

Abstract
Creutzfeldt–Jakob disease (CJD) is a rare neurodegenerative disorder characterized by rapidly progressing dementia, general neurologic deterioration, and death. When the leading symptoms are visual disturbances, it is termed as the Heidenhain variant of CJD (HvCJD). CJD was reported following prion-contaminated pericardium transplants but never after bovine bioprosthetic cardiac valve. In this case report, we describe HvCJD in a patient who had a bovine bioprosthetic cardiac valve implant. An 82-year-old-woman was referred to neuro-ophthalmology clinic for unexplained visual loss that started 1 month previously. Medical history included aortic valve replacement with bovine bioprosthetic valve. On examination, best-corrected visual acuity was 20/120 in the right eye and 20/200 in the left eye; otherwise, the eye examination was normal. Humphrey visual fields revealed complete right homonymous hemianopsia. Magnetic resonance imaging (MRI) demonstrated nonspecific white matter changes. A week later, she was hospitalized due to memory impairment; repeated MRI and total body computed tomography scan showed no significant findings. Electroencephalography recordings and extremely elevated cerebrospinal fluid tau protein were compatible with CJD. The patient died 3 weeks later; autopsy was not performed. The patient had HvCJD. Ophthalmologists being first to see these patients should be aware of this diagnosis. Contaminated bovine bioprosthetic valve might be another source for prion disease. Further research is required to establish this issue.
Keywords: Bovine bioprosthetic cardiac valve, Heidenhain variant of Creutzfeldt–Jakob disease, prion, unexplained visual loss
Creutzfeldt–Jakob disease (CJD) is the most common human prion disease with an incidence of 1 case per 1 million populations per year; it is a rare neurodegenerative disease associated with severe neuronal loss, spongiform changes, and accumulation of the abnormal prion protein. About 85% of CJD cases are sporadic CJD (sCJD), 5%–15% are familial CJD, and <1% are iatrogenic CJD. Six clinical phenotypes of sCJD have been reported; the “classic CJD” phenotype includes myoclonic and Heidenhain variant (MM1 and MV1), accounts for 70% of sCJD cases, and is associated with advanced age at onset, rapidly progressive dementia with early-prominent myoclonus, and a short duration of illness (mean 3.9 months); visual signs may precede severe dementia in about 30% of cases.[1] When sCJD presents with isolated visual symptoms which persist even without any cognitive decline for a few weeks – it is termed the Heidenhain variant.
Case Report
An 82-year-old Jewish woman, Ukrainian descendant, was referred to neuro-ophthalmology clinic for unexplained progressive visual loss over the past month with a significant deterioration in the last few days. The patients and her son denied any behavioral change or memory impairment at this point.
Medical history included hypertension and aortic valve replacement with bovine 19 mm bioprosthetic valve, performed 5 years before her current presentation.
On examination, her best-corrected visual acuity was 20/120 in the right eye and 20/200 in the left eye, pupillary reactions and eye movement were normal, and applanation tonometry was 11/11; after dilating the pupils, both eyes revealed nonsignificant cataract, normal optic disc and retina. Humphrey visual fields revealed complete right homonymous hemianopsia with bilateral macular involvement [Fig. 1]. Brain magnetic resonance imaging (MRI) demonstrated diffuse atrophic and nonspecific white matter changes along with small cystic lesions and anatomical distortion at the basal ganglia level, no cortical pathology [Fig. 2].
Figure 1.

Automated visual fields showing right homonymous hemianopsia with bilateral macular involvement
Figure 2.
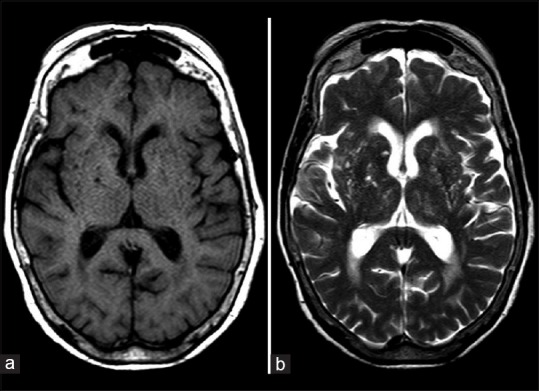
Axial T1 (a) and T2 (b) magnetic resonance imaging demonstrating brain atrophic changes and cystic changes (hypointense lesions in T1 and hyperintense in T2 along with anatomical distortion at the basal ganglia level
One week later, the patient was admitted to the hospital due to memory impairment; repeated complete blood count, chemistry and rheumatologic panel were within normal limits. Electroencephalography (EEG) revealed diffuse slowing and periodic sharp wave discharges along with triphasic wave complexes [Fig. 3]; repeated brain MRI and total body computed tomography showed no significant changes compared to previous studies; cerebrospinal fluid (CSF) revealed extremely elevated tau protein >1200 pg/ml compatible with CJD.
Figure 3.

Electroencephalography showing general slowing with triphasic waves
During her hospitalization, the patient developed rapid loss of short and long memory, disorientation, incontinence, and brisk reflexes. She was discharged home according to her family request and died 3 weeks later. Unfortunately, autopsy was not performed.
Discussion
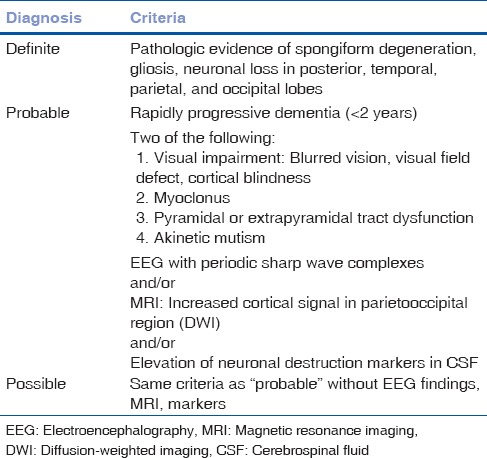
Our patient presented with progressive visual decline over 5 weeks, followed by rapidly progressive dementia and extrapyramidal tract dysfunction. The patient died 2.5 months after her presenting symptoms. According to the World Health Organization, the definite diagnosis of CJD is made by tissue diagnosis not performed in our patient.[2] Considering the disease course, rapidly progressive dementia (<2 years), visual impairment, extrapyramidal tract dysfunction, and the EEG findings, as well as the extremely elevated tau protein, our patient had probable CJD diagnosis [Table 1].
Table 1.
Diagnostic criteria for Creutzfeldt-Jakob disease
Human-to-human transmission of CJD was first reported in 1974 in a 55-year-old woman who developed symptoms 18 months after corneal transplantation from a donor who died of the disease. CJD transmission had been also reported following cadaveric dural graft transplants, radiographic embolization procedures with dura mater, liver transplants, administration of cadaveric human pituitary hormones, and by contaminated neurosurgical instruments. In 1989, a 54-year-old patient who received homograft pericardium for tympanic membrane closure 4 years earlier developed CJD.[3]
Although the definite pathogenesis is unknown, the primary component associated with the infection of CJD is an isoform of a normal host membrane glycoprotein called cellular prion protein (PrPC). The normal prion protein PrPC is protease sensitive, soluble and has a high α-helix content.[4] PrPC can undergo conversion into a conformationally altered isoform (scrapie prion protein [PrPSc]) widely believed to be the pathogenic agent of transmissible spongiform encephalopathies. The PrPSc is protease-resistant, insoluble, forms of amyloid fibrils and has high β-sheet content.[5]
The tissue-specific expression of PrPC is crucial considering that cells expressing high levels of PrPC bear a risk for conversion and accumulation of PrPSc. Quantitative and qualitative analysis of PrPC revealed that widespread tissue expression pattern of PrPC in bovine somatic tissues includes heart and myocardium;[6] it was found also in sheep,[7] hamster, and other nonhuman primates. Furthermore, expression of PrPSc was demonstrated in the hearts of transgenic mice as amyloid deposits that led to myocardial stiffness and cardiac disease.[8] PrPSc has been reported in the heart tissue of one sCJD patient.[9]
Prions exhibit an unusual resistance to conventional disinfection and sterilization methods; the procedures for sterilization of prion-contaminated medical instruments have been controversial for many years. Guidelines for sterilization that appear to be effective have been reported.[10] We presume that all of these guidelines may dismantle any biological tissue.
According to the manufacturer, our patient bioprosthetic aortic valve leaflets were made of bovine pericardium, and no information of bovine PrPSc screening of the pericardium leaflets was available.
We are unable to offer definite proof of the transmission of CJD by the bovine pericardium-made bioprosthetic aortic valve since autopsy was not performed. Still, the interval between the valve replacement surgery and the symptoms of the disease suggests that the pericardium was the conceivable vehicle for the transmission of the patient with CJD.
Conclusion
The patient's diagnosis was probable Heidenhain variant of CJD (HvCJD), suspected to be transmitted by the bovine valve transplantation never reported before. Bovine bioprosthetic valve made of pericardium might be another source for prion disease. Although this is a rare disease, ophthalmologists should be aware of the diagnosis being first to see the HvCJD of the CJD patients. Thorough medical history and good clinical examination and patients’ follow-up may lead to the correct investigation and diagnosis. Further research is required to establish this issue.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- 1.Parchi P, Giese A, Capellari S, Brown P, Schulz-Schaeffer W, Windl O, et al. Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol. 1999;46:224–33. [PubMed] [Google Scholar]
- 2.Brown P, Brunk C, Budka H, Cervenakova L, Collie D, Green A, et al. World Health Organization manual for surveillance of human transmissible spongiform encephalopathies including variant Creutzfeldt-Jakob disease. Geneva, Switzerland: World Health Organization; 2003. [Google Scholar]
- 3.Tange RA, Troost D, Limburg M. Progressive fatal dementia (Creutzfeldt-Jakob disease) in a patient who received homograft tissue for tympanic membrane closure. Eur Arch Otorhinolaryngol. 1990;247:199–201. doi: 10.1007/BF00178983. [DOI] [PubMed] [Google Scholar]
- 4.Glatzel M, Stoeck K, Seeger H, Lührs T, Aguzzi A. Human prion diseases: Molecular and clinical aspects. Arch Neurol. 2005;62:545–52. doi: 10.1001/archneur.62.4.545. [DOI] [PubMed] [Google Scholar]
- 5.Prusiner SB. Prions. Proc Natl Acad Sci U S A. 1998;95:13363–83. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peralta OA, Eyestone WH. Quantitative and qualitative analysis of cellular prion protein(PrP (C)) expression in bovine somatic tissues. Prion. 2009;3:161–70. doi: 10.4161/pri.3.3.9772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Horiuchi M, Yamazaki N, Ikeda T, Ishiguro N, Shinagawa M. A cellular form of prion protein (PrPC) exists in many non-neuronal tissues of sheep. J Gen Virol. 1995;76(Pt 10):2583–7. doi: 10.1099/0022-1317-76-10-2583. [DOI] [PubMed] [Google Scholar]
- 8.Trifilo MJ, Yajima T, Gu Y, Dalton N, Peterson KL, Race RE, et al. Prion-induced amyloid heart disease with high blood infectivity in transgenic mice. Science. 2006;313:94–7. doi: 10.1126/science.1128635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ashwath ML, Dearmond SJ, Culclasure T. Prion-associated dilated cardiomyopathy. Arch Intern Med. 2005;165:338–40. doi: 10.1001/archinte.165.3.338. [DOI] [PubMed] [Google Scholar]
- 10.Rutala WA, Weber DJ. Society for Healthcare Epidemiology of America. Guideline for disinfection and sterilization of prion-contaminated medical instruments. Infect Control Hosp Epidemiol. 2010;31:107–17. doi: 10.1086/650197. [DOI] [PubMed] [Google Scholar]