

Abstract
Tissue-specific enhancers are critical for gene regulation. In this study, we help elucidate the contribution of muscle-associated differential DNA methylation to the enhancer activity of highly muscle-specific genes. By bioinformatic analysis of 44 muscle-associated genes, we show that preferential gene expression in skeletal muscle (SkM) correlates with SkM-specific intragenic and intergenic enhancer chromatin and overlapping foci of DNA hypomethylation. Some genes, e.g., CASQ1 and FBXO32, displayed broad regions of both SkM- and heart-specific enhancer chromatin but exhibited focal SkM-specific DNA hypomethylation. Half of the genes had SkM-specific super-enhancers. In contrast to simple enhancer/gene-expression correlations, a super-enhancer was associated with the myogenic MYOD1 gene in both SkM and myoblasts even though SkM has < 1 percent as much MYOD1 expression. Local chromatin differences in this super-enhancer probably contribute to the SkM/myoblast differential expression. Transfection assays confirmed the tissue-specificity of the 0.3-kb core enhancer within MYOD1’s super-enhancer and demonstrated its repression by methylation of its three CG dinucleotides. Our study suggests that DNA hypomethylation increases enhancer tissue-specificity and that SkM super-enhancers sometimes are poised for physiologically important, rapid up-regulation.
Keywords: DNA methylation, enhancers, promoters, skeletal muscle, heart, MYOD, FBXO32, CASQ1, HOXC gene cluster, PRKAG3, development
Introduction
Enhancers are cis-acting stimulators of transcription that can act at a distance from promoters. They play a critical role in establishing tissue-specific gene expression, and thereby, directing development [1-3]. They often have evolutionarily conserved DNA sequences, including transcription factor binding sites (TFBS), indicating their functional importance [4]. Enhancers may also modulate transcription elongation or differential generation of RNA isoforms [5,6]. Unlike older concepts of mammalian enhancers as mostly being located just upstream or downstream of genes, it is now clear that as many as half of them are within the gene body [7]. Another major new insight into enhancers is that they are often transcribed bidirectionally to give low amounts of small noncoding (nc) RNAs, whose expression is linked to enhancer activity [1,8-10]. Enhancers are implicated not only as major drivers of differentiation and maintenance of tissue-specific differences in transcription, but also as regulators of differential gene expression in response to certain physiological changes [11] and as drivers of disease, including cancer [12].
Enhancers in genome-wide studies are usually identified by characteristic histone modifications [3,13,14]. The most frequently examined modifications are histone H3 lysine-4 monomethylation (H3K4me1) and H3K27 acetylation (H3K27ac), which together are associated with active enhancer chromatin (EnhChr); H3K4me3, which in combination with H3K27ac denotes active promoter-type chromatin; H3K36me3, which in the 3’ half of the gene body indicates transcriptional activity; and H3K27me3 or H3K9me3, which generally designates repressed chromatin. Tests with reporter gene constructs in transfection assays have demonstrated that most EnhChr regions identified by epigenetic marks have enhancer activity [7]. Weak or poised enhancers or promoters can also be identified by histone modifications (H3K4 methylation in the absence of H3K27 acetylation) [7,14].
DNA hypomethylation, which refers to the replacement of some of the 5-methylcytosine (5mC) residues in DNA with unmodified cytosine residues, is implicated in contributing to the generation, activity, or maintenance of many enhancers [15-18]. Typically, high levels of 5mC throughout a promoter or enhancer region are associated with repression of transcription. DNA from mammalian tissues usually contains a very small percentage of 5mC residues in which the methyl group had been oxidized in a genetically programmed enzymatic reaction to a hydroxymethyl group yielding 5-hydroxymethylcytosine (5hmC) residues. Unlike 5mC residues in DNA, enrichment in genomic 5hmC has been positively associated with poised or active enhancers [19,20].
In the present study we analyze the relationship of epigenetics to tissue-specific gene expression focusing on a comparison of skeletal muscle (SkM) to various other human tissues, including heart (cardiac muscle), and brain, and to myoblasts and myotubes (untransformed SkM progenitor cell cultures). We selected a set of genes that has much higher steady-state levels of RNA in SkM than in non-muscle tissues and high absolute levels of expression in SkM or myoblasts to determine if highly selective expression in the SkM lineage is tightly associated with intragenic and/or intergenic SkM-specific EnhChr as well as SkM-specific DNA hypomethylation. In addition, using a 0.3-kb SkM-specific enhancer element that regulates expression of the myogenic MYOD1 transcription factor-encoding gene [21], we assessed the effect of targeted DNA methylation on enhancer activity in reporter gene assays.
Methods
Bioinformatics
Databases with epigenetic and RNA-seq profiles used for the analyses are available at the UCSC Genome Browser [22]. From the ENCODE project [23] we used the following UCSC Genome Browser tracks: DNaseI hypersensitivity profiling, Open Chromatin, DNaseI HS, Duke University [24]; RNA-seq (for tissues; not strand-specific), Massachusetts Institute of Technology [25], and the associated tabular database [26]; and Transcription Levels by Long RNA-seq for poly(A)+ whole-cell RNA by strand-specific analysis on > 200 nt poly(A)+ RNA (for various cell cultures), Cold Spring Harbor Laboratories. For visualizing RNA-seq tracks in the UCSC Genome Browser in figures, the vertical viewing ranges were 0 to 30 for cultured cells and 0 to 2 for tissues.
From the UCSC Genome Browser Track Data Hubs, we used a hub for DNA Methylation, Methylomes from Bisulfite Sequencing Data [3], with data analysis by Song et al. [27]. Another hub was used for Roadmap Epigenomics chromatin state segmentation analysis (chromHMM, AuxilliaryHMM) [3,28]. The color code for chromatin state segmentation in the figures was slightly simplified from the original [28] as shown in the color keys in the figures.
For quantification of RNA-seq data from myoblasts, we used ENCODE tracks for Transcription Levels by RNA-seq, non-strand-specific, on > 200 nt poly(A)+ RNA, California Institute of Technology [22], and the Cufflinks CuffDiff tool [29]. To determine preferential gene expression in myoblasts vs. many non-muscle cell cultures, our previously described results from microarray expression analysis were used [30]. For identifying super-enhancers, unless otherwise specified, the dbSUPER [31] database was used.
The same psoas SkM sample had been used for chromatin state segmentation analysis and bisulfite-seq, namely, a mixture of tissues from a 3 y male and a 34 y male [3]. For DNaseI-hypersensitivity profiling, the SkM sample was a mixture of psoas muscle from five individuals (male and one female) aged 22 to 35 [24]. Another SkM sample had been used for bisulfite-seq (one 72 y female; the type of SkM tissue unidentified) [3], and pooled SkM samples had been used for RNA-seq (multiple SkM tissues not classified as to type, age, or gender) [26]. The myoblasts used for the epigenetic and transcriptome profiles had been derived from the minor fraction of muscle satellite cells in post-natal SkM biopsy samples and represent activated satellite cells of the type used to repair muscle. For identification of potential MYOD binding sites, orthologous sequences to murine C2C12 Mb and Mt binding sites from MyoD ChIP-seq [32] were mapped in the human genome.
DNA Constructs, Transfection, and in vitro DNA Methylation
By fusion PCR, a 386-bp fragment containing the MYOD1 core enhancer was cloned into the vector pCpGfree-promoter-Lucia (InvivoGen), which has a Lucia luciferase gene. The insert for cloning was obtained by PCR on mixed human brain and placenta DNAs using the following primers (lower-case letters are the extensions that were used for fusion cloning; NEBuilder HiFi Assembly Kit, New England Biolabs): ctctacaaatgtggtatgCTACTTGGTAGGTGAGGAG and ggtgaacatattgactgGTGAGAAGCAGGACTCCAG. Before fusion cloning, the plasmid was linearized by reverse PCR using primers for a sequence downstream of the reporter gene’s polyadenylation signal. Transfection into C2C12 or MCF-7 cells utilized a lipid-based reagent (Fast-forward protocol, Effectene reagent, Qiagen). As a reference plasmid for normalizing the transfection efficiency, pCMV-CLuc 2 (New England Biolabs) encoding Cypridina luciferase was co-transfected. Cypridina and Lucia luciferase activity were independently quantified by bioluminescence from cell supernatant aliquots (BioLux Cypridina Luciferase assay kit, New England Biolabs; Quanti-Luc, InvivoGen) harvested 48 or 72 h after transfection. The plasmids were methylated at only the enhancer insert by incubating 1 μg with 4 units of SssI methylase and 160 μM S-adenosylmethionine (New England Biolabs) for 4 h at 37ºC. A mock-methylated control used identical incubation conditions except for the absence of S-adenosylmethionine. As a check for the degree of methylation, another plasmid construct that contained three CGCG sites was similarly methylated and shown thereafter to be fully resistant to BstUI cleavage.
Results
Skeletal Muscle-associated Genes have Skeletal Muscle-specific Intragenic/intergenic Enhancers and Often Super-enhancers
To examine the epigenetics of SkM-associated genes, we first identified 30 genes that are expressed highly and preferentially in SkM compared with non-muscle tissues. From RNA-seq data [26], these genes have an absolute RPKM in SkM of > 50 and RPKM ratios for SkM to lung of > 50 and SkM to heart of > 5 (Table 1). An additional 14 SkM lineage-associated genes were chosen based upon preferential expression in myoblasts vs. non-muscle cell cultures and also in SkM vs. non-muscle tissues. These genes have ≥ 4 times as much signal from myoblasts relative to the average signal from 35 diverse non-muscle cell cultures in expression microarray analyses [30] and RPKM ratios for SkM to lung of > 50 and SkM to heart of > 5 but absolute SkM RPKM values of < 50 (Table 2). We then examined chromatin in and around these genes using publicly available whole-genome profiles [22,28] that indicate active, weak, or poised enhancers or promoters in SkM (psoas and an undefined SkM sample), heart (separate left and right ventricle samples), peripheral blood mononuclear cells (PBMC), spleen, lung, liver, and brain (various subregions). These chromatin state predictions are based upon algorithms using characteristic histone modification profiles (H3K4me1, H3K4me3, H3K27ac, H3K27me3, H3K9me3, H3K36me3) previously determined by ChIP-seq [3,14].
Table 1. Expression Levels and Summary of Enhancer and Promoter Epigenetics for 30 Genes Highly and Preferentially Expressed in Skeletal Muscle.
| RNA in SkM or Mb (RPKM or FPKM) | Ratio of SkM RPKM to RPKM of lung, brn or hrt | Presence of SkM-only Enh or Enh specific to SkM and hrt or brn | SkM-only DNA hypomethylation | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene symbol | Type of SkM function | RNA isoform | SkM | Mb | SkM/lung | SkM/brn | SkM/hrt | Intragenic Enha | Intergenic Enha | Super enhancer | At SkM-only Enh (broad or focal hypometh; SkM-only DHS overlap) | At Pr region (CGI overlap) | At regions other than Pr or SkM-only Enh |
| MYBPC1 | Contraction | NM_206820 | 2159 | 0.2 | 42343 | 658 | 11426 | SkM | SkM | SkM | y (broad) | y | y |
| TPM2 | Contraction | NM_003289 | 1669 | 234 | 52 | 90 | 11 | SkM, hrt | SkM | SkM | y (focal) | n (CGI) | yc |
| NEB | Structural | NM_001164508 | 1461 | 4.0 | 6581 | 3699 | 1022 | SkM | SkM | SkM | y (focal) | y | y |
| TNNC2 | Contraction | NM_003279 | 1084 | 54 | 2905 | 1218 | no txn hrt | SkM | SkM | SkM | y (focal; DHS) | n | y |
| TNNI2b | Contraction | NM_001145829 | 896 | 12 | 617 | 17566 | 5933 | SkM | n | n | y (focal; DHS) | y | y |
| KLHL41 | Myofibrils | NM_006063 | 688 | 103 | 1464 | 1084 | 29 | SkM, hrt | SkM | SkM | y (focal) | y | yc |
| TNNT1 | Contraction | NM_001126133 | 447 | 95 | no txn lung | 272 | 322 | SkM, hrt | n | n | n | y | y |
| MYH2b | Contraction | NM_017534 | 405 | 9.8 | 7233 | 67511 | 6984 | SkM | SkM | SkM | y (focal) | y | y |
| CKM | Signalling | NM_001824 | 343 | 53 | 6348 | no txn brn | 6 | SkM, hrt | SkM | SkM | y (focal) | n (CGI) | yc |
| ENO3 | Metabolism | NM_053013 | 318 | 37 | 796 | 1133 | 7 | SkM, hrt | SkM | n | n | y | yc |
| TNNT3b | Contraction | NM_001042782 | 286 | 10 | 855 | 1272 | 187 | SkM, hrt | SkM | SkM, hrt | y (focal) | n | yc |
| MYH1b | Contraction | NM_005963 | 225 | 12 | 5930 | no txn brn | 5634 | SkM | n | n | n | y | y |
| MYOZ1 | Signalling | NM_021245 | 220 | 0.6 | 1091 | 1118 | 396 | SkM | SkM | n | y (focal; DHS) | y | n |
| MYLPF | Signalling | NM_013292 | 183 | 345 | no txn lung | no txn brn | no txn hrt | SkM, brn | n | SkM | y (focal) | n | y |
| FBXO32 | Stabilization | NM_058229 | 179 | 9.9 | 58 | 20 | 8 | SkM, hrt, brn | SkM | n | y (focal) | n (CGI) | yc |
| SLN | Contraction | NM_003063 | 178 | 6.6 | 2310 | 180 | 9 | SkM | SkM | SkM | y (focal; DHS) | y | n |
| ATP2A1 | Signalling | NM_173201 | 131 | 7.4 | 2015 | 1505 | 26193 | SkM | SkM | n | y (focal; DHS) | y | n |
| PYGM | Metabolism | NM_001164716 | 127 | 0.3 | 2157 | 98 | 43 | SkM | SkM, hrt, brn | SkM | y (focal) | y | yc |
| SMTNL1 | Contraction | NM_001105565 | 107 | 0.1 | 280 | 976 | 593 | SkM | SkM | n | y (focal) | y | n |
| ATP1A2b | Signalling | NM_000702 | 101 | 7.9 | 114 | 1.3 | 6 | SkM, hrt, brn | SkM, hrt, brn | SkM, hrt | n | n | yc |
| MYOT | Structural | NM_006790 | 96 | 0.5 | 1278 | 134 | 15 | SkM, hrt, brn | SkM, hrt, brn | SkM, brn | n | y | yc |
| AMPD1 | Signalling | NM_000036 | 95 | 0.0 | 868 | no txn brn | no txn hrt | SkM | SkM | SkM | y (focal) | y | n |
| MYL1 | Signalling | NM_079422 | 85 | 64 | no txn lung | no txn brn | no txn hrt | SkM | SkM | n | n | n | y |
| RYR1 | Contraction | NM_001042723 | 78 | 8.8 | 1184 | 90 | 797 | SkM | SkM | n | y (focal) | n (CGI) | y |
| PDLIM3 | Structural | NM_014476 | 75 | 121 | 77 | 119 | 8 | SkM | SkM, hrt | SkM | y (focal) | n (CGI) | yc |
| TMOD4 | Structural | NM_013353 | 73 | 0.1 | no txn lung | 853 | 2620 | SkM | n | n | y (focal) | y | n |
| CASQ1b | Signalling | NM_001231 | 63 | 0.5 | 1462 | 58 | 24 | SkM, hrt | SkM | n | y (focal; DHS) | n | yc |
| DDIT4L | Signalling | NM_145244 | 61 | 0.2 | 266 | 87 | 207 | n | SkM | n | y (focal) | n (CGI) | y |
| STAC3 | Contraction | NM_145064 | 60 | 50 | 697 | 869 | 405 | SkM | SkM | n | y (focal) | y | y |
| MYF6b | TF | NM_002469 | 59 | 24 | no txn lung | no txn brn | no txn hrt | n | SkM | SkM | y (focal) | y | y |
aAll genes with SkM, hrt or SkM, brn designations for intragenic or intergenic Enh had at least one SkM-only Enh region in addition to a shared SkM- and heart- or brain-specific Enh region
bGenes from this table or Table 2 that are neighbors in the genome: TNNI2 & TNNT3; MYH2 & MYH1; ATP1A2 & CASQ1; MYF6 & MYF5
cThe designated genes displayed SkM-only DNA hypomethylation in Enh regions shared specifically by SkM and heart
Table 2. Expression Levels and Summary of Enhancer and Promoter Epigenetics for 14 Genes Preferentially Expressed in Skeletal Muscle and Myoblasts.
| RNA in SkM or Mb (RPKM or FPKM) | Ratio of SkM RPKM to RPKM of lung, brn or hrt | Presence of SkM-only Enh or Enh specific to SkM and hrt or brn | SkM-only DNA hypomethylation | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene symbol | Type of SkM function | RNA isoform | SkM | Mb | SkM/lung | SkM/brn | SkM/hrt | Intragenic Enha | Intergenic Enha | Super enhancer | At SkM-only Enh (broad or focal hypometh; SkM-only DHS overlap) | At Pr region (CGI overlap) | At regions other than Pr or SkM-only Enh |
| CAP2 | TF | NM_006366 | 48 | 43 | 103 | 2.1 | 5.1 | SkM, hrt | SkM | SkM | y (focal) | y | yb |
| TNNI1 | Contraction | NM_003281 | 45 | 208 | 1222 | no txn brn | 143 | SkM, hrt | SkM, hrt | SkM | y (focal; DHS) | y | yb |
| CHRNA1 | Signalling | NM_000079 | 21 | 218 | 91 | 90 | 130 | SkM | SkM | n | y (focal; DHS) | y | y |
| HOXC10 | TF | NM_017409 | 18 | 17 | no txn lung | no txn brn | no txn hrt | SkM | SkM | SkM | constit. low meth. | n (CGI) | n |
| SGCA | Structural | NM_001135697 | 12 | 71 | 57 | 339 | 5.1 | SkM, hrt | SkM, hrt | n | n | y | yb |
| ASB5 | Signalling | NM_080874 | 10 | 147 | 1001 | 770 | 113 | SkM, hrt | SkM | SkM | y (broad) | y | y |
| IP6K3 | Signalling | NM_001142883 | 6.6 | 14 | 60 | 45 | 7.6 | SkM, hrt | SkM, hrt | SkM | y (broad) | y | yb |
| CHRND | Signalling | NM_000751 | 3.1 | 5.7 | 97 | no txn brn | no txn hrt | SkM | SkM | n | y (focal) | n | n |
| PITX2 | TF | NM_153427 | 2.9 | 17 | no txn lung | 11 | no txn hrt | SkM | n | SkM | n | n (CGI) | y |
| SIM1 | TF | NM_005068 | 2.1 | 1.1 | 102 | 11 | no txn hrt | SkM | SkM | n | y (focal) | n (CGI) | n |
| PRKAG3 | Metabolism | NM_017431 | 0.3 | 4.7 | no txn lung | no txn brn | no txn hrt | SkM | SkM | SkM | y (focal; DHS) | y | n |
| CDH15c | Signalling | NM_004933 | 0.2 | 139 | no txn lung | no txn brn | no txn hrt | SkM | n | n | y (broad) | n (CGI) | n |
| MYOD1 | TF | NM_002478 | 0.1 | 82 | no txn lung | no txn brn | no txn hrt | n | SkM | SkM | y (broad; DHS) | n (CGI) | n |
| MYF5 | TF | NM_005593 | 0.1 | 134 | no txn lung | no txn brn | no txn hrt | n | SkM | SkM | y (broad) | n (CGI) | n |
Abbreviations for Tables 1 and 2: SkM, skeletal muscle; Mb, myoblasts,hrt, heart; FPKM or RPKM, analogous terms for fragments or reads, respectively, per kilobase of transcript per million mapped reads in RNA-seq for the Mb [23] and tissue [26] RNA-seq databases, respectively; Enh, active enhancer-like chromatin enriched in H3K27ac and H3K4me1; SkM-only DNA hypomethylation at SkM-only Enh, DNA hypomethylated subregion in an Enh seen in SkM but not other examined tissues and classified as either broad or focal; SkM-only DHS overlap, SkM-specific DNaseI hypersensitive site overlapping SkM-only DNA hypomethylation; Pr, promoter region, 1 kb upstream or downstream of the 5' end of the gene; CGI, CpG island; n, no; y, yes; no txn lung, no txn brn, or no txn heart, no detectable transcription in lung, brain, or heart, respectively; TF, transcription factor; constit. low meth., region that had low methylation in all studied tissues. Bn, brain: for RNA-seq the brain sample was hypothalmus and for the enhancer chromatin determinations it was prefrontal cortex, hippocampus, anterior caudate, and cingulate gyrus (all of which gave similar results). Hypothalmus data were not available for enhancer chromatin determinations.
aAll genes with SkM, hrt or SkM, brn designations for intragenic or intergenic Enh had at least one SkM-only Enh region in addition to shared SkM- and heart- or brain-specific Enh
bThe designated genes displayed SkM-only DNA hypomethylation in Enh regions shared specifically by SkM and heart
cBrain refers to hypothalmus, which has no detectable expression of CDH15 although cerebellum has high expression
As shown in Tables 1 and 2, all 44 genes had at least one SkM-specific EnhChr region (SkM-only EnhChr) that was intragenic (in the gene body more than 1 kb downstream of the transcription start site, TSS) or intergenic (between the studied gene and its nearest upstream and downstream RefSeq neighbors and more than 1 kb upstream from the gene’s TSS). Even genes with preferential expression in SkM but at very low absolute expression levels (up to10,000 times lower than for other examined genes; Table 2 vs. Table 1) displayed some SkM-only EnhChr. The amounts of SkM-only EnhChr were often unusually large (3 to 18 kb) and present in both intragenic and intergenic portions of the genome. Seventeen genes also displayed regions of SkM- and heart-specific EnhChr. These genes were expressed at significantly higher levels in heart (p < 0.00001, t-test comparing the log means for the two groups) than were genes that did not have SkM- and heart-specific EnhChr (Figure 1). As expected for the 44 SkM genes, active promoter chromatin was almost always observed at or adjacent to the first transcribed exon in SkM, and often there was intragenic EnhChr adjacent to the promoter.
Figure 1.
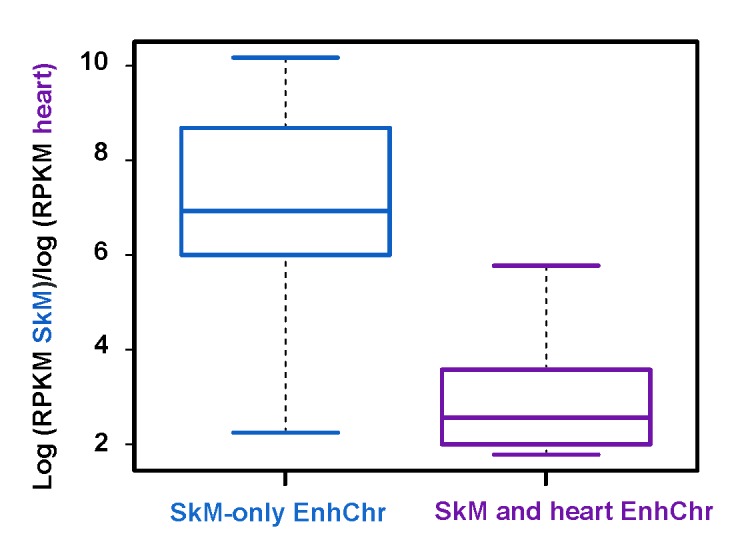
Overlap of heart enhancer chromatin with SkM enhancer chromatin is associated with significantly higher expression in heart. Boxplots displaying the distribution of log ratios for RNA-seq RPKM for skeletal muscle relative to heart for genes with or without tissue-specific heart enhancer chromatin overlapping SkM-associated enhancer chromatin.
Many of the observed EnhChr segments are part of super-enhancers (Tables 1 and 2), defined as broad clusters of similarly tissue-specific enhancers associated with a gene [8,33]. These super-enhancers were identified using a database [31] for many human tissues (including SkM) that detected large clusters of high H3K27ac signal associated with a given gene. Twenty-two of the genes overlapped super-enhancers observed only in SkM, and three had super-enhancers specifically in SkM and heart or brain (Tables 1 and 2). Eight of the genes linked to super-enhancers were found to be located on the genome adjacent to or only one gene removed from another gene in the set of 44 SkM genes (TNNI and TNNT3; MYH2 and MYH1; ATP1A2 and CASQ1; MYF6 and MYF5).
Skeletal Muscle-specific DNA Hypomethylation was Often Embedded in Subregions of Enhancer Chromatin
We looked for subregions in the SkM-associated EnhChr that displayed DNA hypomethylation only in SkM (SkM-only hypomethylation). They were identified as low DNA methylation regions (LMRs) in SkM that did not overlap LMRs from the other above-mentioned tissues (SkM-only LMRs) [22]. The LMRs had been mapped as DNA regions with significantly less methylation than in the rest of the given genome as assessed by analysis of that sample’s bisulfite-seq methylome profile [27]. Some of the SkM-only LMRs could be confirmed using data from our previous methylome study identifying regions of significant SkM-specific DNA hypomethylation from reduced representation bisulfite sequencing (RRBS) profiles of two SkM samples and 14 types of non-muscle tissues [34]. However, RRBS profiles cover only ~5 percent of the CG dinucleotides in the human genome [35], and so RRBS was uninformative for most of the SkM-only LMRs determined from bisulfite-seq profiles.
Thirty-five of the 44 genes displayed SkM-only LMRs over part of their SkM-only EnhChr. These LMRs usually overlapped only a minor portion of the EnhChr and are listed in Tables 1 and 2 as regions of focal hypomethylation. However, in six genes that had EnhChr regions of > 3 kb, more than half of the DNA in the EnhChr was hypomethylated (Tables 1 and 2, listed as regions with broad hypomethylation). The length of SkM-only LMRs that overlapped a contiguous segment of SkM-only EnhChr varied from 0.2 kb (TNNI2) to 18 kb (MYBPC1) with a median length of 2.8 kb. For the 17 genes that exhibited overlapping SkM- and heart-specific EnhChr, all but two (TNNT1 and ASB5) had SkM-only DNA hypomethylation in a subregion of this EnhChr (Tables 1 and 2, last column). Only one of the 44 genes, HOXC10, had no SkM-only DNA hypomethylation in some intragenic or intragenic region. Instead, HOXC10 had constitutive LMRs in its SkM-only EnhChr (Table 2).
We looked for overlapping SkM specificity of open chromatin (DNaseI hypersensitive sites, DHS [24,34]), LMRs, and EnhChr regions because tissue-specific DHS outside of promoter regions are often associated with tissue-specific enhancers [36]. We compared DHS in SkM, heart, brain, and B-cells. Ten genes exhibited SkM-only DHS in hypomethylated regions of SkM-only EnhChr (Tables 1 and 2; SkM-only DHS overlap). Many genes displayed DHS elsewhere in SkM EnhChr (data not shown).
Lastly, we determined the prevalence of SkM-only hypomethylation in promoter regions among the 44 genes (Tables 1 and 2, Pr region column). Twenty-five genes were hypomethylated at their promoter regions (immediately upstream or downstream of the TSS) specifically in SkM. Unlike most human gene promoters [37], none of these 25 promoter regions overlaps a CpG-rich region (CpG island, CGI).
CASQ1, FBXO32, and MYOD1 Illustrate Skeletal Muscle-specific DNA Hypomethylation at Muscle-specific Enhancer Chromatin
To further study SkM-specific epigenetics of enhancers, we focused on CASQ1, FBXO32, and MYOD1 (Figures 2-4), as SkM-associated genes that differ in their relative expression in SkM vs. myoblasts and SkM vs. heart (Tables 1 and 2). CASQ1 encodes calsequestrin-1, a sarcoplasmic Ca2+-binding protein associated with a rare mild autosomal dominant muscle disorder [38,39]. Expression of CASQ1 is strong in SkM, moderate-to-low in heart, and very low in lung, lymph nodes, and myoblasts (Table 1 and Figure 2a). Consistent with this expression pattern, much intragenic EnhChr was found in CASQ1 in the two examined SkM samples while little or no active-type EnhChr was in non-muscle samples and only small regions of EnhChr were seen in myoblasts (Figure 2b). In heart, there was a somewhat smaller region of EnhChr than in SkM. Unexpectedly, despite the gene’s high level of expression in SkM and the presence of transcribed chromatin (H3K36me3) in the 3’ half of the gene in both of the SkM samples, the promoter region in one of the SkM samples displayed EnhChr but no active promoter-type chromatin (Figure 2b). This anomaly might be due to the interconversion of enhancer and promoter chromatin in vivo [40] or to more plasticity in the functional assignments of H3K4 methylation to enhancers and promoters [41] than commonly appreciated.
Figure 2.

CASQ1 SkM-specific and SkM- and heart-specific enhancer chromatin overlaps regions of SkM-only DNA hypomethylation. (a) RefSeq gene depictions and RNA-seq data for the CASQ1 gene region and the ends of the neighboring genes at chr1:160,155,131-160,173,488. For cultured cells, only the transcribed strand of CASQ1 is shown. (b) Chromatin state segmentation; Enh, enhancer chromatin; Pr, promoter chromatin; LCL, lymphoblastoid cell line; lung fib., lung fibroblasts; skeletal muscle #1 and #2, psoas muscle and an undesignated type of SkM sample, respectively; heart, left ventricle; PBMC, peripheral blood mononuclear cells. (c) Bisulfite-seq profiles for the indicated samples with blue bars above each profile indicating low-methylation regions (LMRs), which had significantly lower methylation than the rest of the genome [27]; skeletal muscle, psoas. (d) CpG islands and human/mouse conserved transcription factor binding sites. (e) DNaseI hypersensitivity peaks. (f) Human/mouse DNA sequence conservation. All tracks are from the hg19 reference genome in the UCSC Genome Browser [22] and have been aligned in this and the subsequent two figures. See text for designations of boxed regions.
There was a SkM-only LMR overlapping SkM-only EnhChr immediately upstream of the promoter region of CASQ1 (Figure 2c, red box on the left). Especially important to note is the ~2-kb region of EnhChr seen downstream of the TSS in both SkM and heart that is hypomethylated only in SkM (Figure 2c, red dotted box). This region displayed small DHS peaks significantly over background in SkM but not in heart (Figure 2e, box). There was also SkM- and heart-specific hypomethylation overlapping the promoter as well as a constitutively unmethylated DNA region overlapping SkM- and heart-specific EnhChr (Figure 2c, dotted black lines). The rest of the EnhChr in SkM was mostly methylated (Figure 2c, gray highlighting). The 5’ end of CASQ1 is only about 47 kb from the 3’ end of ATP1A2, another of the studied genes with high expression specifically in SkM (Table 1). Between ATP1A2 and CASQ1 is the ATP1A4 gene (Figure 2a), which is largely testes-specific and is expressed only very weakly in SkM [38].
FBXO32 encodes atrogin-1, an E3 ubiquitin ligase found predominantly in SkM. It plays a critical role in protein degradation, especially in SkM, e.g., during aging and cachexia [42]. It has also been implicated in cardiac myopathy [43]. It is expressed at high levels in SkM, at moderate levels in Mb and heart, and at low levels in some non-muscle tissues [38] and cell cultures (Table 1 and Figure 3a) consistent with the larger amounts of EnhChr found in muscle than in non-muscle samples (Figure 3b). With the exception of a small EnhChr region upstream of the gene in SkM (Figure 3b, red box), SkM and heart were similar to each other in the distribution of EnhChr in the vicinity of FBXO32 despite the eight-fold higher level of FBXO32 RNA in SkM relative to heart. However, as for CASQ1, there was a subregion of the SkM-and-heart specific EnhChr that was significantly hypomethylated only in SkM (Figure 3c, red dotted box). Importantly, there was also an EnhChr subregion in heart that was significantly hypomethylated just in that tissue (Figure 3c, blue box).
Figure 3.

FBXO32 enhancer chromatin overlaps foci of SkM-only DNA hypomethylation and heart-specific DNA hypomethylation foci. (a) Ref-Seq genes and RNA-seq; (b) chromatin state segmentation; (c) bisulfite-seq; (d) CpG islands and conserved TFBS; (e) DNase-seq; (f) human/mouse DNA sequence conservation as described for Figure 2. The region shown is chr8:124,506,725-124,564,335.
MYOD1 encodes the myogenesis-specific subunit of one of the most important SkM-lineage specific transcription factors (TFs) [44], namely, the MYOD protein. This gene has very low expression in SkM, no detectable expression in other tissues, and very high expression specifically in myoblasts and myotubes (Table 1, Figure 4a). In mouse myotubes, a ~36-kb super-enhancer situated mostly upstream of MyoD1 was defined on the basis of dense binding of the MyoD TF protein [33]. Two super-enhancer prediction programs using H3K27ac profiling [31,45] identified a 40 kb MYOD1 super-enhancer in this region in human SkM (Figure 4b, dashed purple line), which displays much DNA sequence conservation between the human and mouse genomes (Figure 4f). Human myoblasts and myotubes exhibited a similar H3K27ac-based super-enhancer but with additional 2-kb and 6-kb EnhChr regions centered at ~45 and 67 kb upstream of MYOD1, respectively (Figure 4b dotted black box and [46]). These two EnhChr regions were missing in SkM tissue (Figure 4b, dotted green box and a further upstream region that is not shown). These regions can extend the super-enhancer further upstream specifically in myogenic progenitor cells. Bisulfite-seq data are available for a SkM sample but not myoblasts or myotubes. In SkM, we observed four SkM-only LMRs upstream of MYOD1 in the SkM super-enhancer (Figure 4c, boxes). They also overlap four SkM-only DHS (Figure 4e, boxes). One of these contains the strong, independent 258-bp core enhancer for MYOD1/MyoD1 [47,48], which is 20 kb upstream of the gene (Figure 4c, highlighting and gray arrow). Another overlaps the independent 0.7-kb enhancer 5 kb upstream of the gene (-5kb enhancer, Figure 4c, box on right). The core enhancer was situated in one of the subregions with a cluster of predicted [22] transcription factor binding sites (TFBS) conserved between mouse and human genomes (Figure 4d).
Figure 4.

Similar MYOD1 super-enhancer regions are seen in SkM tissue, myoblasts, and myotubes despite much lower expression in SkM. (a) Ref-Seq genes (including an ncRNA gene LOC102723330 expressed specifically in myoblasts [46]) and RNA-seq; (b) chromatin state segmentation; (c) bisulfite-seq with the distance for three of the SkM-only LMRs from MYOD1 indicated; the fourth SkM-only LMR overlaps the 0.3-kb core enhancer, which is 20 kb upstream of MYOD1; (d) CpG islands and conserved TFBS; (e) DNase-seq; (f) human/mouse DNA sequence conservation as described for Figure 2. The region shown is chr11:17,690,340-17,754,520 region and does not include the previously described [46] myoblast- and myotube-specific EnhChr region at 67 kb upstream of MYOD1 but does show the myoblast- and myotubes-specific EnhChr region at 45 kb upstream of the gene (dotted black box, panel b).
Like MYOD1, PRKAG3 is expressed much less in SkM than in myoblasts but nonetheless displayed a super-enhancer specifically in SkM and even more EnhChr in SkM than in myoblasts and myotubes (Table 2 and data not shown). This gene encodes a SkM-associated subunit of an AMP-activated protein kinase that plays a key role in controlling cellular energy metabolism in SkM in response to physiological changes [49]. The PRKAG3 super-enhancer in SkM contained a SkM-specific LMR and DHS embedded in SkM-only EnhChr (Table 2). However, the DHS over the smaller EnhChr region in myoblasts were much more prominent than in SkM (data not shown).
Methylation of Only Three CpGs in the MYOD1 Core Enhancer Strongly Represses its Activity
We tested the effect of DNA methylation on the enhancer activity of the 258-kb MYOD1/Myod1 core enhancer [47,48] because this enhancer is highly methylated in vivo in non-muscle tissues and cells but mostly or completely unmethylated in SkM and myoblasts (Figure 4c, highlighting; [50,51]. We cloned a 381-bp human DNA fragment containing the core enhancer and inserted it downstream of a luciferase reporter gene in a vector with a minimal promoter (EEF1A1) engineered to be free of CpGs like the rest of the vector (pCpGfree-promoter-Lucia; Figure 5a). The only CpGs in the final construct were the three in the MYOD1 core-enhancer insert. Upon transfection into a mouse myoblast cell line (C2C12) and assay 48 or 72 h after transfection, luciferase activity was just barely detectable from the vector-only plasmid and was stimulated more than 1000-fold by the MYOD1 core enhancer insert (Figure 5b, untreated vs. vector). CpG methylation (M.SssI) gave an eight-fold decrease in luciferase activity relative to the mock-methylated plasmid (Figure 5b). When we compared reporter gene activity 24, 48, and 72 h after transfection, the methylation-induced silencing of luciferase activity was no less at the longer incubation periods (data not shown). Using a non-myogenic cell line (MCF-7, breast cancer cells) as the host cells for transfection of the unmethylated plasmid, we obtained almost 100-fold lower luciferase activity relative to myoblast host cells, but low amounts of enhancer activity were nonetheless clearly detected in multiple transfection assays (Figure 5c, untreated vs. vector). Methylation of the enhancer abolished detectable enhancer activity in MCF-7 cells (Figure 5c).
Figure 5.

Loss of most activity for a MYOD1 core enhancer construct upon transfection into a non-muscle cell line or upon in vitro methylation of its three CpGs. (a) Schematics showing the unmethylated and methylated construct to test MYOD1 core enhancer activity and the vector; (b) and (c) Reporter gene activity for the C2C12 myoblast cell line and MCF-7 breast cancer line is given as the averages of normalized Lucia bioluminescent signal plus or minus the standard error from eight independent transfection experiments with technical duplicates. Note the different scales for luminescence in (b) and (c). The p-values for the reduction of reporter gene activity in methylated vs. mock-methylated enhancers were p < .01 (T-test) upon transient transfection into either C2C12 or MCF-7.
Discussion
Many recent studies of mammalian epigenetics used whole-genome data to look for significant associations with tissue-specific expression. In this report, we address the related, but different, question of whether all, most, or just many of the genes that we selected for highly preferential expression in SkM display SkM-specific EnhChr and overlapping SkM-specific DNA hypomethylation when compared with heart and non-muscle tissues. We found that SkM-only intragenic or intergenic EnhChr was in all of the 44 studied genes and so probably plays a major role in their SkM-specific expression. EnhChr should reflect and facilitate specific TF binding, which underlies its role in upregulating gene expression [52]. The SkM-only EnhChr regions that we observed were unusually large. Half of the genes were linked to a super-enhancer specifically in SkM (Tables 1 and 2). Super-enhancers and the related term “stretch enhancers” [53] have been characterized as large, strong tissue-specific enhancers associated with expression of key cell-identity genes [33,52]. It was reported that some super-enhancers might act on distant genes [52]. Among the four neighboring pairs of SkM-associated genes that we found, three gene-pairs had only one gene that overlapped or was adjacent to a super-enhancer. This suggests that higher-order SkM-specific multi-genic chromatin structures are involved in activating these genes in SkM from a single super-enhancer per pair.
Some of the EnhChr in the 44 genes was specific to both SkM and heart. This dual-specificity EnhChr was significantly correlated with elevated expression in heart (Figure 1). The SkM/heart epigenetic overlap is not surprising because many genes encoding contractile proteins are selectively expressed in both heart and SkM [54]. The shared chromatin epigenetics suggests that TFs establishing this SkM/heart EnhChr are common to both tissue types.
There were also correlations between epigenetics, transcription, and developmental stage within the muscle lineage. For example, CASQ1 (Figure 2b), TNNC2 and TNNI2 had much more extensive SkM-lineage specific EnhChr in SkM relative to myoblasts and correspondingly more expression in SkM while the opposite was the case for CHRNA1 (Tables 1 and 2 and data not shown). High-level expression of the myopathy-linked CASQ1 [55] in SkM tissue, but not in myoblasts, is consistent with its encoding a sarcoplasmic protein. In myoblasts, CASQ1 had only two small EnhChr regions (Figure 2). The broad EnhChr encompassing these sites in SkM is likely to have been derived from spreading of EnhChr subsequent to the myogenic progenitor stage.
Many reports provide evidence for the involvement of DNA hypomethylation in the establishment or maintenance of enhancers [16,18,56-58]. In our study, SkM-only DNA hypomethylation was observed overlapping SkM-only EnhChr in 80 percent of the 44 SkM-associated genes. These hypomethylated regions in SkM EnhChr usually were just foci of hypomethylated DNA surrounded by high or partial [59] DNA methylation, similar to enhancers described in a recent study of a cancer cell line [60]. The importance of the SkM-only DNA-hypomethylated foci within EnhChr is suggested by our finding that almost all of the genes displaying EnhChr regions shared by SkM and heart contained foci of SkM-only DNA hypomethylation (Tables 1 and 2). These hypomethylated foci within EnhChr may increase the enhancer’s SkM specificity, probably by facilitating tissue-specific TF binding [61,62], and can also reflect the consequences of specific TF binding [63].
In addition to EnhChr hypomethylation, we also observed frequent SkM-only promoter hypomethylation but only at CpG-poor promoters (non-CGI promoters, Tables 1 and 2). Tissue-specific promoter hypomethylation at non-CGI promoters is much less common throughout the genome than is constitutive unmethylation at CGI promoters [37]. There were fewer SkM-preferentially expressed genes displaying SkM-only promoter hypomethylation than enhancer hypomethylation in the studied EnhChr regions (57 and 89 percent of the genes, respectively). Both SkM promoter hypomethylation and enhancer hypomethylation are likely to be related to the establishment or maintenance of differential gene expression.
HOXC10 is exceptional among the 44 examined SkM-associated genes because it was the only one that did not display some SkM-specific hypomethylation in its vicinity but instead the HOXC10 gene region was constitutively unmethylated (Table 2). It is part of the tightly co-regulated HOXC gene cluster. We previously reported that this gene cluster displays a continuous multigenic promoter/enhancer region specifically in myoblasts and myotubes compared with non-myogenic cell cultures [64]. The expression of many adjacent genes in this gene cluster is specific for SkM tissue and myogenic progenitor cells (Table 2). Furthermore, the HOXC gene cluster overlaps a multigenic super-enhancer and is bordered by hypermethylated DNA specifically in SkM (this study) as well as in myoblasts and myotubes [64]. These results suggest that SkM tissue and myogenic progenitor cells both use a similar DNA hypermethylated border around this multigenic cluster [20] to co-regulate expression of these developmental TF-encoding homeobox genes. This DNA hypermethylation might help prevent the spreading of activating histone modifications to genes on the periphery of the cluster as well as the intrusion of repressive H3K27me3 marks from outside the cluster [64].
The completely SkM-lineage specific expression of MYOD1, a myogenic TF-encoding gene [54], is due to its strong upstream tissue-specific enhancer elements [47,48]. It has only a weak and non-specific promoter while its upstream super-enhancer in myogenic progenitor cells [33,46] encompasses the well-known SkM lineage-specific 258-bp core enhancer and 0.7-kb enhancer [47] located about 20 and 5 kb upstream of the mouse and human MyoD1/MYOD1 genes (Figure 4). Both human SkM tissue and myogenic progenitor cells have a ~40-kb super-enhancer even though MYOD1 is expressed at only very low levels in SkM, unlike in myoblasts and myotubes (Figure 4 and Table 2; [46]). Although super-enhancers are usually associated with high-level expression of the associated genes [33], there are differences between the epigenetics of the MYOD1 super-enhancer in SkM and myogenic progenitor cells that can help explain their very large differences in expression of MYOD1. In myoblasts and myotubes, we reported [46] that the MYOD1 super-enhancer extends further upstream than previously noted [33] and includes EnhChr regions 45 and 67 kb upstream of MYOD1. These EnhChr regions are close to the ear-specific OTOG gene, the nearest upstream gene to MYOD1. In the present study, we found that EnhChr is missing from these two regions in SkM tissue (-45kb in Figure 4b and -67kb, data not shown). The -67kb region, like the core enhancer and the -5kb enhancer, binds the Myod/MYOD TF at orthologous mouse/human DNA sequences [32,46]. MYOD1/Myod1 is as an autoregulatory TF gene [65]. The autoregulation probably includes binding of the MYOD enhancer-regulatory TF [32,66] to the most distant EnhChr region within the MYOD1 super-enhancer of myogenic progenitor cells.
The positive MYOD1 autoregulatory circuit can help explain the maintenance (although not the establishment) of much lower MYOD1 super-enhancer activity in SkM than in myogenic progenitor cells. Our finding of less open chromatin at the core enhancer and the -67kb EnhChr in SkM vs. in myogenic progenitor cells (Figure 4e, arrow and [46]) could reflect less binding of MYOD protein to this enhancer in muscle fibers than in myoblasts and myotubes. Why the specific DHS at the -5kb enhancer was equally large in both types of samples remains to be determined but is consistent with the -5kb enhancer playing more of a role in maintaining MYOD1/MyoD1 expression in differentiated cells than does the core enhancer, which is more associated with fetal myogenesis [21]. However, it has been proposed that the core and -5kb enhancers can cooperate with one another as well as being able to act independently of each other [47]. They probably cooperate within the context of a super-enhancer, as proposed for constituent enhancers in other super-enhancers [12].
There still remain major questions about the MYOD1 super-enhancer. Why is its core enhancer situated so far from the MYOD1 gene? Why is this 258-bp constituent enhancer surrounded by tens of kilobases of super-enhancer chromatin despite being highly active on its own? What is the function of its tissue-specific DNA hypomethylation? The MYOD1 core enhancer is hypomethylated in SkM and myogenic cell cultures relative to non-muscle samples [50,51], as confirmed for SkM tissue in the present study. Our transfection results show that in vitro methylation targeted to the MYOD1 core enhancer in a reporter plasmid used for transfection of mouse myoblasts reduced its activity about eightfold even though it contains only three CpGs. (Figure 5b). DNA hypomethylation in the MYOD1 super-enhancer in SkM tissue may be part of the epigenetic memory [17,67] that facilitates increased activation of this super-enhancer at physiologically relevant times in postnatal muscle.
However, demethylation of the MYOD1 core enhancer in the SkM lineage, while probably part of core enhancer activation, is not sufficient for its activity [68]. We observed that a reporter gene construct with a downstream unmethylated core enhancer transfected into a breast cancer line (MCF-7) gave only about 1 percent the reporter gene activity than it did in myoblasts (Figure 5b and c), probably, in part, due to the absence of MYOD/Myod TF in the cells. However, even in MCF-7 cells, there was reproducibly about three times more reporter gene activity in the presence of the core enhancer relative to the vector-only control plasmid. Given the absolute specificity of expression of MYOD1 for the SkM lineage in vivo, embedding the core enhancer in so much regulatory chromatin might be necessary to completely repress MYOD1 expression in non-muscle cells and to fine-tune the timing and level of expression of this gene prenatally and postnatally.
Postnatally, SkM tissue is unusually dynamic in its inter-conversion from one fiber type to another in response to physiological demands and environmental signals. SkM also requires postnatal repair. Differential expression of MYOD1 at the transcription level is involved in these processes [69-71]. PRKAG3, another of the genes we studied, is linked to ameliorating muscle fatigue and controlling a metabolic signaling cascade [72]. Like MYOD1, PRKAG3 is implicated in responses to changing usage of SkM, including fiber-type switching [49]. PRKAG3 is also similar to MYOD1 in exhibiting a SkM- specific super-enhancer with DNA hypomethylated subregions despite much lower expression in SkM than myoblasts (Table 2). Because there is only low expression of MYOD1 and PRKAG3 in normal SkM, we propose that their super-enhancers are poised for upregulation. Poised EnhChr is typically categorized as displaying H3K4me1 enrichment but little or no H3K27ac in the vicinity of other genes [2,73,74]. In contrast, the MYOD1 and PRKAG3 super-enhancers in SkM are enriched in both H3K27ac and H3K4me1. SkM fibers may harbor pre-existing, poised or weak super-enhancers for these genes to allow their rapid upregulation in response to environmental signals similar to what has been proposed for some inflammation-activated super-enhancers in mouse macrophages [75]. For the MYOD1 super-enhancer, such up-regulation may involve increased levels of active MYOD protein (possibly including post-translational activation of the MYOD TF [76,77]) positively controlling the enhancer activity as part of the above-mentioned autoregulation.
Methylated DNA surrounded a focal region of SkM-only DNA hypomethylation in most of the studied genes. However, on the basis of bisulfite-seq profiles, which were used to track DNA methylation in this study, we cannot distinguish 5mC from the much less abundant but more dynamic 5hmC [78]. Unlike 5mC, 5hmC has been reported to be enriched in active enhancers [20]. Using a quantitative assay (Epimark) for 5hmC and 5mC at specific CpG sites, we recently demonstrated that 5hmC replaces some of the 5mC in specific regions of super-enhancer chromatin of MYOD1 in SkM, but not in heart or leukocytes [46]. In addition, some of the 5mC near the core enhancer of myoblasts and myotubes is replaced by 5hmC. We propose that high levels of DNA methylation or hydroxymethylation around foci of DNA hypomethylation in enhancers are needed to set up borders to prevent spreading or contraction of these foci [20] as well as to regulate repressive elements within the enhancer [75] and thereby help fine-tune enhancer activity.
Acknowledgments
We thank M. Badoo and the Tulane Cancer Center (COBRE grant NIGMS P20GM103518) for help with the Cufflinks analysis of RNA-seq data for myoblasts. This research was funded in part by NIH Grant NS04885 and a Louisiana Cancer Research Center Grant to M.E.
Glossary
- SkM
skeletal muscle
- SkM-only
in SkM but not in other examined tissues
- 5mC
5-methylcytosine
- 5hmC
5-hydroxymethylcytosine
- RNA-seq
whole-genome next-generation sequencing of cDNA
- FPKM or RPKM
analogous terms for fragments or reads, respectively, per kilobase of transcript per million mapped reads in RNA-seq
- H3K4me1
histone H3 lysine-4 monomethylation
- H3K27ac
H3K27acetylation
- H3K4me3
H3K4 trimethylation
- H3K36me3
H3 lysine-36 trimethylation
- ChIP-seq
chromatin immunoprecipitation/next-generation sequencing
- EnhChr
enhancer-type chromatin enriched in H3K27ac and H3K4me1
- LMR
low methylated DNA region
- TSS
transcription start site
- PBMC
peripheral blood mononuclear cells
- LCL
lymphoblastoid cell line
- bisulfite-seq
whole-genome profiling of DNA methylation from bisulfite-treated DNA
- RRBS
reduced representation bisulfite sequencing
- DHS
DNaseI-hypersensitive sites
- TF
transcription factor
- TFBS
transcription factor binding sites
- RefSeq
the Reference Sequence gene database
Author Contributions
ME directed the research and wrote the manuscript; KE prepared the DNA constructs, did the transfections, helped with project design and manuscript writing; KE and HLP did the bioinformatics analyses; ML did the biostatistical analyses.
References
- Wu H, Nord AS, Akiyama JA. et al. Tissue-specific RNA expression marks distant-acting developmental enhancers. PLoS Genet. 2014;10(9):e1004610. doi: 10.1371/journal.pgen.1004610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Romanoski CE, Benner C. et al. The selection and function of cell type-specific enhancers. Nat Rev Mol Cell Biol. 2015;16(3):144–154. doi: 10.1038/nrm3949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundaje A, Meuleman W, Ernst J. et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518(7539):317–330. doi: 10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emera D, Yin J, Reilly SK. et al. Origin and evolution of developmental enhancers in the mammalian neocortex. Proc Natl Acad Sci U S A. 2016;113(19):E2617–E2626. doi: 10.1073/pnas.1603718113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadhouders R, van den Heuvel A, Kolovos P. et al. Transcription regulation by distal enhancers: who's in the loop? Transcription. 2012;3(4):181–186. doi: 10.4161/trns.20720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onodera CS, Underwood JG, Katzman S. et al. Gene isoform specificity through enhancer-associated antisense transcription. PLoS One. 2012;7(8):e43511. doi: 10.1371/journal.pone.0043511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintzman ND, Ren B. Finding distal regulatory elements in the human genome. Curr Opin Genet Dev. 2009;19(6):541–549. doi: 10.1016/j.gde.2009.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulakanti K, Pinello L, Stelloh C. et al. Enhancer transcribed RNAs arise from hypomethylated, Tet-occupied genomic regions. Epigenetics. 2013;8(12):1303–1320. doi: 10.4161/epi.26597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mousavi K, Zare H, Dell'orso S. et al. eRNAs promote transcription by establishing chromatin accessibility at defined genomic loci. Mol Cell. 2013;51(5):606–617. doi: 10.1016/j.molcel.2013.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arner E, Daub CO, Vitting-Seerup K. et al. Transcribed enhancers lead waves of coordinated transcription in transitioning mammalian cells. Science. 2015;347(6225):1010–1014. doi: 10.1126/science.1259418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray JM, Kim TK, West AE. et al. Genomic views of transcriptional enhancers: Essential determinants of cellular identity and activity-dependent responses in the CNS. J Neurosci. 2015;35(41):13819–13826. doi: 10.1523/JNEUROSCI.2622-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz D, Schuijers J, Lin CY. et al. Convergence of developmental and oncogenic signaling pathways at transcriptional super-enhancers. Mol Cell. 2015;58(2):362–370. doi: 10.1016/j.molcel.2015.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barski A, Cuddapah S, Cui K. et al. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129(4):823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- Ernst J, Kheradpour P, Mikkelsen TS. et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473(7345):43–49. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidl C, Klug M, Boeld TJ. et al. Lineage-specific DNA methylation in T cells correlates with histone methylation and enhancer activity. Genome Res. 2009;19(7):1165–1174. doi: 10.1101/gr.091470.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiench M, John S, Baek S. et al. DNA methylation status predicts cell type-specific enhancer activity. EMBO J. 2011;30(15):3028–3039. doi: 10.1038/emboj.2011.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hon GC, Rajagopal N, Shen Y. et al. Epigenetic memory at embryonic enhancers identified in DNA methylation maps from adult mouse tissues. Nat Genet. 2013;45(10):1198–1206. doi: 10.1038/ng.2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blattler A, Yao L, Witt H. et al. Global loss of DNA methylation uncovers intronic enhancers in genes showing expression changes. Genome Biol. 2014;15(9):469. doi: 10.1186/s13059-014-0469-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serandour AA, Avner S, Oger F. et al. Dynamic hydroxymethylation of deoxyribonucleic acid marks differentiation-associated enhancers. Nucleic Acids Res. 2012;40(17):8255–8265. doi: 10.1093/nar/gks595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich M, Ehrlich KC. DNA cytosine methylation and hydroxymethylation at the borders. Epigenomics. 2014;6(6):563–566. doi: 10.2217/epi.14.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JC, Goldhamer DJ. The core enhancer is essential for proper timing of MyoD activation in limb buds and branchial arches. Dev Biol. 2004;265(2):502–512. doi: 10.1016/j.ydbio.2003.09.018. [DOI] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, UCSC Genome Browser [Internet] 2016. [June 2016]. Available from: http://ucsc.genome.edu .
- Myers RM, Stamatoyannopoulos J, Snyder M. et al. A user's guide to the encyclopedia of DNA elements (ENCODE). PLoS Biol. 2011;9(4):e1001046. doi: 10.1371/journal.pbio.1001046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song L, Zhang Z, Grasfeder LL. et al. Open chromatin defined by DNaseI and FAIRE identifies regulatory elements that shape cell-type identity. Genome Res. 2011;21(10):1757–1767. doi: 10.1101/gr.121541.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ET, Sandberg R, Luo S. et al. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456(7221):470–476. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krupp M, Marquardt JU, Sahin U. et al. RNA-Seq Atlas--a reference database for gene expression profiling in normal tissue by next-generation sequencing. Bioinformatics. 2012;28(8):1184–1185. doi: 10.1093/bioinformatics/bts084. [DOI] [PubMed] [Google Scholar]
- Song Q, Decato B, Hong EE. et al. A reference methylome database and analysis pipeline to facilitate integrative and comparative epigenomics. PLoS One. 2013;8(12):e81148. doi: 10.1371/journal.pone.0081148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roadmap Epigenetics Consortium [Internet] 2016. Available from: http://genome.ucsc.edu/cgi-bin/hgTrackUi?hgsid=492885615_Yg1tPNyBQ5YaWPbdnAAZoOA4y03u&c=chr10&g=hub_24125_RoadmapConsolidatedAssaya27004.
- Trapnell C, Roberts A, Goff L. et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7(3):562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsumagari K, Chang S-C, Lacey M. et al. Gene expression during normal and FSHD myogenesis. BMC Medical Genomics. 2011;4:67. doi: 10.1186/1755-8794-4-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan A, Zhang X. dbSUPER: a database of super-enhancers in mouse and human genome. Nucleic Acids Res. 2016;44(D1):D164–D171. doi: 10.1093/nar/gkv1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y, Yao Z, Sarkar D. et al. Genome-wide MyoD binding in skeletal muscle cells: a potential for broad cellular reprogramming. Dev Cell. 2010;18(4):662–674. doi: 10.1016/j.devcel.2010.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte WA, Orlando DA, Hnisz D. et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153(2):307–319. doi: 10.1016/j.cell.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsumagari K, Baribault C, Terragni J. et al. Early de novo DNA methylation and prolonged demethylation in the muscle lineage. Epigenetics. 2013;8(3):317–332. doi: 10.4161/epi.23989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith ZD, Gu H, Bock c. et al. High-throughput bisulfite sequencing in mammalian genomes. Methods. 2009;48(3):226–232. doi: 10.1016/j.ymeth.2009.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi H, Shulha HP, Lin JM. et al. Identification and characterization of cell type-specific and ubiquitous chromatin regulatory structures in the human genome. PLoS Genet. 2007;3(8):e136. doi: 10.1371/journal.pgen.0030136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deaton AM, Bird A. CpG islands and the regulation of transcription. Genes Dev. 2011;25(10):1010–1022. doi: 10.1101/gad.2037511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebhan M, Chalifa-Caspi V, Prilusky J. GeneCards [Internet] 2016. Available from: http://www.genecards.org .
- Canato M, Capitanio P, Reggiani C. et al. The disorders of the calcium release unit of skeletal muscles: what have we learned from mouse models? J Muscle Res Cell Motil. 2015;36(1):61–69. doi: 10.1007/s10974-014-9396-7. [DOI] [PubMed] [Google Scholar]
- Leung D, Jung I, Rajagopal N. et al. Integrative analysis of haplotype-resolved epigenomes across human tissues. Nature. 2015;518(7539):350–354. doi: 10.1038/nature14217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekowska A, Benoukraf T, Zacarias-Cabeza J. et al. H3K4 tri-methylation provides an epigenetic signature of active enhancers. Embo J. 2011;30(20):4198–4210. doi: 10.1038/emboj.2011.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukari A, Muqbil I, Mohammad RM. et al. F-BOX proteins in cancer cachexia and muscle wasting: Emerging regulators and therapeutic opportunities. Semin Cancer Biol. 2016;36:95–104. doi: 10.1016/j.semcancer.2016.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Yacoub N, Shaheen R, Awad SM. et al. FBXO32, encoding a member of the SCF complex, is mutated in dilated cardiomyopathy. Genome Biol. 2016;17:2. doi: 10.1186/s13059-015-0861-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Jones PA. Potentiation of MyoD1 activity by 5-aza-2'-deoxycytidine. Cell Growth Differ. 1990;1(8):383–392. [PubMed] [Google Scholar]
- Wei Y, Zhang S, Shang S. et al. SEA: a super-enhancer archive. Nucleic Acids Res. 2016;44(D1):D172–D179. doi: 10.1093/nar/gkv1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra S, Terragni J, Zhang G. et al. Tissue-specific epigenetics in gene neighborhoods: myogenic transcription factor genes. Hum Mol Genet. 2015;24(16):4660–4673. doi: 10.1093/hmg/ddv198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JC, Love CM, Goldhamer DJ. Two upstream enhancers collaborate to regulate the spatial patterning and timing of MyoD transcription during mouse development. Dev Dyn. 2001;221(3):274–288. doi: 10.1002/dvdy.1138. [DOI] [PubMed] [Google Scholar]
- Goldhamer DJ, Brunk BP, Faerman A. et al. Embryonic activation of the myoD gene is regulated by a highly conserved distal control element. Development. 1995;121(3):637–649. doi: 10.1242/dev.121.3.637. [DOI] [PubMed] [Google Scholar]
- Canto C, Jiang LQ, Deshmukh AS. et al. Interdependence of AMPK and SIRT1 for metabolic adaptation to fasting and exercise in skeletal muscle. Cell Metab. 2010;11(3):213–219. doi: 10.1016/j.cmet.2010.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunk BP, Goldhamer DJ, Emerson CP, Jr.. Regulated demethylation of the myoD distal enhancer during skeletal myogenesis. Dev Biol. 1996;177(2):490–503. doi: 10.1006/dbio.1996.0180. [DOI] [PubMed] [Google Scholar]
- Zhang F, Pomerantz JH, Sen G. et al. Active tissue-specific DNA demethylation conferred by somatic cell nuclei in stable heterokaryons. Proc Natl Acad Sci U S A. 2007;104(11):4395–4400. doi: 10.1073/pnas.0700181104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quang DX, Erdos MR, Parker SC. et al. Motif signatures in stretch enhancers are enriched for disease-associated genetic variants. Epigenetics Chromatin. 2015;8:23. doi: 10.1186/s13072-015-0015-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker SC, Stitzel ML, Taylor DL. et al. Chromatin stretch enhancer states drive cell-specific gene regulation and harbor human disease risk variants. Proc Natl Acad Sci U S A. 2013;110(44):17921–17926. doi: 10.1073/pnas.1317023110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassoon D, Lyons G, Wright WE. et al. Expression of two myogenic regulatory factors myogenin and MyoD1 during mouse embryogenesis. Nature. 1989;341(6240):303–307. doi: 10.1038/341303a0. [DOI] [PubMed] [Google Scholar]
- D'Adamo MC, Sforna L, Visentin S. et al. A calsequestrin-1 mutation associated with a skeletal muscle disease alters sarcoplasmic Ca2+ release. PLoS One. 2016;11(5):e0155516. doi: 10.1371/journal.pone.0155516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W, Briones V, Lister R. et al. CG hypomethylation in Lsh-/- mouse embryonic fibroblasts is associated with de novo H3K4me1 formation and altered cellular plasticity. Proc Natl Acad Sci U S A. 2014;111(16):5890–5895. doi: 10.1073/pnas.1320945111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lay FD, Liu Y, Kelly TK. et al. The role of DNA methylation in directing the functional organization of the cancer epigenome. Genome Res. 2015;25(4):467–477. doi: 10.1101/gr.183368.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reizel Y, Spiro A, Sabag O. et al. Gender-specific postnatal demethylation and establishment of epigenetic memory. Genes Dev. 2015;29(9):923–933. doi: 10.1101/gad.259309.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaidatzis D, Burger L, Murr R. et al. DNA sequence explains seemingly disordered methylation levels in partially methylated domains of mammalian genomes. PLoS Genet. 2014;10(2):e1004143. doi: 10.1371/journal.pgen.1004143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlet J, Duymich CE, Lay FD. et al. Bivalent regions of cytosine methylation and H3K27 acetylation suggest an active role for DNA methylation at enhancers. Mol Cell. 2016;62(3):422–431. doi: 10.1016/j.molcel.2016.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spruijt CG, Gnerlich F, Smits AH. et al. Dynamic readers for 5-(hydroxy)methylcytosine and its oxidized derivatives. Cell. 2013;152(5):1146–1159. doi: 10.1016/j.cell.2013.02.004. [DOI] [PubMed] [Google Scholar]
- Liu H, Liu X, Zhang S. et al. Systematic identification and annotation of human methylation marks based on bisulfite sequencing methylomes reveals distinct roles of cell type-specific hypomethylation in the regulation of cell identity genes. Nucleic Acids Res. 2016;44(1):75–94. doi: 10.1093/nar/gkv1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsankov AM, Gu H, Akopian V. et al. Transcription factor binding dynamics during human ES cell differentiation. Nature. 2015;518(7539):344–349. doi: 10.1038/nature14233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsumagari K, Baribault C, Terragni J. et al. DNA methylation and differentiation: HOX genes in muscle cells. Epigen Chromatin. 2013;6(1):25. doi: 10.1186/1756-8935-6-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thayer MJ, Tapscott SJ, Davis RL. et al. Positive autoregulation of the myogenic determination gene MyoD1. Cell. 1989;58(2):241–248. doi: 10.1016/0092-8674(89)90838-6. [DOI] [PubMed] [Google Scholar]
- Blum R, Vethantham V, Bowman C. et al. Genome-wide identification of enhancers in skeletal muscle: the role of MyoD1. Genes Dev. 2012;26(24):2763–2779. doi: 10.1101/gad.200113.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng YQ, Desprat R, Fu H. et al. DNA methylation supports intrinsic epigenetic memory in mammalian cells. PLoS Genet. 2006;2(4):e65. doi: 10.1371/journal.pgen.0020065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taberlay PC, Kelly TK, Liu CC. et al. Polycomb-repressed genes have permissive enhancers that initiate reprogramming. Cell. 2011;147(6):1283–1294. doi: 10.1016/j.cell.2011.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes SM, Taylor JM, Tapscott SJ. et al. Selective accumulation of MyoD and myogenin mRNAs in fast and slow adult skeletal muscle is controlled by innervation and hormones. Development. 1993;118(4):1137–1147. doi: 10.1242/dev.118.4.1137. [DOI] [PubMed] [Google Scholar]
- Hennebry A, Berry C, Siriett V. et al. Myostatin regulates fiber-type composition of skeletal muscle by regulating MEF2 and MyoD gene expression. Am J Physiol Cell Physiol. 2009;296(3):C525–C534. doi: 10.1152/ajpcell.00259.2007. [DOI] [PubMed] [Google Scholar]
- Dilworth FJ, Blais A. Epigenetic regulation of satellite cell activation during muscle regeneration. Stem Cell Res Ther. 2011;2(2):18. doi: 10.1186/scrt59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes BR, Glund S, Long YC. et al. 5'-AMP-activated protein kinase regulates skeletal muscle glycogen content and ergogenics. Faseb J. 2005;19(7):773–779. doi: 10.1096/fj.04-3221com. [DOI] [PubMed] [Google Scholar]
- Creyghton MP, Cheng AW, Welstead GG. et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci U S A. 2010;107(50):21931–21936. doi: 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rada-Iglesias A, Bajpai R, Swigut T. et al. A unique chromatin signature uncovers early developmental enhancers in humans. Nature. 2011;470(7333):279–283. doi: 10.1038/nature09692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hah N, Benner C, Chong LW. et al. Inflammation-sensitive super enhancers form domains of coordinately regulated enhancer RNAs. Proc Natl Acad Sci U S A. 2015;112(3):E297–E302. doi: 10.1073/pnas.1424028112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling BM, Bharathy N, Chung TK. et al. Lysine methyltransferase G9a methylates the transcription factor MyoD and regulates skeletal muscle differentiation. Proc Natl Acad Sci U S A. 2012;109(3):841–846. doi: 10.1073/pnas.1111628109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartorelli V, Puri PL, Hamamori Y. et al. Acetylation of MyoD directed by PCAF is necessary for the execution of the muscle program. Mol Cell. 1999;4(5):725–734. doi: 10.1016/s1097-2765(00)80383-4. [DOI] [PubMed] [Google Scholar]
- Stroud H, Feng S, Morey Kinney S. et al. 5-Hydroxymethylcytosine is associated with enhancers and gene bodies in human embryonic stem cells. Genome Biol. 2011;12(6):R54. doi: 10.1186/gb-2011-12-6-r54. [DOI] [PMC free article] [PubMed] [Google Scholar]