA polymerase bound to the vapA promoter. At lower VirR concentrations, a single band shift was observed in band shift assays, whereas a second band shift became apparent at higher VirR concentrations. This has been observed in DNA binding studies of other LTTRs. For example, CbbR, an LTTR that controls the expression of CO2 fixation genes in Xanthobacter flavus, binds as a dimer to a promoter-distal high-affinity binding site, giving rise to a band shift with a high mobility. A second CbbR dimer is subsequently recruited by cooperative binding to a promoter-proximal low-affinity binding site, leading to the formation of a second DNA-protein complex with a lower mobility (44, 45). Whether a similar scenario is true for VirR remains to be established.
Although VirR is required for vapA expression, it is not sufficient to express vapA to wild-type levels. The introduction of the complete virR operon, together with vapA, in a virulence plasmid-free strain did lead to wild-type levels of VapA protein, as judged by Western blotting. The VirR operon contains four additional genes. One of these, vapH (ORF6), is a vapA homologue. The observed increase in vapA expression was not due to a cross reaction of the VapA monoclonal antibody, which is specific for VapA, with VapH (4). The most likely explanation for the increased expression of vapA is the presence of ORF8, which encodes a two-component response regulator. The virulence plasmid does not encode a sensor kinase, suggesting that the response regulator encoded by ORF8 interacts with a chromosomally encoded sensor kinase. In recent years, there have been several reports of genes regulated by both LTTRs and response regulators. In E. coli, the LTTR NhaR and the response regulator RcsB were shown to directly but independently regulate the osmC gene (37), while LhrA, a LysR homologue, was found to be involved with the response regulator SprE in a pathway promoting the degradation of the global regulator RpoS (11).
The present study shows that the LTTR VirR is required for transcription of the vapA gene. Whether this protein is also required for the expression of other genes located in the pathogenicity island and whether the response regulator is indeed involved in controlling the expression of vapA are currently being studied in our laboratory.
Acknowledgments
This study was supported by grants from the Health Research Board (RP55/2001) and Enterprise Ireland (SC/00/442).
We thank Shinji Takai for providing R. equi ATCC 33701 (P−) and VapA monoclonal antibodies and John Prescott for making pRE7 available.
REFERENCES
- 1.Agaisse, H., and D. Lereclus. 1996. STAB-SD: a Shine-Dalgarno sequence in the 5′ untranslated region is a determinant of mRNA stability. Mol. Microbiol. 20:633-643. [DOI] [PubMed] [Google Scholar]
- 2.Benoit, S., A. Benachour, S. Taouji, Y. Auffray, and A. Hartke. 2002. H2O2, which causes macrophage-related stress, triggers induction of expression of virulence-associated plasmid determinants in Rhodococcus equi. Infect. Immun. 70:3768-3776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Birnboim, H., and J. Doly. 1979. A rapid alkaline extraction procedure for screening recombinant plasmid DNA. Nucleic Acids Res. 7:1513-1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Byrne, B. A., J. F. Prescott, G. H. Palmer, S. Takai, V. M. Nicholson, D. C. Alperin, and S. A. Hines. 2001. Virulence plasmid of Rhodococcus equi contains inducible gene family encoding secreted proteins. Infect. Immun. 69:650-656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen, L. H., S. A. Emory, A. L. Bricker, P. Bouvet, and J. G. Belasco. 1991. Structure and function of a bacterial mRNA stabilizer: analysis of the 5′ untranslated region of ompA mRNA. J. Bacteriol. 173:4578-4586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Christman, M. F., G. Storz, and B. N. Ames. 1989. OxyR, a positive regulator of hydrogen peroxide-inducible genes in Escherichia coli and Salmonella typhimurium, is homologous to a family of bacterial regulatory proteins. Proc. Natl. Acad. Sci. USA 86:3484-3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cole, S. T., R. Brosch, J. Parkhill, T. Garnier, C. Churcher, D. Harris, S. V. Gordon, K. Eiglmeier, S. Gas, C. E. I. Barry, F. Tekaia, K. Badcock, D. Basham, D. Brown, T. Chillingworth, R. Connor, R. Davies, K. Devlin, T. Feltwell, S. Gentles, N. Hamlin, S. Holroyd, T. Hornsby, K. Jagels, and B. G. Barrell. 1998. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393:537-544. [DOI] [PubMed] [Google Scholar]
- 8.Coynault, C., V. Robbe-Saule, M. Y. Popoff, and F. Norel. 1992. Growth phase and SpvR regulation of transcription of Salmonella typhimurium spvABC virulence genes. Microb. Pathog. 13:133-143. [DOI] [PubMed] [Google Scholar]
- 9.Donisi, A., M. G. Suardi, S. Casari, M. Longo, G. P. Cadeo, and G. Carosi. 1996. Rhodococcus equi infection in HIV-infected patients. AIDS 10:359-362. [DOI] [PubMed] [Google Scholar]
- 10.Doty, S. L., M. Chang, and E. W. Nester. 1993. The chromosomal virulence gene, chvE, of Agrobacterium tumefaciens is regulated by a LysR family member. J. Bacteriol. 175:7880-7886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gibson, K. E., and T. J. Silhavy. 1999. The LysR homolog LrhA promotes RpoS degradation by modulating activity of the response regulator sprE. J. Bacteriol. 181:563-571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Giguère, S., M. K. Hondalus, J. A. Yager, P. Darrah, D. M. Mosser, and J. F. Prescott. 1999. Role of the 85-kilobase plasmid and plasmid-encoded virulence-associated protein A in intracellular survival and virulence of Rhodococcus equi. Infect. Immun. 67:3548-3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goethals, K., M. Van Montagu, and M. Holsters. 1992. Conserved motifs in a divergent nod box of Azorhizobium caulinodans ORS571 reveal a common structure in promoters regulated by LysR-type proteins. Proc. Natl. Acad. Sci. USA 89:1646-1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grob, P., and D. G. Guiney. 1996. In vitro binding of the Salmonella dublin virulence plasmid regulatory protein SpvR to the promoter regions of spvA and spvR. J. Bacteriol. 178:1813-1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harris, S. J., Y. L. Shih, S. D. Bentley, and G. P. Salmond. 1998. The hexA gene of Erwinia carotovora encodes a LysR homologue and regulates motility and the expression of multiple virulence determinants. Mol. Microbiol. 28:705-717. [DOI] [PubMed] [Google Scholar]
- 16.Heffernan, E. J., J. Fierer, G. Chikami, and D. Guiney. 1987. Natural history of oral Salmonella dublin infection in BALB/c mice: effect of an 80-kilobase-pair plasmid on virulence. J. Infect. Dis. 155:1254-1259. [DOI] [PubMed] [Google Scholar]
- 17.Hondalus, M. K., and D. M. Mosser. 1994. Survival and replication of Rhodococcus equi in macrophages. Infect. Immun. 62:4167-4175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Iwanicka-Nowicka, R., and M. M. Hryniewicz. 1995. A new gene, cbl, encoding a member of the LysR family of transcriptional regulators belongs to Escherichia coli cys regulon. Gene 166:11-17. [DOI] [PubMed] [Google Scholar]
- 19.Jain, S., B. R. Bloom, and M. K. Hondalus. 2003. Deletion of vapA encoding virulence associated protein A attenuates the intracellular actinomycete Rhodococcus equi. Mol. Microbiol. 50:115-128. [DOI] [PubMed] [Google Scholar]
- 20.Johansson, J., P. Mandin, A. Renzoni, C. Chiaruttini, M. Springer, and P. Cossart. 2002. An RNA thermosensor controls expression of virulence genes in Listeria monocytogenes. Cell 110:551-561. [DOI] [PubMed] [Google Scholar]
- 21.Kang, J. G., M. Y. Hahn, A. Ishihama, and J. H. Roe. 1997. Identification of sigma factors for growth phase-related promoter selectivity of RNA polymerases from Streptomyces coelicolor A3(2). Nucleic Acids Res. 25:2566-2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim, S. I., C. Jourlin-Castelli, S. R. Wellington, and A. L. Sonenshein. 2003. Mechanism of repression by Bacillus subtilis CcpC, a LysR family regulator. J. Mol. Biol. 334:609-624. [DOI] [PubMed] [Google Scholar]
- 23.Kohl, O., and H. H. Tillmanns. 2002. Cerebral infection with Rhodococcus equi in a heart transplant recipient. J. Heart Lung Transplant. 21:1147-1149. [DOI] [PubMed] [Google Scholar]
- 24.Kovacikova, G., and K. Skorupski. 1999. A Vibrio cholerae LysR homolog, AphB, cooperates with AphA at the tcpPH promoter to activate expression of the ToxR virulence cascade. J. Bacteriol. 181:4250-4256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kulakova, A. N., L. A. Kulakov, N. V. Akulenko, V. N. Ksenzenko, J. T. Hamilton, and J. P. Quinn. 2001. Structural and functional analysis of the phosphonoacetate hydrolase (phnA) gene region in Pseudomonas fluorescens 23F. J. Bacteriol. 183:3268-3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680-685. [DOI] [PubMed] [Google Scholar]
- 27.Mangan, M. W., and W. G. Meijer. 2001. Random insertion mutagenesis of the intracellular pathogen Rhodococcus equi using transposomes. FEMS Microbiol. Lett. 205:243-246. [DOI] [PubMed] [Google Scholar]
- 28.Muraoka, S., R. Okumura, N. Ogawa, T. Nonaka, K. Miyashita, and T. Senda. 2003. Crystal structure of a full-length LysR-type transcriptional regulator, CbnR: unusual combination of two subunit forms and molecular bases for causing and changing DNA bend. J. Mol. Biol. 328:555-566. [DOI] [PubMed] [Google Scholar]
- 29.Nagy, I., G. Schoofs, F. Compernolle, P. Proost, J. Vanderleyden, and R. de Mot. 1995. Degradation of the thiocarbamate herbicide EPTC (S-ethyl dipropylcarbamothioate) and biosafening by Rhodococcus sp. strain NI86/21 involve an inducible cytochrome P-450 system and aldehyde dehydrogenase. J. Bacteriol. 177:676-687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Paradkar, A. S., K. A. Aidoo, and S. E. Jensen. 1998. A pathway-specific transcriptional activator regulates late steps of clavulanic acid biosynthesis in Streptomyces clavuligerus. Mol. Microbiol. 27:831-843. [DOI] [PubMed] [Google Scholar]
- 31.Park, H. J., and E. S. Kim. 2003. An inducible Streptomyces gene cluster involved in aromatic compound metabolism. FEMS Microbiol. Lett. 226:151-157. [DOI] [PubMed] [Google Scholar]
- 32.Prescott, J. F. 1991. Rhodococcus equi: an animal and human pathogen. Clin. Microbiol. Rev. 4:20-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ren, J., and J. F. Prescott. 2003. Analysis of virulence plasmid gene expression of intra-macrophage and in vitro grown Rhodococcus equi ATCC 33701. Vet. Microbiol. 94:167-182. [DOI] [PubMed] [Google Scholar]
- 34.Sambrook, J., and D. W. Russell. 2001. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 35.Schell, M. A. 1993. Molecular biology of the LysR family of transcriptional regulators. Annu. Rev. Microbiol. 47:597-626. [DOI] [PubMed] [Google Scholar]
- 36.Strohl, W. R. 1992. Compilation and analysis of DNA sequences associated with apparent streptomycete promoters. Nucleic Acids Res. 20:961-974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sturny, R., K. Cam, C. Gutierrez, and A. Conter. 2003. NhaR and RcsB independently regulate the osmCp1 promoter of Escherichia coli at overlapping regulatory sites. J. Bacteriol. 185:4298-4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Takai, S., N. Fukunaga, K. Kamisawa, Y. Imai, Y. Sasaki, and S. Tsubaki. 1996. Expression of virulence-associated antigens of Rhodococcus equi is regulated by temperature and pH. Microbiol. Immunol. 40:591-594. [DOI] [PubMed] [Google Scholar]
- 39.Takai, S., S. A. Hines, T. Sekizaki, V. M. Nicholson, D. A. Alperin, M. Osaki, D. Takamatsu, M. Nakamura, K. Suzuki, N. Ogino, T. Kakuda, H. Dan, and J. F. Prescott. 2000. DNA sequence and comparison of virulence plasmids from Rhodococcus equi ATCC 33701 and 103. Infect. Immun. 68:6840-6847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Takai, S., T. Sekizaki, T. Ozawa, T. Sugawara, Y. Watanabe, and S. Tsubaki. 1991. Association between a large plasmid and 15- to 17-kilodalton antigens in virulent Rhodococcus equi. Infect. Immun. 59:4056-4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tan, C., J. F. Prescott, M. C. Patterson, and V. M. Nicholson. 1995. Molecular characterization of a lipid-modified virulence-associated protein of Rhodococcus equi and its potential in protective immunity. Can. J. Vet. Res. 59:51-59. [PMC free article] [PubMed] [Google Scholar]
- 42.Tkachuk-Saad, O., and J. Prescott. 1991. Rhodococcus equi plasmids: isolation and partial characterization. J. Clin. Microbiol. 29:2696-2700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.van den Bergh, E. R., L. Dijkhuizen, and W. G. Meijer. 1993. CbbR, a LysR-type transcriptional activator, is required for expression of the autotrophic CO2 fixation enzymes of Xanthobacter flavus. J. Bacteriol. 175:6097-6104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van Keulen, G., L. Girbal, E. R. van den Bergh, L. Dijkhuizen, and W. G. Meijer. 1998. The LysR-type transcriptional regulator CbbR controlling autotrophic CO2 fixation by Xanthobacter flavus is an NADPH sensor. J. Bacteriol. 180:1411-1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.van Keulen, G., A. N. Ridder, L. Dijkhuizen, and W. G. Meijer. 2003. Analysis of DNA binding and transcriptional activation by the LysR-type transcriptional regulator CbbR of Xanthobacter flavus. J. Bacteriol. 185:1245-1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Weinstock, D. M., and A. E. Brown. 2002. Rhodococcus equi: an emerging pathogen. Clin. Infect. Dis. 34:1379-1385. [DOI] [PubMed] [Google Scholar]
- 47.Zaim, J., and A. M. Kierzek. 2003. The structure of full-length LysR-type transcriptional regulators. Modeling of the full-length OxyR transcription factor dimer. Nucleic Acids Res. 31:1444-1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zheng, H., O. Tkachuk-Saad, and J. F. Prescott. 1997. Development of a Rhodococcus equi-Escherichia coli plasmid shuttle vector. Plasmid 38:180-187. [DOI] [PubMed] [Google Scholar]