

Abstract
An understanding of epigenetics is indispensable to our understanding of gene regulation under normal and pathological states. This knowledge will help with designing better therapeutic approaches in regenerative tissue medicine. Epigenetics allows us to parse out the mechanisms by which transcriptional regulators gain access to specific gene loci thereby imprinting epigenetic information affecting chromatin function. This epigenetic memory forms the basis of cell lineage specification in multicellular organisms. Post-translational modifications to DNA and histones in the nucleosome core form characteristic epigenetic codes which are distinct for self-renewing and primed progenitor cell populations. Studies of chromatin modifiers and modifications in renal development and disease have been gaining momentum. Both congenital and adult renal diseases have a gene-environment component, which involves alterations to the epigenetic information imprinted during development. This epigenetic memory must be characterized to establish optimal treatment of both acute and chronic renal diseases.
Keywords: Epigenetics, Kidney, HATs, HDACs, chromatin modifiers, histone modifications, bivalent histone code, nephron progenitors, kidney disease
Introduction
Regenerative medicine will benefit from an understanding of the mechanisms by which pluripotent/multipotent cells give rise to differentiated progeny. Multicellular organisms despite having cells with identical genetic information have morphologically and functionally distinct tissues. We now know that cellular differentiation and tissue morphogenesis in these organisms is brought about by epigenetic and transcriptional regulation of genes and post-translational modification of proteins [1]. The DNA in cells is organized into higher order structures via their association with highly basic histone proteins. Heterogeneous dimers of histones H3/H4 and H2A/H2B form an 8 histone core around which is wrapped 147bp of DNA, together forming the nucleosome [2,3]. The H1 and avian H5 histone [4,5] serve as linkers between successive nucleosomes to establish the highly organized chromatin structure. The amino terminal ends of the histones in the nucleosome core are subject to several post-translational modifications primarily on specific Lysine (K) and Arginine (R) residues. These modifications greatly influence the local accessibility of the DNA and hence the expressivity of the genes [6,7]. Heterochromatin is an inactive chromatin, whereas the condensation and decondensation of chromatin at gene promoters generally occurs in euchromatin [8]. This review will summarize the epigenetic mechanisms prevalent in the developing kidney with a view on form and function.
Congenital abnormalities of the kidney and urinary tract (CAKUT) affect one in 500 newborns and account for 20 to 30 percent of birth defects [9,10]. CAKUT predisposes children to chronic kidney disease and hypertension associated with significant morbidity and mortality [11]. A vast majority of CAKUT cases are non-syndromic whose etiopathogenesis have yet to be fully characterized. These non-syndromic forms of CAKUT exhibit phenotypic variability and are suspected to have an epigenetic basis [12,13]. Thus, unravelling the epigenetic mechanisms of normal renal development could prove useful in both the prevention and treatment of CAKUT.
Epigenetic Modifiers and Modifications
Epigenetics refers to dynamic, covalent modifications to the DNA and associated histone proteins that affect gene expression and are involved in chromatin remodeling [1,13]. Although heritable, these modifications are not encoded in the DNA sequence but form a part of the cellular memory [12]. They are vital to the establishment and maintenance of cellular identities in differentiated cell lineages. The generation of induced pluripotent stem cells from adult cells involves erasing this cellular memory [14,15]. Some of the most well-known histone modifications are acetylation, methylation, phosphorylation, and ubiquitination.
HATs and HDACs
Histone acetyl transferases (HATs) and Histone deacetylases (HDACs) mediate the addition and removal of acetyl groups from various histone and non-histone proteins [16,17]. Lacking a DNA binding domain, HDACs access their target genes through their association with several protein complexes (i.e., Sin3 complex, NuRD complex, Co-REST complex, and SMRT/N-CoR complex) [18]. This association with different protein complexes is what confers the spatial and temporal specificity of HDAC-mediated gene expression. Some of the non-histone proteins that are subject to HDAC regulation are p53, STAT3, YY1, GATA1, E2F1, tubulin, and Hsp90 [19-21]. HDACs are broadly grouped into four classes based on their homology to yeast homologues_ Class I: constitutes HDACs 1-3 and 8; Class II: HDACs 4-7, 9, and 10; Class III: Sirtuins 1-7; Class IV: HDAC 11 [22]. Histone acetyltransferases CBP and p300 are essential for maintaining the morphological integrity of the kidney [23].
Expression Profile of HDACs and their Roles in the Developing Kidney
In the kidney, HDAC 1-4, 7, and 9 expression is elevated prenatally but declines upon maturation into the adult kidney [22,24,25]. Immunostaining localizes HDAC 1, 2, and 3 to the undifferentiated metanephric mesenchyme, dichotomizing ureteric branches, and the stroma. HDAC3 also localizes to podocytes. By contrast, HDAC 5, 6, and 8 are constitutively expressed. The renal microvasculature expresses HDAC 7, 8, and 9. Despite, the spatially and temporally restricted distribution of HDACs the global acetylation levels on histones H3 and H4 remain unchanged during kidney development. This could be indicative of the tight coupling of HAT and HDAC functions during development.
The pan HDAC inhibitor, Scriptaid, causes widespread cellular arrest and apoptosis impairing both nephron formation and ureteric bud branching morphogenesis in ex vivo cultures of embryonic kidney explants [24]. Genetic profiles of these Scriptaid-treated explants revealed changes in approximately 12 percent of the gene transcripts probed. The genes most significantly altered belonged either to the cell cycle, canonical Wnt, TGF-b/Smad, cancer, or PI3K/AKT pathways. These assays revealed marked upregulation of CDK inhibitors and tumor suppressors (e.g., p21, p15, p19, Bop1, and Htra1). By contrast, several of the well-known oncogenes including c-myc, N-myc, cyclin D1, cyclin J, cyclin B2, and thymidylate synthase showed significant downregulation. Notably, the expression of several of the regulators of renal morphogenesis _Osr1, Eya1, Pax2 and 8,Gdnf, WT1, Emx2, Wnt9b, Wnt4, Sfrp1 and 2, Lhx1 and FoxD1 is contingent upon normal HDAC activity. This finding from explant cultures is further exemplified in Scriptaid treated MK4 cells_ an immortalized cell line reminiscent of committed metanephric mesenchyme of the developing kidney. In MK4 cell lines, pursuant to HDAC inhibition the promoters or enhancers of developmental regulators Pax2, Pax8, Gdnf, Sfrp1, and p21 become hyper-acetylated. It follows that HDACs are essential for cell survival, cell proliferation, and differentiation processes in the embryonic kidney. De Groh et al. [26] found that HDAC inhibitors were capable of supporting the expansion of renal progenitor cells in the zebrafish pronephros. The establishment of the pronephric kidney field in zebrafish requires the morphogen, retinoic acid. In the absence of this morphogen, retinoic acid receptor dimers bind co-repressor complexes bearing HDACs in order to silence its target genes. The inhibition of HDACs is believed to lower the threshold of retinoic acid needed to activate target genes like cyp26a1 and allow renal progenitor cell expansion. In conclusion, HDACs modulate retinoic acid signaling to establish the early kidney field. The apparent contradiction in the outcome of HDAC inhibition on cellular proliferation may be intrinsic to differences in pronephros versus metanephros morphogenesis.
Gene deletion and overexpression studies have assigned a developmental role for HDACs 1, 2, and 5 [27-29]. The conditional loss of HDAC1 or HDAC2 is well tolerated in multiple tissues and the mice are viable. However, co-deletion of HDACs 1 and 2 is detrimental in all tissues examined [29]. This is borne out in the metanephric kidney. Combined loss of HDACs 1 and 2 either from the ureteric bud or nephron progenitors causes renal cystic hypodysplasia and early postnatal death [22,24, and manuscript in preparation]. The cap mesenchyme shows premature depletion in the neonates. Absence of HDACs 1 and 2 in the ureteric bud lineage disrupts branching morphogenesis early owing to reduced cell proliferation and elevated apoptosis. In the nephron progenitors, HDACs 1 and 2 do not impart a survival role but instead support cell proliferation. Nephron formation arrests at the renal vesicle stage in these mutants resulting in fewer nephron numbers. Increased acetylation of p53 was noted in both conditional models following the loss of HDACs 1 and 2. The renal developmental pathways most affected were the Wnt, Sonic Hedgehog, and p53 pathways.
Gene-environment interactions: The intrauterine environment can alter the epigenetic landscape enough to impact fetal development [13]. These aberrant epigenetic changes established in utero and perpetuated through multiple cellular generations may form the basis of certain diseases. Fetal malnutrition or utero-placental insufficiency cause fetal growth restriction and low birth weight. Low birth weight is associated with a higher risk for chronic kidney disease, hypertension, diabetes, and cardiovascular disease [13,30,31]. The epigenetic contribution of HDACs in the context of gene-environment interaction is exemplified by the bradykinin B2 receptor (Bdkrb2) null mice [32-34]. The progeny of Bdkrb2 null mice have no overt phenotype when their mothers are fed a normal salt diet (0.3 percent NaCl) but exhibit renal dysgenesis and perinatal lethality when exposed to high salt stress in utero (5 percent NaCl). Using genetic rescue models and chromatin immunoprecipitation assays a molecular network involving HDACs, p53, and E-cadherin was discovered. The Bdkrb2 mutant mice under salt stress show direct repression of E-cadherin by phosphorylated-p53Ser23 following the deacetylation of E-cadherin promoter by HDACs. Apart from E-cadherin, the cooperativity between HDACs and p53 extends to the polycystic kidney disease 1 gene [35]. Therefore, intra-uterine epigenetic reprogramming of gene expression and function is a potential contributing factor in chronic kidney diseases. Phenotypically it can manifest as low nephron endowment, glomerular sclerosis, and compensatory glomerular hypertrophy [30].
Marumo et al. [36] examined the contribution of HATs and HDACs to regeneration following ischemic injury of the kidney. They found that ischemia reduces cellular levels of ATP and HATs causing a decline in histone acetylation levels in the proximal tubular cells of the outer medulla. Subsequently, the inhibition of HDAC5 was essential for the restoration of histone acetylation levels and the induction of BMP7 during the regeneration of the tubular epithelia. HDAC inhibition was also found to improve the therapeutic outcome of polycystic kidney disease in genetic mouse models of the disease [37,38]. In these instances, HDAC5 and HDAC6 inhibition contributed to fewer cysts and less compromised kidney function. HDAC6 has been implicated in ciliary biogenesis, EGFR trafficking, and the regulation of Wnt signaling via its effects on b-catenin. Inhibition of HDAC6 prevents nuclear translocation of b-catenin and lower c-Myc expression which prevents expansion of cystic epithelia [39]. Since, lowering of HDAC levels is permissive for kidney regeneration and polycystic kidney disease management the therapeutic potential of its inhibitors warrants a thorough investigation.
Histone Methylation and Methyltransferases in the Kidney
The link between epigenetic regulation and development became evident from studies on Drosophila. The histone methyltransferases and their co-factors were subsequently grouped under two main categories: Polycomb or Trithorax. Methylation of histones is mediated by histone methyltransferases (HMTs) and histone demethylases (HDMs). Tri-methylation of lysines 4 and 36 (H3K4me3 and H3K36me3) is commonly indicative of gene activation while di- and tri-methylation of lysines 9 and 27 (H3K9me2/3 and H3K27me2/3) generally signify gene repression [40,41]. Mouse models of diabetic nephropathy have elevated levels of H3K9 and H3K23 acetylation, H3K4 di-methylation and H3 Ser10 phosphorylation [42]. Histone methyltransferase SET7/9 plays a role in inflammation and diabetes [43].
The transcription factors Osr1, Lhx1, Pax2 and Pax8 have significant roles in specifying the renal lineage from the intermediate mesoderm. The expression of Pax2 is essential for the specification of renal epithelia and the formation of the nephric duct [44-46]. The DNA-binding protein, Pax2 helps recruit the Trithorax-like protein complex, MLL3/4 via the adaptor protein PTIP (Pax-transactivation domain interacting protein) (Figure 1). The MLL3/4 complex is believed to add H3K4me modifications making the chromatin active and permissive [47]. A similar association between Pax5 and PTIP allows chromatin looping at the Immunoglobulin heavy chain locus in B-cells [48]. Pax2 in binding co-repressors Grg4/Tle4 is able to recruit the Polycomb complex which adds repressive modifications to chromatin [49]. Thus, based on the choice of epigenetic co-factors that it binds Pax2 can toggle between activator and repressor of chromatin states.
Figure 1.
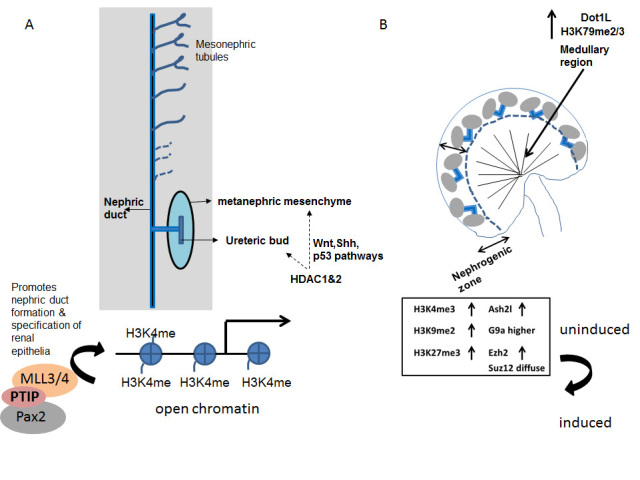
Renal cell fate is influenced by epigenetic modifications. (A) Specification of the nephric duct and renal epithelia requires the recruitment of the MLL complex by Pax2 via the PTIP adapter protein. (B) Loss of bivalent epigenetic code in nephron-fated induced mesenchyme. The cap mesenchyme represents the progenitors of all nephrons in the kidney. The promoters of lineage specific genes in this cell population are bivalent and carry both activation (H3K4me3) and repressive (H3K9me3, H3K27me3) methylation marks. Upon commitment to the formation of nephrons the promoters of renal-lineage genes lose the repressive marks (H3K9me3 and H3K27me3). The expression of lysine methyl transferases (Ash2l, G9a, Ezh2 and Suz12) coincide with that of their corresponding epigenetic modifications.
McLaughlin et al. [50] examined the epigenetic states of nephron progenitors residing in the cap mesenchyme. The self-renewing cap mesenchyme cells are Six2-positive and are capable of committing to the nephrogenic lineage in response to Wnt signals emanating from the ureteric bud. Upon commitment to the nephrogenic lineage the cells begin to express Wnt4, Lhx1, Pax8, and Fgf8. Stage-specific nephron differentiation strongly correlates with dynamic changes in epigenetic modifications to histones both spatially and temporally (Figure 1). Notably, at E15.5, the mouse nephron progenitors in the cap mesenchyme show an equal enrichment of active and repressive marks (H3K4me3, H3K9me3 and H3K27me3). In the nascent nephrons, however, the repressive marks H3K9me3 and H3K27me3 are greatly downregulated. During mid-organogenesis (E15.5) the activation mark, H3K79me3 shows a stronger expression in the inner cortical tubules relative to the nascent nephron and cap mesenchyme residing in the outer cortex. In the adult kidney, H3K79me3 is enriched in the differentiating segments of nephrons and collecting ducts. The abundance of H3K4me3 and H3K79me3 were reciprocal in nature (Figure 2). The former is more abundant in the developing kidney while the latter increases in expression postnatally coinciding with nephron differentiation. The temporal expression levels of H3K9ac, H3K9me3, and H3K27me3 remains unchanged during kidney development [50].
Figure 2.

Reciprocal nature of H3K4me and H3K79me expression during development. The H3K4me3 marks are more abundant in the prenatal phase of the developing kidney while H3K79me3 levels increase during postnatal development of the kidney.
Not surprisingly the distribution of histone lysine methyltransferases (KMT) and histone lysine demethylases (KDM) overlaps that of the corresponding histone marks (Figure 1). Accordingly, the expression of H3K4 KMT, Ash2l resembles that of H3K4me3 within the nephrogenic zone of the E15.5 kidney. Ezh2, a component of the Polycomb repressor complex 2 (PRC2) has a pattern of expression similar to H3K27me3. Both are high in the Six2+ cap mesenchyme. Another member of the PRC2 complex, Suz12 is diffusely present in the cap mesenchyme and nascent nephrons. The H3 lysine 9 KMT, G9a is high in the cap mesenchyme relative to the nascent nephrons. It was also found expressed in the distal segment of the S-shaped nephrons. Similarly, the postnatal drop in levels of the H3K27me3 KDMs, Jmjd3 and Utx explains the steady state levels of this histone mark in the whole kidney pre- and postnatally. Two of the H3K27 KMTs, Ezh1 and Ezh2 are reciprocal in their spatial expression within the developing kidney and may account for the steady state levels of H2K27me3 during embryonic kidney development. The postnatal increase in H3K79me3 levels is similar to that of its KMT, Dot1L [50].
In addition to lysine residues McLaughlin et al also established the distribution of methylated arginine residues 2, 8, and 17 on histone H3. They reported that di-methylated forms of H3R2 (H3R2me2) and H3R17 (H3R17me2) are abundant in the cap mesenchyme and nascent nephrons. By contrast, H3R8me2 expression is stronger in the nascent nephrons [50]. Therefore, HMTs other than KMTs also have specific roles in renal development.
Chromatin bivalency in nephron progenitors: Comparative studies using clonal mesenchyme cell lines, MK3 (reminiscent of uninduced nephron progenitors) and MK4 (reminiscent of Wnt-induced progenitors) lend a better perspective to the distribution of histone modifications and modifiers in the developing kidney [51]. In un-induced MK3 cells, the promoters of renal progenitor genes Six2 and Osr1 have strong H3K4me3 peaks around the transcription start sites. In the more committed MK4 cell lines the promoters of these same genes are silenced by the loss of H3K4me3 and gain of the H3K4 demethylase, Kdm5b. The nephrogenic genes (e.g., Pax8, Jag1, Lef1, and Ccnd1) in MK3 cells are “bivalent,” harboring both activating (H3K4me3) and repressive (H3K27me3) histone marks. This epigenetic “bivalency” is a feature shared with embryonic stem cells (ESCs). A good many regulatory genes in ESCs are said to be poised for differentiation since they carry both activating and repressive epigenetic modifications [1]. When induced to differentiate into specific cell lineages this bivalency gets resolved by the loss or gain of the appropriate modifications. In MK4 cells or MK3 cell lines induced to differentiate with Wnt3a some of the nephrogenic gene promoters (e.g., Pax2 and Lhx1) show gain of H3K4me3 and loss of H3K27me3. Although the resolution of this “bivalent code” is significant for some it does not apply to all lineage specific genes for example Wnt4 and Notch2. Genome wide ChIP-seq analysis of MK3 and MK4 cells lines were also used to compare the epigenetic signatures of certain candidate genes associated with CAKUT. Four recurring themes of epigenetic signatures emerged from these comparisons: genes like Hoxa11 and Fgfrl1 with high H3K4me3 in both cell lines; loss of repressive mark H3K27me3 and gain of active mark H3K4me3 as seen with Blk and Wnt7b genes; loss of H3K4me3 seen in Hoxa13 and Tsc2 and finally gain of H3K4me3 on promoters of Lrp2 and Setdb1 genes [51].
DNA Methylation
In eukaryotes, DNA methylation occurs on CpG dinucleotides which are clustered in CpG islands commonly occurring on gene promoters. Aside from CpG islands, these dinucleotides are also found in regions with repetitive DNA sequences and in centromeric regions [52]. They may be distributed within genes or in the intergenic regions. The addition of methyl groups to the cytosine on CpG dinucleotides can modify gene function. The methylation of DNA may prevent the binding of the transcriptional machinery. Alternatively, methylated DNA is bound by methyl-CpG-binding domain proteins (MBDs) which can recruit specific chromatin modifiers to these sites. In cancer, methyl-CpG-binding domain protein 2 (MBD2 or MeCP2) causes transcriptional silencing of hypermethylated genes and has a role in Rett syndrome and autism [53,54]. Several of the tumor suppressor genes are transcriptionally silenced by DNA methylation.
Epigenetics of Disease
Cell fate is regulated largely by epigenetic mechanisms governed by cis-acting regulatory marks (on DNA and basic histone proteins) and trans-acting factors/enzymes. The epigenetic code is heritable through successive cell divisions and constitutes “cellular memory.” Changes in the density of DNA methylation both locally and globally, incorrect modifications on histones, perturbation of function and distribution of chromatin modifying enzymes represent genetic lesions with distinct pathological outcomes [55].
Loss of Genomic Imprinting and Epigenetic Lesions
Genomic imprinting is a mechanism by which addition of methylation marks bring about silencing of an allele at a given gene locus. In women, dosage compensation is accomplished by X-chromosome inactivation of imprinted genes [56]. DNA methyltransferase enzymes, DNMT3a and 3b are involved in setting up de novo methylation patterns very early in the development of an organism. DNMT1 functions to copy DNA methylation patterns on to daughter strands as the DNA replicates [57]. This accounts for the perpetuation of methylation patterns though multiple cell divisions. In the kidney, DNA methylation plays a direct role in the genesis of renal fibrosis. Inhibition of DNA methylation using 5’azacytidine protected mice from renal fibrosis [58]. Aberrant DNA methylation was reported in IUGR (intrauterine growth restriction) subjects by Einstein et al [59]. They found that DNA methylation at the HNF4a promoter of IUGR subjects increased their susceptibility to Type 2 diabetes.
Monogenic disorders of imprinted genes and components of the epigenetic machinery represent two classes of epigenetic disorders. Loss of normal imprinting on chromosome 11 p15 (H19/IGF2) results in Beckwith-Wiedemann syndrome characterized by tissue overgrowth and elevated cancer risk [60,61]. Similarly, Prader Willi Syndrome and Angelman syndrome results from micro-deletions in imprinted genes SNRPN (small nuclear ribonucleoprotein polypeptide N) and UBE3A (ubiquitin protein ligase E3A) on chromosome 15 [62]. Rett Syndrome and ICF (Immunodeficiency, centromeric instability, facial anomalies) syndrome are examples of monogenic disorders affecting components of the epigenetic machinery [63,64]. Rett syndrome patients have mutations in MeCP2 (methyl-CpG-binding protein 2) gene causing disruption of epigenetic silencing resulting in postnatal neurodegeneration in children. ICF syndrome is caused by mutations in the DNA methyltransferase gene DNMT3B and is characterized by defects of the immune system and developmental disorders. Alteration in the global or local methylation of DNA is commonly encountered in cancers. Many tumors show hypomethylation at promoters of growth promoting genes (e.g., HRAS, cyclin D2, maspin, S100A4, MAGE gene family, PAX2) or silencing of tumor suppressor genes (RB, p16, VHL, APC, E-cadherin). The loss of imprinting of a normally silent allele of a gene (IGF2) is another mechanism which contributes to cancer pathology (Wilm’s tumor) [55]. Over-expression of the H3K27 methyltransferase EZH2 has been reported in the etiology of metastatic prostate cancer [65]. Lymphoma and colorectal cancers show loss of H4K16 aceytlation and H4K20 tri-methylation marks [66]. MLL-1, the H3K4 methyltransferase is over-expressed in acute lymphoblastic leukemia [67]. Mutations in CBP (CREB-binding protein), a transcription factor with histone acetytransferase activity is the cause of Rubinstein-Tabi syndrome typified by skeletal, cardiac, and neural developmental disorders [68].
Epigenetics in Acute or Chronic Kidney Disease
Chronic kidney disease (CKD) is marked by a progressive decline in kidney function and culminates in end stage renal disease (ESRD) [69]. Its etiology is highly varied involving genetic and epigenetic components. Environmental insults can lead to altered metabolic states (e.g., hyperglycemia in diabetes and uremia in CKD) which in turn can trigger changes in chromatin modifications and gene expression [70]. Hyperglycemia in rat mesangial cells causes elevated expression of the H3K4 methyltransferase, SET7/9 and the corresponding active histone marks (H3K4me1-3) on the promoters of extracellular matrix/fibrotic genes _connective tissue growth factor, collagen1, plasminogen activator inhibitor-1 downstream of TGF-b1 signaling [71]. In this model of diabetic nephropathy the elevation in fibrotic gene expression was reversed by siRNA silencing of SET7/9 or by function perturbing antibodies against TGF-b1 which reduces SET7/9 and H3K4 association/occupancy on the promoters of these genes. The reversible nature of epigenetic modifications makes them highly amenable to therapeutics applications. This potential is exemplified by Losartan which reverses the epigenetic changes in db/db diabetic mice [72]. The identification of epigenetic markers associated with CKD will enable risk estimation and allow the optimization of treatment options for personalized medicine.
Epigenetic regulation of gene expression via micro RNAs (RNA interference) ensures normal kidney development and homeostasis [73]. The inactivation of dicer, an enzyme central to micro RNA synthesis in mouse podocytes leads to renal failure and lethality [74,75]. Diabetic nephropathy in humans shows a correlation between TGF-b1 induction of miR-192 and fibrosis [76]. In mouse models of diabetic kidney disease both fibrosis and proteinuria were reversed following miR-192 inhibition [77]. For a more comprehensive listing of miRNAs associated with kidney diseases the readers are directed to the review by Smyth et al [70] and the references therein. Multiple large-scale association studies (GWAS) have uncovered DNA methylation targets of CKD. Some that promote renal fibrosis are NPHP4 (nephronophthisis 4), IQSEC1, TCF3, nitric oxide synthase 3 (NOS3), NFkBIL2 (NF-κB light polypeptide gene enhancer in B cells inhibitor-like2), TGFb3, and TGFb1 [78,79].
It has been demonstrated that epigenetic modifications established during the diseased state persists long after the initial insult has been removed [13]. From a therapeutic standpoint, the treatment of diseases can benefit from the knowledge of altered epigenetic states at pathogenic gene loci. Elevated glucose levels cause oxidative stress owing to enhanced expression of the pro-inflammatory gene, thioredoxin-interacting protein (TXNIP). The oxidative injury to mesangial cells and podocytes under hyperglycemic conditions can be attenuated by the knockdown of TXNIP or the upregulation of histone methyltransferase EZH2 [41,80,81]. Histone modifiers EZH2, Suv39h1 and their modifications H3K27me3 and H3K9me3 were decreased at the promoters of inflammatory genes during diabetic nephropathy. High glucose mediated upregulation of TXNIP expression can be countered by inhibiting histone acetylation. Also, the pathogenesis of renal fibrosis is driven by DNMT1 mediated hypermethylation of Rasal1 and can be reversed by TET3-mediated upregulation of Rasal 1. Thus, addressing the epigenetic etiology of chronic kidney diseases could offer novel therapeutic avenues.
Therapeutic Applications of Epigenetics
The knowledge of epigenetics offers both prognostic and therapeutic value [82]. Malignant tumors typically show hyper-methylation of CpG islands on tumor suppressor genes [83]. Hyper-methylation of KISS1, a suppressor of metastasis, is used in the prognosis of bladder tumors [84]. In other instances, hypo-methylation of DNA (e.g., LINE-1) is used as a bio marker of urothelial cancer risk [85,86]. Micro-RNAs, miR-141, miR-200c, and miR-30b show elevated expression in bladder cancers and can be used in their diagnosis [87]. Drugs that function as inhibitors of DNA methyltransferases (Zebularine) or histone deacetylases (Romidepsin, Vorinostat, Panobinostat) have been employed to restore tumor suppressor functions [88-90]. The HDAC inhibitor Vorinostat has renoprotective properties in addition to its use in anti-cancer therapy. Other histone deacetylase inhibitors such as Lys-CoA (p300/CBP inhibitor), Valproic acid, and SAHA (suberoylanilide hydroxamic acid) could have utility in cancer therapy [91,92] However, efficacy of these inhibitors has been offset owing to the toxicity of these inhibitors.
The success and fidelity of tissue regeneration-based therapies relies on our knowledge of gene regulatory mechanisms. It is now known that induced pluripotent stem cells (iPSCs) retain epigenetic memory of their tissue of origin [93]. A correlation has emerged between persistent hypermethylation in iPSCs depending on its tissue of origin and its plasticity. Cataloging of the epigenetic modifications and modifiers is essential to overcome the barriers to tissue reprogramming and to devise new approaches to ensure the success of tissue regeneration techniques.
Conclusions and Outlook
Epigenetic modifications are important for cell lineage specification. This review enumerates some of the epigenetic aspects that ensure normal kidney development and the pathological outcomes of epigenetic dysfunction. In it we describe how the epigenetic signature of the self-renewing cap mesenchyme cells is distinct from that of the committed nascent nephron derivatives that they give rise to. We enumerate instances where the modifiers of epigenetic information (e.g., HDACs and KMTs) govern distinct aspects of renal development and the impact of environmental insults on cellular memory, epigenetic gene regulation, and disease states.
Recently, several high throughput genome-wide association studies correlating epigenetic landscapes to gene expression data (transcriptome studies) are being undertaken. Patterns of epigenetic modifications are emerging that can define cellular pluripotency, commitment, or pathological states. The advent of targeted epigenomic editors (comprising a “DNA targeting module fused to a chromatin regulator”) has set us on a new path of tissue engineering and drug discovery [94,95]. This field has the potential to offer innovative approaches for treating chronic kidney diseases as an alternative to existing renal replacement therapies such as dialysis and kidney transplantation. However, given the complexity of chromatin structure, the combinatorial nature of the epigenetic code, and the context dependent variation in epigenetic modifications the design and utility of these techniques are fraught with multiple challenges.
Acknowledgments
Research reported in this publication was supported by the National Institutes of Health grants P50 DK096373-03 and P30GM103337.
Glossary
- CAKUT
Congenital abnormalities of the kidney and urinary tract
- HAT
histone acetyltransferase
- HDAC
histone deacetylase
- KMT
histone lysine methyltransferases
- KDM
histone lysine demethylases
- PRC2
Polycomb repressor complex 2
- Ezh2
Enhancer of Zeste
- ChIP
chromatin immunoprecipitation
References
- Harikumar A, Meshorer E. Chromatin remodeling and bivalent histone modifications in embryonic stem cells. EMBO Rep. 2015;16(12):1609–1619. doi: 10.15252/embr.201541011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luger K, Mäder AW, Richmond RK. et al. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;389(6648):251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- Turinetto V, Giachino C. Histone variants as emerging regulators of embryonic stem cell identity. Epigenetics. 2015;10(7):563–573. doi: 10.1080/15592294.2015.1053682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan L, Roberts VA. Complex of linker histone H5 with the nucleosome and its implications for chromatin packing. PNAS. 2016;103(22):8384–8389. doi: 10.1073/pnas.0508951103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou YB, Gerchman SE, Ramakrishnan V. et al. Position and orientation of the globular domain of linker histone H5 on the nucleosome. Nature. 1998;395(6700):402–405. doi: 10.1038/26521. [DOI] [PubMed] [Google Scholar]
- Eslaminejad MB, Fani N, Shahhoseini M. Epigenetic regulation of osteogenic and chondrogenic differentiation of mesenchymal stem cells in culture. Cell J. 2013;15(1):1–10. [PMC free article] [PubMed] [Google Scholar]
- Qiu J. Epigenetics: unfinished symphony. Nature. 2006;441(7090):143–145. doi: 10.1038/441143a. [DOI] [PubMed] [Google Scholar]
- Zhou VW, Goren A, Bernstein BE. Charting histone modifications and the functional organization of mammalian genomes. Nat Rev Genet. 2011;12(1):7–18. doi: 10.1038/nrg2905. [DOI] [PubMed] [Google Scholar]
- Chen S, El-Dahr S. Histone deacetylases in kidney development: implications for disease and therapy. Pediatr Nephrol. 2013;28:689–698. doi: 10.1007/s00467-012-2223-8. [DOI] [PubMed] [Google Scholar]
- Nicolaou N, Renkema KY, Bongers EM. et al. Genetic, environmental, and epigenetic factors involved in CAKUT. Nat Rev Nephrol. 2015;11(12):720–731. doi: 10.1038/nrneph.2015.140. [DOI] [PubMed] [Google Scholar]
- Reidy KJ, Rosenblum ND. Cell and molecular biology of kidney development. Semin Nephrol. 2009;29(4):321–337. doi: 10.1016/j.semnephrol.2009.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dressler GR, Patel SR. Epigenetics in kidney development and renal disease. Transl Res. 2015;165(1):166–176. doi: 10.1016/j.trsl.2014.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woroniecki R, Gaikwad AB, Susztak K. Fetal environment, epigenetics, and pediatric renal disease. Pediatr Nephrol. 2011;26(5):705–711. doi: 10.1007/s00467-010-1714-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu F, Zhang Y. Cell totipotency: molecular features, induction, and maintenance. Natl Sci Rev. 2015;2(2):217–225. doi: 10.1093/nsr/nwv009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Yamanaka S. A decade of transcription factor-mediated reprogramming to pluripotency. Nat Rev Mol Cell Biol. 2016;17(3):183–193. doi: 10.1038/nrm.2016.8. [DOI] [PubMed] [Google Scholar]
- Hassig CA, Schreiber SL. Nuclear histone acetylases and deacetylases and transcriptional regulation: HATs off to HDACs. Curr Opin Chem Biol. 1997;1(3):300–308. doi: 10.1016/s1367-5931(97)80066-x. [DOI] [PubMed] [Google Scholar]
- McKinsey TA, Zhang CL, Olson EN. Control of muscle development by dueling HATs and HDACs. Curr Opin Genet Dev. 2001;11(5):497–504. doi: 10.1016/s0959-437x(00)00224-0. [DOI] [PubMed] [Google Scholar]
- Marks PA, Miller T, Richon VM. Histone deacetylases. Curr Opin Pharmacol. 2003;3(4):344–351. doi: 10.1016/s1471-4892(03)00084-5. [DOI] [PubMed] [Google Scholar]
- Glozak MA, Sengupta N, Zhang X. et al. Acetylation and deacetylation of non-histone proteins. Gene. 2005;363:15–23. doi: 10.1016/j.gene.2005.09.010. [DOI] [PubMed] [Google Scholar]
- Kruse JP, Gu W. Modes of p53 regulation. Cell. 2009;137(4):609–622. doi: 10.1016/j.cell.2009.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spange S, Wagner T, Heinzel T. et al. Acetylation of non-histone proteins modulates cellular signalling at multiple levels. Int J Biochem Cell Biol. 2009;41(1):185–198. doi: 10.1016/j.biocel.2008.08.027. [DOI] [PubMed] [Google Scholar]
- Chen S, Yao X, Li Y. et al. Histone deacetylase 1 and 2 regulate Wnt and p53 pathways in the ureteric bud epithelium. Development. 2015;142(6):1180–1192. doi: 10.1242/dev.113506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez RA, Pentz ES, Jin X. et al. CBP and p300 are essential for renin cell identity and morphological integrity of the kidney. Am J Physiol Heart Circ Physiol. 2009;296(5):H1255–H1262. doi: 10.1152/ajpheart.01266.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Bellew C, Yao X. et al. Histone deacetylase (HDAC) activity is critical for embryonic kidney gene expression, growth, and differentiation. J Biol Chem. 2011;286(37):32775–32789. doi: 10.1074/jbc.M111.248278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, El-Dahr S. Histone deacetylases in kidney development: implications for disease and therapy. Pediatr Nephrol. 2013;28:689–698. doi: 10.1007/s00467-012-2223-8. [DOI] [PubMed] [Google Scholar]
- de Groh ED, Swanhart LM, Cosentino CC. et al. Inhibition of histone deacetylase expands the renal progenitor cell population. J Am Soc Nephrol. 2010;21(5):794–802. doi: 10.1681/ASN.2009080851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang S, McKinsey TA, Zhang CL. et al. Histone deacetylases 5 and 9 govern responsiveness of the heart to a subset of stress signals and play redundant roles in heart development. Mol Cell Biol. 2004;24(19):8467–8476. doi: 10.1128/MCB.24.19.8467-8476.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagger G, O'Carroll D, Rembold M. et al. Essential function of histone deacetylase 1 in proliferation control and CDK inhibitor repression. EMBO J. 2002;21(11):2672–2681. doi: 10.1093/emboj/21.11.2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery RL, Davis CA, Potthoff MJ. et al. Histone deacetylases 1 and 2 redundantly regulate cardiac morphogenesis, growth, and contractility. Genes Dev. 2007;21(14):1790–1802. doi: 10.1101/gad.1563807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luyckx VA, Brenner BM. Low birth weight, nephron number, and kidney disease. Kidney Int Suppl. 2005;97:S68–S77. doi: 10.1111/j.1523-1755.2005.09712.x. [DOI] [PubMed] [Google Scholar]
- Alexander BT. Intrauterine growth restriction and reduced glomerular number: role of apoptosis. Am J Physiol Regul Integr Comp Physiol. 2003;285(5):R933–R934. doi: 10.1152/ajpregu.00446.2003. [DOI] [PubMed] [Google Scholar]
- El-Dahr SS, Aboudehen K, Dipp S. Bradykinin B2 receptor null mice harboring a Ser23-to-Ala substitution in the p53 gene are protected from renal dysgenesis. Am J Physiol Renal Physiol. 2008;295(5):F1404–F1413. doi: 10.1152/ajprenal.90378.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Dahr SS, Harrison-Bernard LM, Dipp S. et al. Bradykinin B2 null mice are prone to renal dysplasia: gene-environment interactions in kidney development. Physiol Genomics. 2000;3(3):121–131. doi: 10.1152/physiolgenomics.2000.3.3.121. [DOI] [PubMed] [Google Scholar]
- Fan H, Harrell JR, Dipp S. et al. A novel pathological role of p53 in kidney development revealed by gene-environment interactions. Am J Physiol Renal Physiol. 2005;288(1):F98–F107. doi: 10.1152/ajprenal.00246.2004. [DOI] [PubMed] [Google Scholar]
- van Bodegom D, Roessingh W, Pridjian A. et al. Mechanisms of p53-mediated repression of the human polycystic kidney disease-1 promoter. Biochim Biophys Acta. 2010;1799(7):502–509. doi: 10.1016/j.bbagrm.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marumo T, Hishikawa K, Yoshikawa M. et al. Epigenetic regulation of BMP7 in the regenerative response to ischemia. J Am Soc Nephrol. 2008;19(7):1311–1320. doi: 10.1681/ASN.2007091040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y, Semanchik N, Lee SH. et al. Chemical modifier screen identifies HDAC inhibitors as suppressors of PKD models. Proc Natl Acad Sci U S A. 2009;106(51):21819–21824. doi: 10.1073/pnas.0911987106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia S, Li X, Johnson T. et al. Polycystin-dependent fluid flow sensing targets histone deacetylase 5 to prevent the development of renal cysts. Development. 2010;137(7):1075–1084. doi: 10.1242/dev.049437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X. Epigenetics and autosomal dominant polycystic kidney disease. Biochim Biophys Acta. 2011;1812(10):1213–1218. doi: 10.1016/j.bbadis.2010.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eissenberg JC, Shilatifard A. Histone H3 lysine 4 (H3K4) methylation in development and differentiation. Dev Biol. 2010;339(2):240–249. doi: 10.1016/j.ydbio.2009.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiqi FS, Majumder S, Thai K. et al. The Histone Methyltransferase Enzyme Enhancer of Zeste Homolog 2 Protects against Podocyte Oxidative Stress and Renal Injury in Diabetes. J Am Soc Nephrol. 2016;27(7):2021–2034. doi: 10.1681/ASN.2014090898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayyed SG, Gaikwad AB, Lichtnekert J. et al. Progressive glomerulosclerosis in type 2 diabetes is associated with renal histone H3K9 and H3K23 acetylation, H3K4 dimethylation and phosphorylation at serine 10. Nephrol Dial Transplant. 2010;25(6):1811–1817. doi: 10.1093/ndt/gfp730. [DOI] [PubMed] [Google Scholar]
- Li Y, Reddy MA, Miao F. et al. Role of the histone H3 lysine 4 methyltransferase, SET7/9, in the regulation of NF-kappaB-dependent inflammatory genes. Relevance to diabetes and inflammation. J Biol Chem. 2008;283(39):26771–26781. doi: 10.1074/jbc.M802800200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchard M, Souabni A, Mandler M. et al. Nephric lineage specification by Pax2 and Pax8. Genes Dev. 2002;16(22):2958–2970. doi: 10.1101/gad.240102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brophy PD, Ostrom L, Lang KM. et al. Regulation of ureteric bud outgrowth by Pax2-dependent activation of the glial derived neurotrophic factor gene. Development. 2001;128(23):4747–4756. doi: 10.1242/dev.128.23.4747. [DOI] [PubMed] [Google Scholar]
- Soofi A, Levitan I, Dressler GR. Two novel EGFP insertion alleles reveal unique aspects of Pax2 function in embryonic and adult kidneys. Dev Biol. 2012;365(1):241–250. doi: 10.1016/j.ydbio.2012.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel SR, Kim D, Levitan I. et al. The BRCT-domain containing protein PTIP links PAX2 to a histone H3, lysine 4 methyltransferase complex. Dev Cell. 2007;13(4):580–592. doi: 10.1016/j.devcel.2007.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab KR, Patel SR, Dressler GR. Role of PTIP in class switch recombination and long-range chromatin interactions at the immunoglobulin heavy chain locus. Mol Cell Biol. 2011;31(7):1503–1511. doi: 10.1128/MCB.00990-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel SR, Bhumbra SS, Paknikar RS. et al. Epigenetic mechanisms of Groucho/Grg/TLE mediated transcriptional repression. Mol Cell. 2012;45(2):185–195. doi: 10.1016/j.molcel.2011.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin N, Wang F, Saifudeen Z. et al. In situ histone landscape of nephrogenesis. Epigenetics. 2014;9(2):222–235. doi: 10.4161/epi.26793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin N, Yao X, Li Y. et al. Histone signature of metanephric mesenchyme cell lines. Epigenetics. 2013;8(9):970–978. doi: 10.4161/epi.25753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boland MJ, Nazor KL, Loring JF. Epigenetic regulation of pluripotency and differentiation. Circ Res. 2014;115(2):311–324. doi: 10.1161/CIRCRESAHA.115.301517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho KL, McNae IW, Schmiedeberg L. et al. MeCP2 binding to DNA depends upon hydration at methyl-CpG. Mol Cell. 2008;29(4):525–531. doi: 10.1016/j.molcel.2007.12.028. [DOI] [PubMed] [Google Scholar]
- LaSalle JM, Yasui DH. Evolving role of MeCP2 in Rett syndrome and autism. Epigenomics. 2009;1(1):119–130. doi: 10.2217/epi.09.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinberg AP. Phenotypic plasticity and the epigenetics of human disease. Nature. 2007;447(7143):433–440. doi: 10.1038/nature05919. [DOI] [PubMed] [Google Scholar]
- Reik W, Dean W, Walter J. Epigenetic reprogramming in mammalian development. Science. 2001;293(5532):1089–1093. doi: 10.1126/science.1063443. [DOI] [PubMed] [Google Scholar]
- Susztak K. Understanding the epigenetic syntax for the genetic alphabet in the kidney. J Am Soc Nephrol. 2014;25(1):10–17. doi: 10.1681/ASN.2013050461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechtel W, McGoohan S, Zeisberg EM. et al. Methylation determines fibroblast activation and fibrogenesis in the kidney. Nat Med. 2010;16(5):544–550. doi: 10.1038/nm.2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Einstein F, Thompson RF, Bhagat TD. et al. Cytosine methylation dysregulation in neonates following intrauterine growth restriction. PLoS One. 2010;5(1):e8887. doi: 10.1371/journal.pone.0008887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBaun MR, Niemitz EL, McNeil DE. et al. Epigenetic alterations of H19 and LIT1 distinguish patients with Beckwith-Wiedemann syndrome with cancer and birth defects. Am J Hum Genet. 2002;70(3):604–611. doi: 10.1086/338934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparago A, Cerrato F, Vernucci M. et al. Microdeletions in the human H19 DMR result in loss of IGF2 imprinting and Beckwith-Wiedemann syndrome. Nat Genet. 2004;36(9):958–960. doi: 10.1038/ng1410. [DOI] [PubMed] [Google Scholar]
- Horsthemke B, Buiting K. Imprinting defects on human chromosome 15. Cytogenet Genome Res. 2006;113(1-4):292–299. doi: 10.1159/000090844. [DOI] [PubMed] [Google Scholar]
- Bienvenu T, Chelly J. Molecular genetics of Rett syndrome: when DNA methylation goes unrecognized. Nat Rev Genet. 2006;7(6):415–426. doi: 10.1038/nrg1878. [DOI] [PubMed] [Google Scholar]
- Xu GL, Bestor TH, Bourc'his D. et al. Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature. 1999;402(6758):187–191. doi: 10.1038/46052. [DOI] [PubMed] [Google Scholar]
- Varambally S, Dhanasekaran SM, Zhou M. et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature. 2002;419(6907):624–629. doi: 10.1038/nature01075. [DOI] [PubMed] [Google Scholar]
- Fraga MF, Ballestar E, Villar-Garea A. et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat Genet. 2005;37(4):391–400. doi: 10.1038/ng1531. [DOI] [PubMed] [Google Scholar]
- Rozenblatt-Rosen O, Rozovskaia T, Burakov D. et al. The C-terminal SET domains of ALL-1 and TRITHORAX interact with the INI1 and SNR1 proteins, components of the SWI/SNF complex. Proc Natl Acad Sci U S A. 1998;95(8):4152–4157. doi: 10.1073/pnas.95.8.4152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrij F, Giles RH, Dauwerse HG. et al. Rubinstein-Taybi syndrome caused by mutations in the transcriptional co-activator CBP. Nature. 1995;376(6538):348–351. doi: 10.1038/376348a0. [DOI] [PubMed] [Google Scholar]
- Shafi T. CKD to ESRD transition: does assessment of kidney function matter? Nephrol Dial Transplant. 2016 doi: 10.1093/ndt/gfw327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth LJ, Duffy S, Maxwell AP. et al. Genetic and epigenetic factors influencing chronic kidney disease. Am J Physiol Renal Physiol. 2014;307(7):F757–F776. doi: 10.1152/ajprenal.00306.2014. [DOI] [PubMed] [Google Scholar]
- Sun G, Reddy MA, Yuan H. et al. Epigenetic histone methylation modulates fibrotic gene expression. J Am Soc Nephrol. 2010;21(12):2069–2080. doi: 10.1681/ASN.2010060633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy MA, Sumanth P, Lanting L. et al. Losartan reverses permissive epigenetic changes in renal glomeruli of diabetic db/db mice. Kidney Int. 2014;85(2):362–373. doi: 10.1038/ki.2013.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagalakshmi VK, Ren Q, Pugh MM. et al. Dicer regulates the development of nephrogenic and ureteric compartments in the mammalian kidney. Kidney Int. 2011;79(3):317–330. doi: 10.1038/ki.2010.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel V, Noureddine L. MicroRNAs and fibrosis. Curr Opin Nephrol Hypertens. 2012;21(4):410–416. doi: 10.1097/MNH.0b013e328354e559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi S, Yu L, Chiu C. et al. Podocyte-selective deletion of dicer induces proteinuria and glomerulosclerosis. J Am Soc Nephrol. 2008;19(11):2159–2169. doi: 10.1681/ASN.2008030312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krupa A, Jenkins R, Luo DD. et al. Loss of MicroRNA-192 promotes fibrogenesis in diabetic nephropathy. J Am Soc Nephrol. 2010;21(3):438–447. doi: 10.1681/ASN.2009050530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putta S, Lanting L, Sun G. et al. Inhibiting microRNA-192 ameliorates renal fibrosis in diabetic nephropathy. J Am Soc Nephrol. 2012;23(3):458–469. doi: 10.1681/ASN.2011050485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth LJ, McKay GJ, Maxwell AP. et al. DNA hypermethylation and DNA hypomethylation is present at different loci in chronic kidney disease. Epigenetics. 2014;9(3):366–376. doi: 10.4161/epi.27161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wing MR, Devaney JM, Joffe MM. et al. DNA methylation profile associated with rapid decline in kidney function: findings from the CRIC study. Nephrol Dial Transplant. 2014;29(4):864–872. doi: 10.1093/ndt/gft537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Marinis Y, Cai M, Bompada P. et al. Epigenetic regulation of the thioredoxin-interacting protein (TXNIP) gene by hyperglycemia in kidney. Kidney Int. 2016;89(2):342–353. doi: 10.1016/j.kint.2015.12.018. [DOI] [PubMed] [Google Scholar]
- Shah A, Xia L, Goldberg H. et al. Thioredoxin-interacting protein mediates high glucose-induced reactive oxygen species generation by mitochondria and the NADPH oxidase, Nox4, in mesangial cells. J Biol Chem. 2013;288(10):6835–6848. doi: 10.1074/jbc.M112.419101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harb-de la Rosa A, Acker M, Kumar RA. et al. Epigenetics application in the diagnosis and treatment of bladder cancer. Can J Urol. 2015;22(5):7947–7951. [PubMed] [Google Scholar]
- Dudziec E, Miah S, Choudhry HM. et al. Hypermethylation of CpG islands and shores around specific microRNAs and mirtrons is associated with the phenotype and presence of bladder cancer. Clin Cancer Res. 2011;17(6):1287–1296. doi: 10.1158/1078-0432.CCR-10-2017. [DOI] [PubMed] [Google Scholar]
- Cebrian V, Fierro M, Orenes-Piñero E. et al. KISS1 methylation and expression as tumor stratification biomarkers and clinical outcome prognosticators for bladder cancer patients. Am J Pathol. 2011;179(2):540–546. doi: 10.1016/j.ajpath.2011.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patchsung M, Boonla C, Amnattrakul P. et al. Long interspersed nuclear element-1 hypomethylation and oxidative stress: correlation and bladder cancer diagnostic potential. PLoS One. 2012;7(5):e37009. doi: 10.1371/journal.pone.0037009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo HD, Kim J. Global DNA hypomethylation in peripheral blood leukocytes as a biomarker for cancer risk: a meta-analysis. PLoS One. 2012;7(4):e34615. doi: 10.1371/journal.pone.0034615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahdavinezhad A, Mousavi-Bahar SH, Poorolajal J. et al. Evaluation of miR-141, miR-200c, miR-30b Expression and Clinicopathological Features of Bladder Cancer. Int J Mol Cell Med. 2015;4(1):32–39. [PMC free article] [PubMed] [Google Scholar]
- Cheng JC, Matsen CB, Gonzales FA. et al. Inhibition of DNA methylation and reactivation of silenced genes by zebularine. J Natl Cancer Inst. 2003;95(5):399–409. doi: 10.1093/jnci/95.5.399. [DOI] [PubMed] [Google Scholar]
- O'Rourke CJ, Knabben V, Bolton E. et al. Manipulating the epigenome for the treatment of urological malignancies. Pharmacol Ther. 2013;138(2):185–196. doi: 10.1016/j.pharmthera.2013.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Cheng X, Connolly BA. et al. Zebularine: a novel DNA methylation inhibitor that forms a covalent complex with DNA methyltransferases. J Mol Biol. 2002;321(4):591–599. doi: 10.1016/S0022-2836(02)00676-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer NG, Ozdag H, Caldas C. p300/CBP and cancer. Oncogene. 2004;23(24):4225–4231. doi: 10.1038/sj.onc.1207118. [DOI] [PubMed] [Google Scholar]
- Phiel CJ, Zhang F, Huang EY. et al. Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J Biol Chem. 2001;276(39):36734–36741. doi: 10.1074/jbc.M101287200. [DOI] [PubMed] [Google Scholar]
- Kim K, Doi A, Wen B. et al. Epigenetic memory in induced pluripotent stem cells. Nature. 2010;467(7313):285–290. doi: 10.1038/nature09342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou VW, Goren A, Bernstein BE. Charting histone modifications and the functional organization of mammalian genomes. Nat Rev Genet. 2011;12(1):7–18. doi: 10.1038/nrg2905. [DOI] [PubMed] [Google Scholar]
- Park M, Keung AJ, Khalil AS. The epigenome: the next substrate for engineering. Genome Biol. 2016;17(1):183. doi: 10.1186/s13059-016-1046-5. [DOI] [PMC free article] [PubMed] [Google Scholar]