

Abstract
Purpose/Aim
Negative feedback controls in endocrine regulatory systems are well recognized. The incretins and their role in glucose regulation have been of major interest recently. Whether the same negative control system applies to the regulation of incretin secretion is not clear. We sought to examine the hypothesis that exogenous administration of glucagon like peptide-1, GLP-1 (7-36) amide or its metabolite GLP-1 (9-36)amide, reduces the endogenous basal release of this incretin.
Materials and Methods
We evaluated the endogenous basal release of GLP-1 using two separate study designs. In protocol A we examined the GLP-1 (7-36)amide levels during the infusion of GLP-1 (9-36) amide. In protocol B, we used PYY and GLP-2 as biomarkers for the endogenous basal release of GLP-1 (7-36) amide and assessed the endogenous basal release of these two hormones during the GLP-1 (7-36) infusion. Twelve lean and 12 obese subjects were enrolled in protocol A and 10 obese volunteers in protocol B.
Results
The plasma levels of GLP-1(7-36) amide in protocol A and PYY and GLP-2 in protocol B remained unchanged during the exogenous infusion of GLP-1(9-36) and GLP-1(7-36) amide respectively.
Conclusions
The negative feedback control system as described by inhibition of the release of endogenous hormone while infusing it exogenously was not observed for the basal secretion of GLP-1(7-36) amide.
Introduction
Negative feedback control by hormones on their own secretion is a common feature of endocrine regulation. Insulin secretion is one such example. When insulin is administered during a hyperinsulinemic-euglycemic clamp, plasma C-peptide levels fall and C-peptide levels return toward the basal level following the discontinuation of insulin infusion in the absence of change in plasma glucose levels (1). In addition, studies on the isolated perfused human pancreas demonstrate an inhibition of glucose-stimulated insulin release by insulin (2). During euglycemia, glucose infusion does not have any influence on insulin secretory kinetics. Therefore these results demonstrate that there is feedback inhibition of both basal and stimulated insulin secretion by insulin itself.
The possible negative feedback of other gluco-regulatory hormones is less clear. However, the phenomenon of negative feed-back has now become clinically relevant because of the introduction of dipeptidyl peptidase-4 (DPP-4) inhibitors to treat type 2 diabetes mellitus (T2DM). These agents prevent the acute degradation of the two incretin peptides, glucagon–like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP), and thereby increase the postprandial biologically active concentrations of both peptides. It is felt that the increases in intact, insulinotropically active GLP-1 and GIP are sufficient to explain the antidiabetic effects seen in T2DM patients during DPP-4 inhibition. Additionally, mice lacking both GIP and GLP-1 receptors do not display any glucose-lowering effect upon DPP-4 inhibition (3). This would seem to preclude any non-specific effects of DPP-4 inhibition, unrelated to incretin action. However, it is also clear that during DPP-4 inhibition, secretion of incretins is reduced. Vildagliptin, an orally active DPP-4 inhibitor, caused a 72% reduction in GLP-1 secretion and a 26% reduction in GIP secretion the postprandial state (4). Similar but lesser effects have been shown with sitagliptin, another orally active DPP-4 inhibitor (5). Saxagliptin, another DPP-4 inhibitor, has more recently been approved by FDA and also has similar effects (6).
These results led us to infer that GLP-1, GIP or their metabolites can inhibit their own secretion. In this study, we have carried out infusions of GLP-1(9-36)amide, the insulinotropically inactive metabolite of GLP-1, to ascertain if the metabolite directly influences basal GLP-1 secretion. Additionally, we have carried out GLP-1(7-36)amide infusions and evaluated Peptide YY (PYY) and glucagon–like peptide-2 (GLP-2) secretion, which are co-secreted with GLP-1 from secretory granules (7), to evaluate any influence of insulinotropically active GLP-1 on its own basal secretion. Finally, we assessed the possible effects of GLP-1 receptor blockade on the basal secretion of GLP-1 by infusing exendin (9-39), a selective GLP-1 receptor antagonist, in addition to GLP-1(9-36)amide. Studies were carried out in obese as well as lean volunteers because the insulinomimetic effects of GLP-1 and its metabolites, per se, are more apparent in the obese state (8, 9).
Materials and Methods
The two protocol designs and subject characteristics have been previously described (8, 9). We will refer to the GLP-1(9-36)amide infusion study as protocol A and to the GLP-1(7-36)amide infusion study as protocol B. In protocol A, we enrolled 12 lean (body mass index (BMI)=22.33±0.39, age 29±1.86 yrs.) and 12 obese (BMI=37.20±1.50, age 42±2.0 yrs.) volunteers. There were six women and six men in each group, all of whom had normal glucose tolerance. We performed two euglycemic clamps on each volunteer, separated by at least 4 weeks. Saline or GLP-1(9-36)amide (1.5 pmol/Kg/min) was infused from 0 to 60 minutes. In both studies, all parameters were followed for 60 minutes after the infusions. A third clamp was performed in seven lean and six obese volunteers. The known GLP-1(7-36)amide receptor was blocked with the infusion of exendin(9-39) (300 pmol/Kg/min) from −60 to +60 minutes and GLP-1(9-36)amide was infused from 0 to 60 minutes. All parameters were followed for an hour after the infusion of the peptides.
In protocol B we enrolled 10 volunteers, all of whom had normal glucose tolerance, (5 male and 5 female, age: 32.5±3.0 yrs, BMI: 34.6±0.8 Kg/m2). Protocol B was similar to protocol A except that GLP-1(7-36)amide was infused (1.5 pmol/Kg/min) from 0 to 60 min. In a second study, we infused insulin in a manner which reproduced the incretin stimulated plasma insulin levels in each volunteer during the GLP-1(7-36) infusion. In all studies all parameters were followed for another hour after the termination of the hormone infusions.
Analytical methods
Blood samples, obtained every 10 minutes from −30 to 120 minutes, were collected using heparinized syringes and were immediately transferred to pre-chilled tubes containing EDTA, peptidase inhibitors aprotinin (trasylol) and DPP-4 inhibitors) as described previously (10). An aliquot of plasma was saved for determination of both active and total GLP-1 in protocol A and both active and total GLP-1 as well as PYY and GLP-2 in protocol B. Total GLP-1 was estimated with an antibody directed toward the C-terminal moiety of the peptide and we measured active GLP-1 by ELISA (Linco Research, Inc.) with a detection limit of 5 pmol/liter. PYY was assayed with a RIA (Linco Research, Inc., now marketed by Millipore), which recognizes both 1–36 and 3–36 forms of human PYY. The limit of sensitivity and the limit of linearity of the PYY assay are 2.32 and 297 pmol/l, respectively (interassay variation, <5%). In addition, we checked the cross-reactivity of the PYY antibody with pancreatic polypeptide (PP) and neuropeptide Y (NPY) by assay of serial dilutions of PYY-(1–36), PP, and NPY synthetic peptides in human plasma with the same kit and did not find any cross-reactivity between PYY and either PP or NPY as described previously (7). GLP-2 was assayed with an ELISA (Alpco Immunoassays, Inc.). The range of the assay is 1.6 to 378 pmol/l.
The trapezoidal rule was used to calculate integrated responses and was divided by time interval which resulted in a mean concentration for that period. Changes between basal levels, infusion period and recovery period for total GLP-1, active GLP-1, GLP-2 and PYY, and difference for those time intervals between total and active GLP-1 were evaluated using paired t-test, using the Bonferroni adjustment. The same method was used to evaluate the changes in GLP-2 and PYY levels in protocol B.
Results
In protocol A, fasting total GLP-1 levels for the lean group were similar in all three clamps (~12.2±1.4 pmol/l). Fasting levels for the obese groups were also similar for all three clamps and were approximately 5.0±0.7 pmol/l. The infusion of GLP-1(9-36)amide created a square wave and the mean concentration of total GLP-1 during GLP-1(9-36)amide and saline infusions for the 0–60 min period for lean group were 145.5±13.0 and 12.8±1.3 pmol/l respectively. The 60–120 min concentrations were 28.6±2.7 and 13.1±1.3 pmol/l. The 0–60 min mean total GLP-1 concentrations for the obese group during GLP-1(9-36) amide and saline infusion were 160.7±12.5 and 4.3±0.7 pmol/l. The 60–120 min concentrations were 33.7±2.4 and 4.1±0.7 pmol/ l. During the exendin(9-39) and GLP-1(9-36)amide infusion studies the corresponding values for fasting, 0–60 min and 60–120 min for both lean and obese groups were very similar to the levels reported above (Fig 1).
Figure 1.
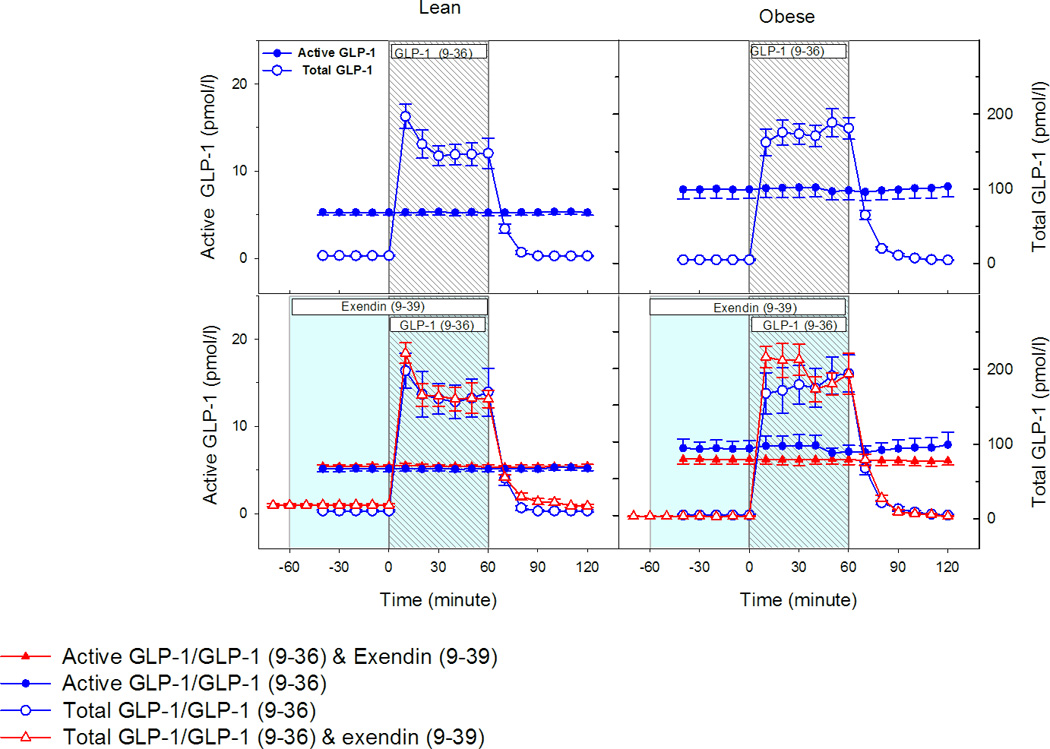
Left panel represents levels from the lean group and right panel from the obese group (N=12 in each group). The top panels display active and total GLP-1 levels during GLP-1 (9-36) amide infusions (N=12 in each group). The bottom panels display active and total GLP-1 levels during GLP-1 (9-36) infusion with and without exendin (9-39) infusion (N=7 in lean and N=6 in obese group).
The baseline values for active GLP-1 in the lean group were approximately 5.28±0.30 pmol/l for all three clamps. It remained unchanged following the infusion of GLP-1(9-36)amide or saline. During the recovery hour no significant changes occurred. In the obese group, the corresponding values for the basal, infusion and recovery period were similar to those of the lean group and remained unchanged. Similar active GLP-1 levels of the lean and the obese groups were observed despite the three fold lower total GLP-1 levels of the obese group. The values for active GLP-1 during GLP-1(9-36)amide and exendin(9-39) infusions were also very similar and did not change in either group.
In protocol B, we measured PYY and GLP-2 for estimation of endogenous GLP-1 release as the antibody to active GLP-1 cannot distinguish endogenously released hormone from exogenously infused hormone. GLP-1 and GLP-2 are colocalized and in some cells GLP-1 and PYY are colocalized; all have similar secretory patterns and reach peak levels at the same time after nutrient stimulation (7). The basal PYY level, the 0–60 minute level and the 60–120 minute level were 17.9±2.8, 16.8±2.6 and 16.3±2.2 pmol/l respectively. The basal GLP-2 level, the 0–60 minute level and the 60–120 minute levels were 72.6±5.3, 66.6±5.4 and 62.5±4.5 pmol/l respectively. PYY and GLP-2 levels remained mostly unchanged from the basal state throughout the study (Figure 2). No effect of exendin(9-39) infusion was observed on basal levels of cosecreted intestinal L-cell peptides (data not shown). Replication of the mild insulinotropic effects of GLP-1(7-36) amide by exogenous insulin infusion also had no effects on the basal levels of co-secreted L-cell peptides (data not shown).
Figure 2.
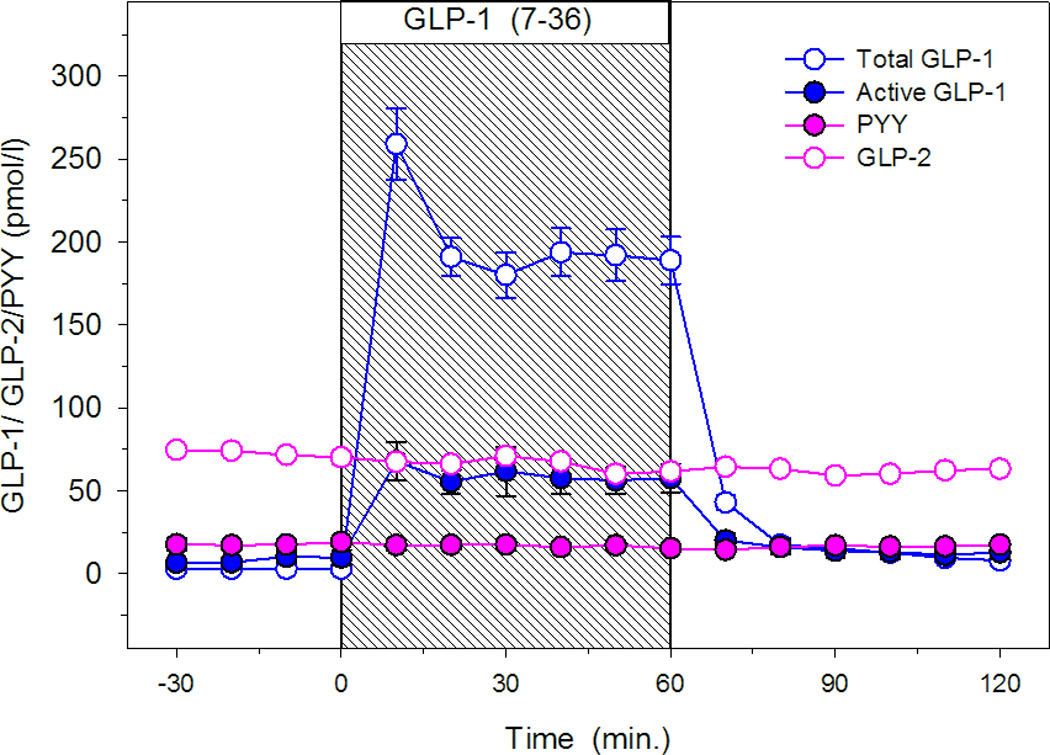
Total and active GLP-1, PYY and GLP-2 levels during GLP-1 (7-36) infusions (N=10).
Discussion
Negative feedback control of basal and stimulated release of peptide hormones by exogenous infusion is well known in many of the endocrine system secretions. It has been shown that GLP-1(7-36)amide secretion could be inhibited by somatostatin in certain species (11–13) Furthermore, GLP-1 infusion has been shown to suppress both GLP-1 and PYY secretion in the post-prandial state (14, 15). However, the magnitude of the suppression was approximately 2 pmol/l. In addition, infusion of the selective GLP-1 receptor antagonist exendin (9-39) has been shown to result in increases in the stimulated level of GLP-1 in patients after Roux-en-Y gastric bypass (16). The presence of negative feedback control in the basal state has not been determined, however. Therefore, the plasma levels of GLP-1(7-36)amide during GLP-1(9-36)amide infusion and PYY and GLP-2 levels during GLP-1(7-36)amide infusion with and without exendin(9-39) infusion were measured to assess this concept.
In the protocols described above, we found no evidence of negative feedback from either exogenous GLP-1(7-36)amide or its metabolite GLP-1(9-36)amide or the blockade of the GLP-1 receptor on the basal secretion of intestinal L cells. When GLP-1 levels are measured with antibodies directed toward the C or the N-terminal of the molecule, it has been demonstrated that approximately 80 % of the circulating levels are the metabolite of the GLP-1(7-36)amide, GLP-1(9-36)amide [see Figure 2] (8). Therefore, it may be argued that the metabolite, the 9-36 moiety, may have a role in the plausible negative feedback control, which we could not confirm. It should be noted that the basal levels of the total GLP-1 in the plasma of the lean group of subjects was approximately three-fold higher than that in the obese group. This difference may be due to a similar three-fold increase in plasma levels of neprilysin in obese compared to lean human subjects (17) Neprilysin has been shown to degrade GLP-1 (18) at several sites (19) and likely would reduce plasma levels of immunoreactive (total) GLP-1 (18) Recently, it has been demonstrated that there is considerable variability in specificity, sensitivity and precision of all measurable isoforms of GLP-1 levels as measured in different commercial assay Kits (20). These investigators report that the Linco (now Millipore) active ELISA GLP-1 kit detect only the active isoform and is capable of recognizing as little as 1 pmol/L differences in GLP-1 (7-36)amide levels.
Our studies have a serious limitation; however, in that we could only assess the effect of exogenous infusions on basal (fasting) hormone release from L cells because the studies were not designed to assess meal-stimulated incretin secretion. Therefore it is possible that negative feedback inhibition of stimulated GLP-1 secretion by GLP-1 may occur. In addition, surrogate markers of intestinal L cell secretion products were assessed during GLP-1 (7-36) infusion in order to infer endogenous GLP-1 secretion, as the GLP-1 antibody used to measure GLP-1 cannot distinguish endogenously produced from exogenously infused GLP-1 levels. Although it is unlikely that the secretion of GLP-1 (7-36) is uncoupled from the co-secretion of PYY and GLP-2 in the basal state, this remains a possibility.
An impetus for undertaking these studies of possible feed-back regulation of GLP-1 secretion by GLP-1(7-36)amide and GLP-1(9-36)amide is the reported finding that in mice the GLP-1 receptor is not expressed in the intestinal L-cells that produce and secrete GLP-1 peptides (21). In addition the receptor is not expressed in the L-cell progenitor cells (21). Yet, GLP-1 receptor agonists stimulate the proliferation of L-cell progenitor cells and increase L-cell mass. These remarkable findings indicate that the effect of GLP-1 on the proliferation of L-cells is indirect via paracrine mechanisms. Since cell proliferative responses would take a much longer time than the one-hour GLP-1 infusions used in our human subjects our failure to observe a modulation of GLP-1 secretion is consistent with the absence of GLP-1 receptors on L-cells. The rationale for analyzing the effects of GLP-1(9-36)amide on the modulation of GLP-1 secretion is based on the observations that the amino-terminal truncated GLP-1(9-36)amide acts via GLP-1 receptor independent mechanisms [9]. Further, as indicated above, GLP-1(9-36)amide is the major form of GLP-1 in the circulation, comprising ~80% of the total immunoreactive GLP-1 in plasma (Figure 2). Our finding of an absence of acute (one hour) feed-back modulation of GLP-1 secretion by either GLP-1(7-36)amide or GLP-1(9-36)amide suggests a lack of direct GLP-1 receptor-mediated or non receptor-mediated secretory responses of L-cells.
There are many theoretical possibilities that could explain how DPP-4 inhibition results in partial inhibition of K and L cell products. Both GIP and GLP-1 lead to insulin secretion and it is possible that increased insulin secretion after DPP-4 inhibition leads to inhibition of the stimulated secretion of incretins. We found no evidence of an insulin-mediated effect on basal GLP-1 release. Postcibal incretin secretion is potentially affected by a variety of factors, including changing circulating levels of the absorbed nutrients. For this reason, we used the euglycemic glucose clamp method to control for changing levels of glucose in our studies. In addition to changing glucose and insulin levels, other meal-associated events in gastrointestinal and pancreatic function may affect changes in the secretion of enteric hormones. For example, altered enteric hormone release would be predicted to be reduced if gastric emptying were impeded by a DPP-4 inhibitor. Although GLP-1 and exendin-4 (exenatide) have been shown to reduce gastric emptying in clinical and laboratory studies (22, 23), Vella et al have shown that DPP-4 inhibition is not associated with changes in gastric emptying (24). There is evidence that vagal mechanisms are involved in mediating incretin secretion. Vagotomy results in diminished incretin secretion (25), and it is theoretically possible that DPP-4 inhibitors influence the active levels of neurotransmitters such as vasoactive intestinal peptide (VIP) and pituitary adenylate cyclase-activating polypeptide (PACAP) that modulate incretin secretion. Finally it is possible that GIP, either directly or indirectly, may exert negative feedback control on GLP-1 secretion. During DPP-4 inhibition, GLP-1 secretion is impacted far more (72% reduction in secretion) than is GIP secretion. A recent study using exogenous GIP (20 ng/kg/min) infusion for 3 hours in type 2 diabetes subjects showed that meal-stimulated GLP-1 secretion was diminished for several hours even after termination of the infusion (26). As plasma GIP levels are elevated during DPP 4 inhibition in both fasting and postprandial states, it is possible that elevated GIP levels are responsible for the meal-related diminution of GLP-1 secretion.
Conclusions
The negative feedback control system as described by inhibition of the release of endogenous hormone while infusing it exogenously was not observed for the basal secretion of GLP-1 (7-36) amide. Further studies are necessary to identify the effectors of the mediation of GLP-1 levels after nutrient ingestion or drug administration.
Acknowledgments
Our appreciation is extended to the men and women who participated in this study. We thank Melissa Scudder for assistance in preparation of this article. This work has been supported in part by the National Institutes of Health (NIH) Grants: AG 00599, AG 623175, DK 30834 and by the Intramural Research Program of NIA. We gratefully acknowledge the support of the Alan McGavin Geriatric Medicine Endowment of the University of British Columbia.
Footnotes
Duality of Interests: Nothing to declare.
References
- 1.Elahi D, Hershlopf RJ, Muller DC, Tobin JD, Blix PM, Rubenstein AH, Unger RH, Andres R. Feedback inhibition of insulin secretion by insulin: relation to the hyperinsulinemia of obesity. N Eng J Med. 1982;308:1196–1202. doi: 10.1056/NEJM198205203062002. [DOI] [PubMed] [Google Scholar]
- 2.Brunicardi FC, Gingerich RL, Andersen DK. Selective effects of arterial insulin on islet cell secretion in the isolated perfused human pancreas. Surg Forum. 1990;41:198–200. [Google Scholar]
- 3.Drucker D. Glucagon-like peptides. Diabetes. 1998;47:159–169. doi: 10.2337/diab.47.2.159. [DOI] [PubMed] [Google Scholar]
- 4.Nauck MA, Heimesaat MM, Behle K, Holst JJ, Nauck MS, Ritzel R, Hufner M, Schmiegel WH. Effects of glucagon-like peptide 1 on counterregulatory hormone responses, cognitive functions, and insulin secretion during hyperinsulinemic, stepped hypoglycemic clamp experiments in healthy volunteers. J Clin Endocrinol Metab. 2002;87:1239–1246. doi: 10.1210/jcem.87.3.8355. [DOI] [PubMed] [Google Scholar]
- 5.Herman GA, Bergman A, Stevens C, Kotey P, Yi B, Zhao P, Dietrich B, Golor G, Schrodter A, Keymeulen B, Lasseter KC, Kipnes MS, Snyder K, Hilliard D, Tanen M, Cilissen C, De Smet M, de Lepeleire I, Van Dyck K, Wang AQ, Zeng W, Davies MJ, Tanaka W, Holst JJ, Deacon CF, Gottesdiener KM, Wagner JA. Effect of single oral doses of sitagliptin, a dipeptidyl peptidase-4 inhibitor, on incretin and plasma glucose levels after an oral glucose tolerance test in patients with type 2 diabetes. J Clin Endocrinol Metab. 2006;91:4612–4619. doi: 10.1210/jc.2006-1009. [DOI] [PubMed] [Google Scholar]
- 6.FDA. FDA Approves New Drug Treatment for Type 2 Diabetes. U.S. Department of Health & Human Services; 2009. [Google Scholar]
- 7.Kim BJ, Carlson OD, Jang HJ, Elahi D, Berry C, Egan JM. Peptide YY is secreted after oral glucose administration in a gender-specific manner. J Clin Endocrinol Metab. 2005;90:6665–6671. doi: 10.1210/jc.2005-0409. [DOI] [PubMed] [Google Scholar]
- 8.Egan JM, Meneilly GS, Habener JF, Elahi D. Glucagon-like peptide-1 augments insulin-mediated glucose uptake in the obese state. J Clin Endocrinol Metab. 2002;87:3768–3773. doi: 10.1210/jcem.87.8.8743. [DOI] [PubMed] [Google Scholar]
- 9.Elahi D, Egan JM, Shannon RP, Meneilly GS, Khatri A, Habener JF, Andersen DK. GLP-1 (9–36) Amide, Cleavage Product of GLP-1 (7–36) Amide, Is a Glucoregulatory Peptide. Obesity (Silver Spring) 2008;16:1501–1509. doi: 10.1038/oby.2008.229. [DOI] [PubMed] [Google Scholar]
- 10.Egan JM, Clocquet AR, Elahi D. The insulinotropic effect of acute exendin-4 administered to humans: comparison of nondiabetic state to type 2 diabetes. J Clin Endocrinol Metab. 2002;87:1282–1290. doi: 10.1210/jcem.87.3.8337. [DOI] [PubMed] [Google Scholar]
- 11.Hansen L, Hartmann B, Bisgaard T, Mineo H, Jorgensen PN, Holst JJ. Somatostatin restrains the secretion of glucagon-like peptide-1 and -2 from isolated perfused porcine ileum. Am J Physiol Endocrinol Metab. 2000;278:E1010–E1018. doi: 10.1152/ajpendo.2000.278.6.E1010. [DOI] [PubMed] [Google Scholar]
- 12.Marco J, Hedo JA, Villaneuva ML. Inhibition of intestinal glucagon-like immunoreactivity (GLI) secretion by somatostatin in man. J Clin Endocrinol Metab. 1977;44:695–698. doi: 10.1210/jcem-44-4-695. [DOI] [PubMed] [Google Scholar]
- 13.Martin PA, Faulkner A. Effects of somatostatin-28 on circulating concentrations of insulin and gut hormones in sheep. J Endocrinol. 1996;151:107–112. doi: 10.1677/joe.0.1510107. [DOI] [PubMed] [Google Scholar]
- 14.Naslund E, Bogefors J, Skogar S, Gryback P, Jacobsson H, Holst JJ, Hellstrom PM. GLP-1 slows solid gastric emptying and inhibits insulin, glucagon, and PYY release in humans. Am J Physiol. 1999;277:R910–R916. doi: 10.1152/ajpregu.1999.277.3.R910. [DOI] [PubMed] [Google Scholar]
- 15.Toft-Nielsen M-B, Masbad S, Holst JJ. Continuous subcutaneous infusion of glucagon-like peptide 1 lowers plasma glucose and reduces appetite in type 2 diabetic patients. Diabetes Care. 1999;22:1137–1143. doi: 10.2337/diacare.22.7.1137. [DOI] [PubMed] [Google Scholar]
- 16.Salehi M, Prigeon RL, D'Alessio DA. Gastric bypass surgery enhances glucagon-like peptide-1-stimulated postprandial insulin secretion in humans. Diabetes. 2011;60:2308–2314. doi: 10.2337/db11-0203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Standeven KF, Hess K, Carter AM, Rice GI, Cordell PA, Balmforth AJ, Lu B, Scott DJ, Turner AJ, Hooper NM, Grant PJ. Neprilysin, obesity and the metabolic syndrome. Int J Obes. 2011;35:1031–1010. doi: 10.1038/ijo.2010.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Palmboeck A, Holst JJ, Carr RD, Deacon CF. Neutral endopeptidase 24.11 and dipeptidyl peptidase IV are both mediators of the degradation of glucagon-like peptide 1 in the anaesthetised pig. Diabetologia. 2005;48:1882–1890. doi: 10.1007/s00125-005-1847-7. [DOI] [PubMed] [Google Scholar]
- 19.Hupe-Sodmann K, McGregor GP, Bridenbaugh R, Göke R, Göke B, Thole H, Zimmermann B, Voigt K. Characterisation of the processing by human neutral endopeptidase 24.11 of GLP-1(7–36) amide and comparison of the substrate specificity of the enzyme for other glucagon-like peptides. Regul Pept. 1995;58:149–56. doi: 10.1016/0167-0115(95)00063-h. [DOI] [PubMed] [Google Scholar]
- 20.Bak MJ, Wewer Albrechtsen NJ, Pedersen J, Knop FK, Vilsbøll T, Jørgensen NB, Hartmann B, Deacon, Dragsted LO, Holst JJ. Specificity and sensitivity of commercially available assays for glucagon-like peptide-1 (GLP-1): implications for GLP-1 measurements in clinical studies. Diabetes Obes Metab. 2014;16:1155–1164. doi: 10.1111/dom.12352. [DOI] [PubMed] [Google Scholar]
- 21.Grigoryan M, Kedees MH, Charron MJ, Guz Y, Teitelman G. 076-88Regulation of mouse intestinal L cell progenitors proliferation by the glucagon family of peptides. Endocrinology. 2012;153:3. doi: 10.1210/en.2012-1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Salehi M, Vahl TP, D'Alessio DA. Regulation of islet hormone release and gastric emptying by endogenous glucagon-like peptide 1 after glucose ingestion. J Clin Endocrinol Metab. 2008;93:4909–4916. doi: 10.1210/jc.2008-0605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Deane AM, Nguyen NQ, Stevens JE, Fraser RJ, Holloway RH, Besanko LK, Burgstad C, Jones KL, Chapman MJ, Rayner CK, Horowitz M. Endogenous glucagon-like peptide-1 slows gastric emptying in healthy subjects, attenuating postprandial glycemia. J Clin Endocrinol Metab. 95:215–221. doi: 10.1210/jc.2009-1503. [DOI] [PubMed] [Google Scholar]
- 24.Vella A, Bock G, Giesler PD, Burton DB, Serra DB, Saylan ML, Dunning BE, Foley JE, Rizza RA, Camilleri M. Effects of dipeptidyl peptidase-4 inhibition on gastrointestinal function, meal appearance, and glucose metabolism in type 2 diabetes. Diabetes. 2007;56:1475–1480. doi: 10.2337/db07-0136. [DOI] [PubMed] [Google Scholar]
- 25.Rocca AS, Brubaker PL. Role of the vagus nerve in mediating proximal nutrient-induced glucagon-like peptide-1 secretion. Endocrinology. 1999;140:1687–1694. doi: 10.1210/endo.140.4.6643. [DOI] [PubMed] [Google Scholar]
- 26.Chia CW, Carlson OD, Kim W, Shin YK, Charles CP, Kim HS, Melvin DL, Egan JM. Exogenous glucose-dependent insulinotropic polypeptide worsens post prandial hyperglycemia in type 2 diabetes. Diabetes. 2009;58:1342–1349. doi: 10.2337/db08-0958. [DOI] [PMC free article] [PubMed] [Google Scholar]