

Abstract
Bladder cancer is one of the most common cancers worldwide, with a high rate of recurrence and poor outcomes as a result of relapse. Bladder cancer patients require lifelong invasive monitoring and treatment, making bladder cancer one of the most expensive malignancies. Lines of evidence increasingly point to distinct genetic and epigenetic alteration patterns in bladder cancer, even between the different stages and grades of disease. In addition, genetic and epigenetic alterations have been demonstrated to play important roles during bladder tumorigenesis. This review will focus on bladder cancer-associated genomic and epigenomic alterations, which are common in bladder cancer and provide potential diagnostic markers and therapeutic targets for bladder cancer treatment.
Keywords: Epigenetics, Genetic; Urinary Bladder Neoplasms; DNA Methylation; Epigenetic Therapy
INTRODUCTION
Bladder cancer is the fifth most common malignancy in the United States, with an estimated 76,960 new cases and 16,390 deaths in 2016 [1,2]. Up to 80% of bladder cancer cases are associated with environmental exposure. Tobacco use is most commonly associated with bladder cancer incidence, as smoking duration and density are directly related to increased risk [3]. Other common risk factors include chronic urinary tract infections, arsenic exposure, and occupational exposure to carcinogens in the rubber and fossil fuel industries [4,5]. More than 90% of bladder cancer cases are diagnosed as transitional cell carcinomas (TCCs), 5% as squamous-cell carcinomas, and 2% as adenocarcinomas [6]. Approximately 25% of new TCCs are muscle invasive bladder cancers (MIBCs, high-grade stages T2–T4), while the majority of TCCs are non-MIBCs (NMIBCs, low-grade stages Tis/carcinoma in situ [CIS], Ta, and T1) [7,8]. MIBC patients have poor outcomes, with 10-year survival rates of only 50% as compared to the 80% 10-year survival rates of NMIBC patients [6]. The standard therapy of MIBC patients is radical cystectomy, followed extended lymph node dissection; this therapeutic regimen results in improved survival [7]. Neoadjuvant chemotherapy is also recommended after radical cystectomy and increases the likelihood of eliminating residual tumor cells.
The prognosis for NMIBC patients is more favorable, with 5-year survival rates of 78% [2], however, 60%–70% of these will relapse and 10%–20% will progress to MIBC [4]. The most challenging aspect of bladder cancer management is predicting and diagnosing tumor recurrence or disease progression. The current gold standard for monitoring bladder cancer recurrence is lifelong cystoscopy and cytology [8,9]. Cystoscopic examination allows for the direct visualization of the inside of the urinary bladder, but is invasive and expensive. Although urine cytology is widely used in the diagnosis of bladder cancer, it is less invasive, has low sensitivity in detecting low-grade tumors, and cannot completely rule out the presence of a tumor. Several new tests have been developed, such as nuclear matrixprotein-22 [10], bladder tumor-associated antigen [11], the ImmunoCyt assay (Scimedx, Denville, NJ, USA) [12], and the Uro Vysion assay (Abbott Molecular Inc., Des Plaines, IL, USA) [13]. However, due to relatively low sensitivities and/or specificities, these methods have not been used in routine clinical tests [14]. Therefore, there is a crucial need not only for reliable, accurate and convenient methods of diagnosis and monitoring for the recurrence or progression of NMIBC, but also for the identification of novel therapeutic targets, especially for MIBC patients. Thus, understanding the mechanisms of bladder cancer genesis is of high importance for guiding clinical decision-making. With the rapid improvement of high-throughput DNA sequencing technologies, increasing numbers of genomic and epigenomic changes have been uncovered.
SOMATIC GENETIC ABERRATIONS
Cancerous cells have growth advantages over normal cells that historically are thought to result from a series of genetic mutations [15]. As with most carcinomas, the exact causes of bladder cancer remain elusive. Somatic genetic mutation is one of the most important leading factors for bladder cancer tumorigenesis and progression. Bladder cancer is typically not inherited, but rather results from an accumulation of somatic mutations in bladder cells over time. The number of the genetic alterations has enormously increased due to the advances of the second-generation DNA sequencing methods [16]. Frequently-mutated genes in MIBCs include TP53 (41%), KDM6A (28%), ARID1A (22%), PIK3CA (18%), MLL2 (17%), CREBBP (15%), RB1 (15%), STAG2 (13%), FGFR3 (13%), EP300 (13%), TSC1 (8%), and HRAS (8%) [17,18].
Recently, Hedegaard et al. [19] reported that NMIBCs can be grouped into 3 subclasses (classes 1, 2, and 3) based on whole genome expression profiles. Class 1 tumors have a lower risk of progression and better prognosis than classes 2 and 3 tumors. Class 1 tumors display upregulation of early cell-cycle genes (CCND1, ID1, and RBL2), while late cell-cycle genes (CDC20, CDC25A, CDKs, and PLK1) and cancer stem cell markers (ALDH1A1, ALDH1A2, PROM1, NES, and THY1) are highly expressed in class 2 tumors. The keratin (KRT) gene family shows increased expression in class 2 and/or class 3 tumors as compared to class 1 tumors. Most MIBCs (86%) display mutations in chromatin remodeling genes, including histone methyltransferases (58% of cases), histone lysine demethylases (54%), SWI/SNF complexes (40%), and histone acetyltransferases (32%). In total, 76% of all primary bladder tumors display mutations in at least 1 chromatin regulatory gene [17]. In essence, several signaling pathways are altered due to these mutations, implying that these pathways play critical roles in controlling normal proliferation of urinary bladder cells.
p53/RB Pathway Signaling
The factors p53 (transcription factor) and RB (retinoblastoma) are the 2 key factors in the cell cycle regulation pathway, which is altered in 93% of bladder cancer cases [17]. Factor p53, which is encoded by TP53, is the most famous and well-studied tumor-suppressor, and is inactivated by somatic mutations in approximately 50% of all human cancers [17,20]. TP53 mutations are highly frequent ( >40%) in MIBCs, in contrast to the NMIBCs, of which only about 8% possess TP53 mutations [19,21]. In addition, TP53 mutations in bladder cancer are likely linked to smoking and also to higher grade and stage [22]. The RB protein is encoded by the RB1 tumor suppressor gene, and functions in several cellular processes by regulating the expression of genes involved in cell proliferation, differentiation and apoptosis by interacting with chromatin, DNA-modifying enzymes and transcription factors [23,24]. Factor p53 induces the expression of p21 [25], a cyclin dependent kinase (CDK) inhibitor, and consequently blocks RB phosphorylation [26]. Somatic mutations in genes involved in the p53/RB signaling pathway have been identified in bladder tumors, and include MDM2 (9%), CDKN1A (14%), CDKN2A (47%), CCND1 (10%), FBXW7 (10%), and E2F3/SOX4 (20%) [17].
FGFR3 and RAS-MAPK Signaling Pathways
NMIBCs also show a high frequency (~80%) of activating mutations in the fibroblast growth factor receptor 3 (FGFR3) signaling pathway [27], which consequently activates the RAS-MAP kinase (RAS-MAPK) pathway and phospholipase Cγ (PLCγ), leading to uncontrolled cell proliferation [28]. The FGFR3 and RAS gene mutations are mutually exclusive in bladder cancer, suggesting that these 2 genes share similar functions and their mutations confer the same phenotype [29]. However, the possibility still remains that the activated FGFR3 and RAS are synthetic lethal, suggesting they control 2 different pathways. Two fusion proteins, FGFR3-TACC3 (transforming acid coiled coil 3) and FGFR3-BAIAP2L1 (BAI1-associated protein 2-like 1), have been identified in bladder cancer [30]. Based on the protein structure analysis, the FGFR3-TACC3 fusion protein is predicted to auto-dimerize and constitutively activate the kinase domain of FGFR3 [17], suggesting that the mutational profile of FGFR3 alone may not be the cause of aberrant FGFR3 signaling.
PI3K/mTOR Pathway Alterations
The phosphatidylinositol 3-kinase (PI3K) pathway is a critical cell-signaling pathway that regulates multiple cellular processes. PI3K pathway alterations are present in 42% of all bladder cancers [17]. This includes the activation of upstream regulators of the PI3K pathway, namely EGFR, ERBB2, and ERBB3. Moreover, upregulation of PIK3CA, AKT1/2/3, and PDK1 expression, as well as loss-of-function mutations in TSC1/2 (tuberous sclerosis 1/2) and PTEN occur in MIBC and NMIBC bladder tumors [31]. Overexpression of EGFR or ERBB2 leads to RAS activation, which in turn activates the PI3K pathway [32]. As a result, the mTOR pathway is activated by the inactivation of the TSC1/TSC2 complex [31], thereby increasing cell proliferation.
Chromosomal Rearrangements
Chromosomal rearrangements, namely the concomitant result of aberrant nonhomologous end joining [33], may result in oncogene formation and therefore may initiate tumorigenesis [30,34] or increasing oncogene expression [35]. MIBCs display more chromosomal alternations than NMIBCs [36,37]. Deletions in both arms of chromosome 9 are frequently observed in both NMIBCs and MIBCs [38], and bladder cancer patients with tumors harboring deletions of 9ptr-p22, 9q22.3, 9q33, or 9q34 had more rapid recurrence than those patients without these deletions [39]. Chromosome 9 deletions also affect some tumor suppressor genes, including cyclin-dependent kinase inhibitor 2A (CDKN2A) and 2B (CDKN2B), as well as TSC1 [40]. Amplifications were frequently detected at 6p22.3 (E2F3), 8p12 (FGFR1), 8q22.2 (CMYC), 11q13 (CCND1, EMS1, INT2), and 19q13.1 (CCNE), and homozygous deletions were detected at 9p21.3, 8p23.1, and 11p13 [41].
EPIGENETIC ABERRATIONS IN BLADDER CANCER
Unlike genetic mutations and copy number variation, epigenetic events regulate gene expression outcome without changing the underlying DNA sequence. Epigenetic regulation includes DNA methylation, histone modifications, microRNA regulation and nucleosome positioning, all of which are distorted in every form of human cancer. Similar to genetic alterations, epigenetic changes also play important roles in altering the transcriptome during cancer initiation and progression [42,43]. In addition, a significant number of genetic mutations of epigenetic regulator genes occur in virtually every cancer type, thereby disturbing the epigenome patterns. These include somatic mutations of genes that encode for DNA methyltransferases, chromatin modifiers, and chromatin remodelers [17,44]. A substantial portion (76%) of all primary bladder tumors displays mutations in at least one chromatin regulatory gene [17]. These mutations cause epigenetic dysregulation in bladder cancers, and are now being investigated using basic scientific experiments, translational studies, and clinical trials.
DNA Methylation (5mC)
In mammalian cells, DNA methylation almost exclusively found at the C5 position of cytosine (5mC) in the context of 5`-CG-3` dinucleotides (CpG). The CpG sites are found throughout the genome, with 28 million CpG sites in the haploid genome. However, since methylated CpGs are prone to spontaneous deamination to uracil more frequently than unmethylated CpGs, CpG content is reduced to 20% of what is expected by sequence prediction alone. As a result, the genome is largely CpG-depleted; however, there are regions of the genome, termed CpG islands (CGIs), which contain their expected CpG content. These CGIs are located at the promoter/5’ regions of more than 50% of all known genes and are normally unmethylated [45]. Promoter CGIs methylation may be associated with gene silencing [46], while CGIs of gene bodies is positively correlated with gene expression [47,48].
In human cells, CpG methylation is catalyzed by 3 DNA methyltransferases (DNMT1, DNMT3A, and 3B), as well as by accessory proteins, such as DNMT3L and UHRF1 [49]. The DNA methyltransferase DNMT1 is mainly responsible for the maintenance of DNA methylation patterns after DNA replication, while DNMT3A and 3B are primarily responsible for de novo DNA methylation as well as for helping to maintain the DNA methylation distributions [50,51].
DNA methylation is critical for mammalian development, and its aberrancies are hallmarks of many human diseases, including cancer [42,43]. Cancer cells, including bladder cancer, show an overexpression of DNMT1, DNMT3A, and 3B, which in turn results in DNA hypermethylation of promoter regions, and the possible subsequent silencing of tumor suppressor genes [52]. Distinguishable DNA methylation differences have been found between NMIBC and MIBC bladder tumors. Specifically, distinct DNA hypomethylation patterns have been found at non-CpG islands in NMIBCs and CpG island DNA hypermethylation patterns in MIBCs [53,54]. For example, promoter CGIs of IPF1, GALR1, TAL1, PENK, and TJP2 display DNA hypermethylation in MIBCs [54]. In addition, DNA methylation alterations in transposons also are common events in bladder cancer. For example, LINE-1 repetitive element DNA hypomethylation, which correlates with activated MET oncogene transcription, has been identified in bladder cancer patients [55]. In bladder cancers, DNA methylation changes with frequencies of 48%–96% are present at several gene promoters, including DNA hypermethylation of A2BP1, NPTX2, SOX11, PENK, NKX62, DBC1, MYO3A, CA10, POU4F2, HOXA9, MEIS1, GDF15, TMEFF2, VIM, STK11, MSH6, BRCA1, TBX2, TBX3, GATA2, ZIC4, PAX5A, MGMT, and IGSF4 [53,56-60]. Additionally, tumor-specific DNA hypermethylation of ZO2 [54], MYOD1 [61], and CDH13 [62] has been detected in adjacent-normal urothelial tissues and is associated with reduced expression, suggesting that DNA methylation alternations are early-driver events in urothelial tumorigenesis. These unique DNA methylation alterations are promising diagnostic biomarkers, especially when multiple markers are combined into a multigene panel. Combining DNA methylation data of several genes shows high sensitivity and specificity for bladder cancer diagnostics. Yu et al. [63] reported 92% sensitivity and 87% specificity for both primary and recurrent cases by monitoring DNA methylation of 11 genes. The detection sensitivity of a 3-gene panel comprised of ZNF671, IRF8, and SFRP1 DNA methylation was 96% and approached 100% for high-grade and recurrent patients, compared to only 58% sensitivity using DNA methylation of ZNF671 alone [64]. DNA methylation of TWIST and NID2 yielded 90% sensitivity and 96% specificity for predicting bladder cancer recurrence [65].
DNA from bladder tumors is released into the urinary tract and can be identified from urine sediments. Measuring tumorderived DNA methylation changes in urine sediments of bladder cancer patients is a promising noninvasive means for early detection of bladder cancer as well as response to therapy and relapse. Analysis of urine sediment DNA methylation showed the same cancer-specific DNA hypermethylation patterns at the promoter regions of apoptosis-associated genes, including DAPK, BCL2, and TERT, as in the corresponding tumor tissues [66]. These loci are unmethylated in normal bladder tissues, suggesting they can be used as early diagnostic biomarkers for bladder cancer. These studies also indicated a noninvasive and viable method for bladder cancer diagnosis. A 3-marker panel, detecting DNA methylation of SOX1, IRAK3, and LINE-1-MET, showed a tumor detection sensitivity and specificity of 89% and 97%, respectively, while reliably predicting recurrence (80%) and the absence of recurrence (74%) in patient urine sediments [67].
DNA methylation at specific gene regions has been shown to be associated with disease progression and patient survival. For example, the RUNX3 gene promoter is commonly silenced by DNA methylation (71%) in bladder tumors [68]. RUNX3-promoter DNA methylation is positively correlated with tumor progression and survival, and may serve a prognostic marker for bladder cancer [69]. In addition, DNA hypermethylation of CDH1, FHIT, LAMC2, RASSF1A, DAPK, MINT31, and SFRP are all related to bladder tumor development and survival [70-72], and have the potential to be prognostic markers.
DNA Hydroxymethylation (5hmC)
The mechanisms of DNA demethylation have been more elusive to characterize, and can be achieved via passive or active processes. Passive DNA demethylation occurs if the newly synthesized DNA strand cannot be methylated (usually in DNMT1-deficient cells) after DNA replication. Active DNA demethylation is mediated by the Ten-eleven Translocation (TET) family of enzymes via the progressive oxidation of 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC), then 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC). Residues of 5fC and 5caC are finally excised by thymine DNA glycosylase, and replaced with an unmethylated cytosine [73,74].
As the intermediate of the DNA active demethylation process, 5hmC levels reflect global DNA methylation levels, as well as the activity of the TET enzymes [75]. Munari et al. [76] reported dramatically reduced 5hmC levels in bladder tumors, suggesting low TET activity. Determining 5hmC levels is experimentally straightforward and has the potential to be a biomarker for diagnosis and prognosis of bladder cancer and other cancers. Vitamin C is a known cofactor of the TET enzymes. Interestingly, Liu et al. [77] recently reported that vitamin C and the DNA methylation inhibitor 5’aza-2’-deoxycytidine (5-azaCdR) have synergistic effects on both DNA demethylation and inhibition of cancer cell proliferation. They showed that vitamin C activates DNA demethylation by TET, and enhances the immune signaling pathway by increasing endogenous retrovirus transcription. In their study, around 92% of the patients had vitamin C levels below normal range, among which 63% had severe vitamin C deficiency. Vitamin C has great promise in enhancing DNA demethylation in epigenetic therapies for cancer patients.
Histone Modifications
Histones are a series of small highly conserved proteins and the key protein component of chromatin. Histone covalent modifications, including methylation, ubiquitination, SUMOylation, acetylation, and phosphorylation, at specific residues on histone N-terminal tails, play crucial roles in regulating fundamental biological processes including gene expression, DNA replication, and DNA damage repair [78]. Acetylation and methylation on lysine residues are the most studied histone modifications. Trimethylation of histone H3 at lysine 4 (H3K4me3), acetylation on H3 lysine 9 (H3K9Ac) and lysine 27 (H3K27Ac) correlate with gene activation, while trimethylation on H3 lysine 9 (H3K9me3) and lysine 27 (H3K27me3) are associated with gene repression [79]. The multiple regional epigenetic silencing (MRES) phenotype in bladder cancer cells is associated with histone H3K9 and H3K27 methylation and histone H3K9 hypoacetylation [80]. The MRES tumors display rare FGFR3 mutations, as well as a gene expression pattern similar to CIS grade tumors, however, most MRES tumors are high-grade (76% of MIBCs). Ellinger et al. [81] detected H3K9me3 and H3K27me3 in bladder cancer by chromatin immunoprecipitation (ChIP) followed by microarray hybridization (ChIP-chip), and discovered a negative correlation between these histone modifications and tumor stage. Most MIBCs (89%) contain at least 1 histone modification alteration [17], suggesting that changes in histone modifications may be promising biomarkers for bladder cancer diagnosis and prognosis, as well as novel therapeutic targets for bladder cancer patients.
The histone modification-associated genes are highly mutated in bladder tumors, and include the lysine-specific demethylase 6A (KDM6A), which removes the H3K27 trimethylation mark, and is mutated in >25% of bladder cancer cases. In addition, ARID1A, which encodes for a component of the SWI/SNF chromatin-remodeling complex, is altered in 22% of bladder tumors, while MLL2, which encodes for an H3K4 methylase, is altered in 17% of bladder cancer patients [18]. Wu et al. [82] reported that mutations in MLL, EP400 (a component of the NuA4 histone acetyltransferase complex) and PRDM2 (a nuclear histone/protein methyltransferase) are associated with bladder cancer relapse. MLL mutations in recurrent bladder cancers result in elevated H3K4me3 levels and increased expression of GATA4 and ETS1 [82]. Interestingly, MLL3, which also encodes for an H3K4 methylase, is exclusively mutated in primary tumors, suggesting distinct histone modification signatures between primary and recurrent tumors [82]. Recently, the coexistence of the active H3K27Ac marker and DNA methylation at a subset of enhancers was reported, suggesting the dual roles of DNA methylation, either alone or via cooperation with histone modifications, is an important aspect of bladder cancer [83].
MicroRNAs
MicroRNAs regulate gene expression and function in numerous biological processes. Some microRNAs have been characterized as oncogenes (such as miR-183, miR-96, miR17-5p, and miR-20a) or tumor suppressors (such as miR-145, miR-143, and miR125b) [84]. Friedman et al. [85] detected the expression of microRNAs in bladder cancer using microarray and quantitative reverse transcription polymerase chain reaction assays, and found that miR-1, miR-101, miR-143, miR-145, miR-29c, and miR-127 were downregulated, whereas miR-224, miR-182, and miR-183 were upregulated, in bladder tumors. Some microRNAs are epigenetically regulated in human cancers. A small subset of microRNAs (5%) are significantly upregulated in the T24 human bladder cancer cell line when treated with the DNMT inhibitor 5-Aza-CdR and the histone demethylase inhibitor SAHA [86], suggesting these miRNAs function as tumor suppressor genes. Specifically, miR-127 represses the expression of the BCL6 oncogene [86].
Additionally, microRNAs also affect other epigenetic regulators. MiR-101 directly represses EZH2 (H3K27 methyltransferase) expression and inhibits cell proliferation [85]. MiR-21, miR-148a, miR-126, and miR-152 target DNMT1, and miR-29a/29b/29c inhibit DNMT3A and 3B [87]. However, it should be noted that even though these data are promising, the diagnostic value of microRNA testing in bladder cancers still remains controversial, due to assay data and relatively low specificity [88].
Nucleosome Positioning
The nucleosome is the basic unit of chromatin, and consists of ~147 bp DNA wrapping around the histone octamer, which is comprised of dimers of histones H2A, H2B, H3, and H4. Generally, nucleosomes function as gene expression repressors by blocking transcription machinery binding to promoter sites [89]. Early studies in Saccharomyces cerevisiae identified nucleosome-depleted regions upstream of transcription start sites, which are accessible to transcription factors and correlate with active gene expression [90]. Lay et al. [91] determined that nucleosome positioning patterns are affected by DNA methylation and histone modifications, and identified regions of nucleosome depletion in HCT116 DKO1 colon cancer cells, in which genetic disruption of DNMT1 and DNMT3B results in dramatically decreased DNA methylation levels (5% HCT116 wild-type cells). Additionally, nucleosome positioning is regulated by chromatin remodelers and histone modifiers, which are also frequently mutated in bladder cancer [18]. Although these studies indicate the importance of nucleosome positioning during tumorigenesis, the role of nucleosome position in bladder cancer has been largely overlooked.
GENETIC AND EPIGENETIC ALTERATIONS AS THERAPEUTIC TARGETS IN BLADDER CANCER
Recent technological advances have allowed for the discovery of genetic and epigenetic alterations, which have led to a better understanding of the mechanisms of bladder cancer at the molecular level, and have provided a tremendous number of specific biological and molecular targets for therapy. As a result, p53, FGFR3, ERBB2, and PI3K have been targeted by immuno-and/or chemotherapy in clinical trials [92,93].
Unlike genetic alterations, epigenetic changes can be reversed via pharmacological treatment. Therefore, epigenetic treatment offers a new strategy for anticancer therapy. Several small molecule inhibitors have been approved by the U.S. Food and Drug Administration, and were shown to be therapeutically efficacious for various cancer types [94]. The epigenetic drugs in clinical use mainly include DNMT inhibitors (5-azacytidine and 5-Aza-2’-deoxycytidine) and histone deacetylases (HDACs) inhibitors (SAHA, valproic acid, and romidepsin) [95]. Tazemetostat (an EZH2 inhibitor) is currently being evaluated in ongoing clinical trials. Bladder cancer has been considered for epigenetic therapy, namely the use of DNMT inhibitors and HDAC inhibitors to treat bladder cancer [44,96]. Clinic trials using these epigenetic drugs on bladder cancer have been ongoing [97].
CONCLUSIONS
In this review, we discussed genetic and epigenetic alterations in bladder cancer; however, it should be noted that, in most cases, multiple genetic and epigenetic changes occur simultaneously or are mutually influenced by each other. As shown above, many genetic mutations disrupt the functions of genes involved in epigenetic regulation, and conversely epigenetic aberrancies lead to alterations of transcription (Fig. 1). Activating FGFR3 mutations are highly frequent in NMIBCs; however, a proportion of NMIBCs with wild-type FGFR3 show DNA hypermethylation and unfavorable prognosis as compared to the FGFR3 mutated tumors, indicating that these 2 tumor subtypes have different genetic backgrounds [98].
Fig. 1.
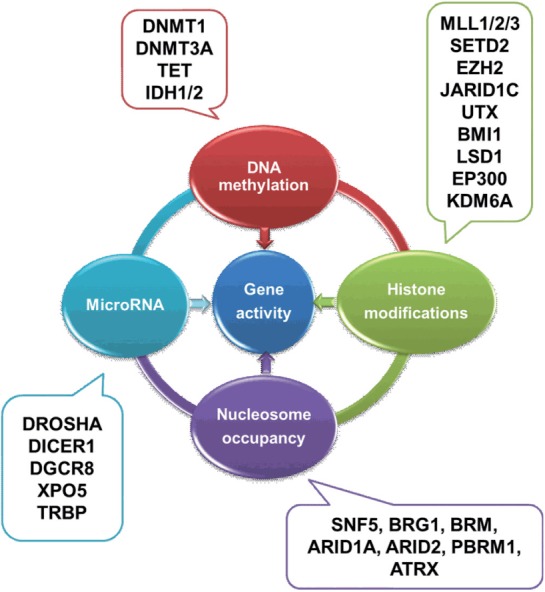
Interaction of the genetic and epigenetic alterations. Whole-genome sequencing studies showed that many genetic mutations disturb the epigenetic regulation pathways in cancer. The 4 types of epigenetic regulation pathways also mutually interacted. The genome transcription is regulated by both genetic and epigenetic factors. Examples of some, but not all, of the mutations are shown.
DNA methylation status alone cannot explain changes in gene expression. Including analyses of chromatin context further highlights the importance of the crosstalk between individual epigenomic regulators. These complex regulation networks make the discovery of key tumorigenic drivers challenging. Combinations of several biomarkers, including genetic and epigenetic markers, have improved sensitivities and specificities for bladder cancer diagnosis and prognosis, compared to the 48% mean sensitivity using traditional urine cytology.
Epigenetic factors have been increasingly recognized for the value of diagnosis, prognosis, and therapy of cancers. DNA methylation has the possibility to be an ideal therapeutic target for cancer. Some DNMT inhibitors, including 5-aza-2’-deoxycytidine, need to be incorporated into the genome in order to inhibit DNA methylation, which means their function relies on the DNA replication. These agents may have a more profound effect on tumor cells, due to their higher proliferation, than normal somatic cells. Vitamin C deficiency is commonly found in patients with multiple cancers [77,99], and may boost the efficiency of DNMT inhibitors, suggesting that dietary supplementation of vitamin C could enhance the efficiency of DNMT1 inhibitor treatment. Epigenetic therapies aim to revert to the normal epigenome in the cancer cells, and consequently the transcriptome. They function more in controlling the abnormal cell proliferation rather than killing tumor cells, and implies that these agents result in fewer side effects and less toxicity to normal cells.
Genome-wide studies of genetic and epigenetic alternations in bladder cancer open the opportunity to develop novel, reliable, sensitive, and specific methods to monitor early tumors or recurrence, and to design personalized therapies.
Acknowledgments
We thank the Vicky Joseph Cancer Research Fund (GL) to support this work.
Footnotes
Conflict of Interest
No potential conflict of interest relevant to this article was reported.
REFERENCES
- 1.Cancer facts & figures 2016 [Internet] Atlanta (GA): American Cancer Society; 2016. [cited 2016 Aug 28]. Available from: http://www.cancer.org/research/cancerfactsstatistics/cancerfactsfigures2016/ [Google Scholar]
- 2.Surveillance, Epidemiology, and End Results. SEER stat fact sheets: bladder cancer [Internet] Bethesda (MD): National Cancer Institute; 2016. [cited 2016 Aug 28]. Available from: http://seer.cancer.gov/statfacts/html/urinb.html. [Google Scholar]
- 3.Freedman ND, Silverman DT, Hollenbeck AR, Schatzkin A, Abnet CC. Association between smoking and risk of bladder cancer among men and women. JAMA. 2011;306:737–45. doi: 10.1001/jama.2011.1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brandau S, Böhle A. Bladder cancer. I. Molecular and genetic basis of carcinogenesis. Eur Urol. 2001;39:491–7. doi: 10.1159/000052494. [DOI] [PubMed] [Google Scholar]
- 5.Feng Z, Hu W, Rom WN, Beland FA, Tang MS. 4-aminobiphenyl is a major etiological agent of human bladder cancer: evidence from its DNA binding spectrum in human p53 gene. Carcinogenesis. 2002;23:1721–7. doi: 10.1093/carcin/23.10.1721. [DOI] [PubMed] [Google Scholar]
- 6.Kaufman DS, Shipley WU, Feldman AS. Bladder cancer. Lancet. 2009;374:239–49. doi: 10.1016/S0140-6736(09)60491-8. [DOI] [PubMed] [Google Scholar]
- 7.Prasad SM, Decastro GJ, Steinberg GD. Urothelial carcinoma of the bladder: definition, treatment and future efforts. Nat Rev Urol. 2011;8:631–42. doi: 10.1038/nrurol.2011.144. [DOI] [PubMed] [Google Scholar]
- 8.Babjuk M, Böhle A, Burger M, Capoun O, Cohen D, Compérat EM, et al. EAU Guidelines on Non-Muscle-invasive Urothelial Carcinoma of the Bladder: Update 2016. Eur Urol. 2016 Jun 17; doi: 10.1016/j.eururo.2016.05.041. [Epub]. http://doi.org/10.1016/j.eururo.2016.05.041. [DOI] [PubMed] [Google Scholar]
- 9.Morgan TM, Keegan KA, Clark PE. Bladder cancer. Curr Opin Oncol. 2011;23:275–82. doi: 10.1097/CCO.0b013e3283446a11. [DOI] [PubMed] [Google Scholar]
- 10.Gupta NP, Sharma N, Kumar R. Nuclear matrix protein 22 as adjunct to urine cytology and cystoscopy in follow-up of superficial TCC of urinary bladder. Urology. 2009;73:592–6. doi: 10.1016/j.urology.2008.04.051. [DOI] [PubMed] [Google Scholar]
- 11.Irani J, Desgrandchamps F, Millet C, Toubert ME, Bon D, Aubert J, et al. BTA stat and BTA TRAK: A comparative evaluation of urine testing for the diagnosis of transitional cell carcinoma of the bladder. Eur Urol. 1999;35:89–92. doi: 10.1159/000019824. [DOI] [PubMed] [Google Scholar]
- 12.Têtu B, Tiguert R, Harel F, Fradet Y. ImmunoCyt/uCyt+ improves the sensitivity of urine cytology in patients followed for urothelial carcinoma. Mod Pathol. 2005;18:83–9. doi: 10.1038/modpathol.3800262. [DOI] [PubMed] [Google Scholar]
- 13.van Rhijn BW, van der Poel HG, van der Kwast TH. Urine markers for bladder cancer surveillance: a systematic review. Eur Urol. 2005;47:736–48. doi: 10.1016/j.eururo.2005.03.014. [DOI] [PubMed] [Google Scholar]
- 14.Mowatt G, Zhu S, Kilonzo M, Boachie C, Fraser C, Griffiths TR, et al. Systematic review of the clinical effectiveness and cost-effectiveness of photodynamic diagnosis and urine biomarkers (FISH, ImmunoCyt, NMP22) and cytology for the detection and follow-up of bladder cancer. Health Technol Assess. 2010;14:1–331. doi: 10.3310/hta14040. [DOI] [PubMed] [Google Scholar]
- 15.Vineis P, Schatzkin A, Potter JD. Models of carcinogenesis: an overview. Carcinogenesis. 2010;31:1703–9. doi: 10.1093/carcin/bgq087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weinberg RA. The biology of cancer. New York: Garland Science; 2007. [Google Scholar]
- 17.Cancer Genome Atlas Research Network Comprehensive molecular characterization of urothelial bladder carcinoma. Nature. 2014;507:315–22. doi: 10.1038/nature12965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gui Y, Guo G, Huang Y, Hu X, Tang A, Gao S, et al. Frequent mutations of chromatin remodeling genes in transitional cell carcinoma of the bladder. Nat Genet. 2011;43:875–8. doi: 10.1038/ng.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hedegaard J, Lamy P, Nordentoft I, Algaba F, Høyer S, Ulhøi BP, et al. Comprehensive transcriptional analysis of early-stage urothelial carcinoma. Cancer Cell. 2016;30:27–42. doi: 10.1016/j.ccell.2016.05.004. [DOI] [PubMed] [Google Scholar]
- 20.Soussi T, Béroud C. Assessing TP53 status in human tumours to evaluate clinical outcome. Nat Rev Cancer. 2001;1:233–40. doi: 10.1038/35106009. [DOI] [PubMed] [Google Scholar]
- 21.Bakkar AA, Wallerand H, Radvanyi F, Lahaye JB, Pissard S, Lecerf L, et al. FGFR3 and TP53 gene mutations define two distinct pathways in urothelial cell carcinoma of the bladder. Cancer Res. 2003;63:8108–12. [PubMed] [Google Scholar]
- 22.Wallerand H, Bakkar AA, de Medina SG, Pairon JC, Yang YC, Vordos D, et al. Mutations in TP53, but not FGFR3, in urothelial cell carcinoma of the bladder are influenced by smoking: contribution of exogenous versus endogenous carcinogens. Carcinogenesis. 2005;26:177–84. doi: 10.1093/carcin/bgh275. [DOI] [PubMed] [Google Scholar]
- 23.Robertson KD, Ait-Si-Ali S, Yokochi T, Wade PA, Jones PL, Wolffe AP. DNMT1 forms a complex with Rb, E2F1 and HDAC1 and represses transcription from E2F-responsive promoters. Nat Genet. 2000;25:338–42. doi: 10.1038/77124. [DOI] [PubMed] [Google Scholar]
- 24.Korenjak M, Brehm A. E2F-Rb complexes regulating transcription of genes important for differentiation and development. Curr Opin Genet Dev. 2005;15:520–7. doi: 10.1016/j.gde.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 25.Dulić V, Kaufmann WK, Wilson SJ, Tlsty TD, Lees E, Harper JW, et al. p53-dependent inhibition of cyclin-dependent kinase activities in human fibroblasts during radiation-induced G1 arrest. Cell. 1994;76:1013–23. doi: 10.1016/0092-8674(94)90379-4. [DOI] [PubMed] [Google Scholar]
- 26.De Luca A, MacLachlan TK, Bagella L, Dean C, Howard CM, Claudio PP, et al. A unique domain of pRb2/p130 acts as an inhibitor of Cdk2 kinase activity. J Biol Chem. 1997;272:20971–4. doi: 10.1074/jbc.272.34.20971. [DOI] [PubMed] [Google Scholar]
- 27.Cappellen D, De Oliveira C, Ricol D, de Medina S, Bourdin J, Sastre-Garau X, et al. Frequent activating mutations of FGFR3 in human bladder and cervix carcinomas. Nat Genet. 1999;23:18–20. doi: 10.1038/12615. [DOI] [PubMed] [Google Scholar]
- 28.di Martino E, L’Hôte CG, Kennedy W, Tomlinson DC, Knowles MA. Mutant fibroblast growth factor receptor 3 induces intracellular signaling and cellular transformation in a cell type- and mutation-specific manner. Oncogene. 2009;28:4306–16. doi: 10.1038/onc.2009.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jebar AH, Hurst CD, Tomlinson DC, Johnston C, Taylor CF, Knowles MA. FGFR3 and Ras gene mutations are mutually exclusive genetic events in urothelial cell carcinoma. Oncogene. 2005;24:5218–25. doi: 10.1038/sj.onc.1208705. [DOI] [PubMed] [Google Scholar]
- 30.Williams SV, Hurst CD, Knowles MA. Oncogenic FGFR3 gene fusions in bladder cancer. Hum Mol Genet. 2013;22:795–803. doi: 10.1093/hmg/dds486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Knowles MA, Platt FM, Ross RL, Hurst CD. Phosphatidylinositol 3-kinase (PI3K) pathway activation in bladder cancer. Cancer Metastasis Rev. 2009;28:305–16. doi: 10.1007/s10555-009-9198-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tamaskovic R, Schwill M, Nagy-Davidescu G, Jost C, Schaefer DC, Verdurmen WP, et al. Intermolecular biparatopic trapping of ErbB2 prevents compensatory activation of PI3K/AKT via RASp110 crosstalk. Nat Commun. 2016;7:11672. doi: 10.1038/ncomms11672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ghezraoui H, Piganeau M, Renouf B, Renaud JB, Sallmyr A, Ruis B, et al. Chromosomal translocations in human cells are generated by canonical nonhomologous end-joining. Mol Cell. 2014;55:829–42. doi: 10.1016/j.molcel.2014.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shtivelman E, Lifshitz B, Gale RP, Canaani E. Fused transcript of abl and bcr genes in chronic myelogenous leukaemia. Nature. 1985;315:550–4. doi: 10.1038/315550a0. [DOI] [PubMed] [Google Scholar]
- 35.Mayr C, Hemann MT, Bartel DP. Disrupting the pairing between let-7 and Hmga2 enhances oncogenic transformation. Science. 2007;315:1576–9. doi: 10.1126/science.1137999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Balbás-Martínez C, Sagrera A, Carrillo-de-Santa-Pau E, Earl J, Márquez M, Vazquez M, et al. Recurrent inactivation of STAG2 in bladder cancer is not associated with aneuploidy. Nat Genet. 2013;45:1464–9. doi: 10.1038/ng.2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Morrison CD, Liu P, Woloszynska-Read A, Zhang J, Luo W, Qin M, et al. Whole-genome sequencing identifies genomic heterogeneity at a nucleotide and chromosomal level in bladder cancer. Proc Natl Acad Sci U S A. 2014;111:E672–81. doi: 10.1073/pnas.1313580111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hartmann A, Schlake G, Zaak D, Hungerhuber E, Hofstetter A, Hofstaedter F, et al. Occurrence of chromosome 9 and p53 alterations in multifocal dysplasia and carcinoma in situ of human urinary bladder. Cancer Res. 2002;62:809–18. [PubMed] [Google Scholar]
- 39.Simoneau M, LaRue H, Aboulkassim TO, Meyer F, Moore L, Fradet Y. Chromosome 9 deletions and recurrence of superficial bladder cancer: identification of four regions of prognostic interest. Oncogene. 2000;19:6317–23. doi: 10.1038/sj.onc.1204022. [DOI] [PubMed] [Google Scholar]
- 40.Weber RG, Hoischen A, Ehrler M, Zipper P, Kaulich K, Blaschke B, et al. Frequent loss of chromosome 9, homozygous CDKN2A/p14(ARF)/CDKN2B deletion and low TSC1 mRNA expression in pleomorphic xanthoastrocytomas. Oncogene. 2007;26:1088–97. doi: 10.1038/sj.onc.1209851. [DOI] [PubMed] [Google Scholar]
- 41.Veltman JA, Fridlyand J, Pejavar S, Olshen AB, Korkola JE, DeVries S, et al. Array-based comparative genomic hybridization for genome-wide screening of DNA copy number in bladder tumors. Cancer Res. 2003;63:2872–80. [PubMed] [Google Scholar]
- 42.Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis. 2010;31:27–36. doi: 10.1093/carcin/bgp220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shen H, Laird PW. Interplay between the cancer genome and epigenome. Cell. 2013;153:38–55. doi: 10.1016/j.cell.2013.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Han H, Wolff EM, Liang G. Epigenetic alterations in bladder cancer and their potential clinical implications. Adv Urol. 2012;2012:546917. doi: 10.1155/2012/546917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Reinert T. Methylation markers for urine-based detection of bladder cancer: the next generation of urinary markers for diagnosis and surveillance of bladder cancer. Adv Urol. 2012;2012:503271. doi: 10.1155/2012/503271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jones PA, Liang G. Rethinking how DNA methylation patterns are maintained. Nat Rev Genet. 2009;10:805–11. doi: 10.1038/nrg2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kulis M, Heath S, Bibikova M, Queirós AC, Navarro A, Clot G, et al. Epigenomic analysis detects widespread gene-body DNA hypomethylation in chronic lymphocytic leukemia. Nat Genet. 2012;44:1236–42. doi: 10.1038/ng.2443. [DOI] [PubMed] [Google Scholar]
- 48.Yang X, Han H, De Carvalho DD, Lay FD, Jones PA, Liang G. Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell. 2014;26:577–90. doi: 10.1016/j.ccr.2014.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Duymich CE, Charlet J, Yang X, Jones PA, Liang G. DNMT3B isoforms without catalytic activity stimulate gene body methylation as accessory proteins in somatic cells. Nat Commun. 2016;7:11453. doi: 10.1038/ncomms11453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen T, Hevi S, Gay F, Tsujimoto N, He T, Zhang B, et al. Complete inactivation of DNMT1 leads to mitotic catastrophe in human cancer cells. Nat Genet. 2007;39:391–6. doi: 10.1038/ng1982. [DOI] [PubMed] [Google Scholar]
- 51.Liang G, Chan MF, Tomigahara Y, Tsai YC, Gonzales FA, Li E, et al. Cooperativity between DNA methyltransferases in the maintenance methylation of repetitive elements. Mol Cell Biol. 2002;22:480–91. doi: 10.1128/MCB.22.2.480-491.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002;3:415–28. doi: 10.1038/nrg816. [DOI] [PubMed] [Google Scholar]
- 53.Reinert T, Modin C, Castano FM, Lamy P, Wojdacz TK, Hansen LL, et al. Comprehensive genome methylation analysis in bladder cancer: identification and validation of novel methylated genes and application of these as urinary tumor markers. Clin Cancer Res. 2011;17:5582–92. doi: 10.1158/1078-0432.CCR-10-2659. [DOI] [PubMed] [Google Scholar]
- 54.Wolff EM, Chihara Y, Pan F, Weisenberger DJ, Siegmund KD, Sugano K, et al. Unique DNA methylation patterns distinguish noninvasive and invasive urothelial cancers and establish an epigenetic field defect in premalignant tissue. Cancer Res. 2010;70:8169–78. doi: 10.1158/0008-5472.CAN-10-1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wolff EM, Byun HM, Han HF, Sharma S, Nichols PW, Siegmund KD, et al. Hypomethylation of a LINE-1 promoter activates an alternate transcript of the MET oncogene in bladders with cancer. PLoS Genet. 2010;6:e1000917. doi: 10.1371/journal.pgen.1000917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chung W, Bondaruk J, Jelinek J, Lotan Y, Liang S, Czerniak B, et al. Detection of bladder cancer using novel DNA methylation biomarkers in urine sediments. Cancer Epidemiol Biomarkers Prev. 2011;20:1483–91. doi: 10.1158/1055-9965.EPI-11-0067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kandimalla R, van Tilborg AA, Zwarthoff EC. DNA methylation-based biomarkers in bladder cancer. Nat Rev Urol. 2013;10:327–35. doi: 10.1038/nrurol.2013.89. [DOI] [PubMed] [Google Scholar]
- 58.Costa VL, Henrique R, Danielsen SA, Duarte-Pereira S, Eknaes M, Skotheim RI, et al. Three epigenetic biomarkers, GDF15, TMEFF2, and VIM, accurately predict bladder cancer from DNA-based analyses of urine samples. Clin Cancer Res. 2010;16:5842–51. doi: 10.1158/1078-0432.CCR-10-1312. [DOI] [PubMed] [Google Scholar]
- 59.Kandimalla R, van Tilborg AA, Kompier LC, Stumpel DJ, Stam RW, Bangma CH, et al. Genome-wide analysis of CpG island methylation in bladder cancer identified TBX2, TBX3, GATA2, and ZIC4 as pTa-specific prognostic markers. Eur Urol. 2012;61:1245–56. doi: 10.1016/j.eururo.2012.01.011. [DOI] [PubMed] [Google Scholar]
- 60.Agundez M, Grau L, Palou J, Algaba F, Villavicencio H, Sanchez-Carbayo M. Evaluation of the methylation status of tumour suppressor genes for predicting bacillus Calmette-Guérin response in patients with T1G3 high-risk bladder tumours. Eur Urol. 2011;60:131–40. doi: 10.1016/j.eururo.2011.04.020. [DOI] [PubMed] [Google Scholar]
- 61.Rideout WM 3rd, Eversole-Cire P, Spruck CH, 3rd, Hustad CM, Coetzee GA, Gonzales FA, et al. Progressive increases in the methylation status and heterochromatinization of the myoD CpG island during oncogenic transformation. Mol Cell Biol. 1994;14:6143–52. doi: 10.1128/mcb.14.9.6143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yamamoto E, Toyota M, Suzuki H, Kondo Y, Sanomura T, Murayama Y, et al. LINE-1 hypomethylation is associated with increased CpG island methylation in Helicobacter pylori-related enlarged-fold gastritis. Cancer Epidemiol Biomarkers Prev. 2008;17:2555–64. doi: 10.1158/1055-9965.EPI-08-0112. [DOI] [PubMed] [Google Scholar]
- 63.Yu J, Zhu T, Wang Z, Zhang H, Qian Z, Xu H, et al. A novel set of DNA methylation markers in urine sediments for sensitive/specific detection of bladder cancer. Clin Cancer Res. 2007;13:7296–304. doi: 10.1158/1078-0432.CCR-07-0861. [DOI] [PubMed] [Google Scholar]
- 64.Yeh CM, Chen PC, Hsieh HY, Jou YC, Lin CT, Tsai MH, et al. Methylomics analysis identifies ZNF671 as an epigenetically repressed novel tumor suppressor and a potential non-invasive biomarker for the detection of urothelial carcinoma. Oncotarget. 2015;6:29555–72. doi: 10.18632/oncotarget.4986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Renard I, Joniau S, van Cleynenbreugel B, Collette C, Naômé C, Vlassenbroeck I, et al. Identification and validation of the methylated TWIST1 and NID2 genes through real-time methylation-specific polymerase chain reaction assays for the noninvasive detection of primary bladder cancer in urine samples. Eur Urol. 2010;58:96–104. doi: 10.1016/j.eururo.2009.07.041. [DOI] [PubMed] [Google Scholar]
- 66.Friedrich MG, Weisenberger DJ, Cheng JC, Chandrasoma S, Siegmund KD, Gonzalgo ML, et al. Detection of methylated apoptosis-associated genes in urine sediments of bladder cancer patients. Clin Cancer Res. 2004;10:7457–65. doi: 10.1158/1078-0432.CCR-04-0930. [DOI] [PubMed] [Google Scholar]
- 67.Su SF, de Castro Abreu AL, Chihara Y, Tsai Y, Andreu-Vieyra C, Daneshmand S, et al. A panel of three markers hyper- and hypomethylated in urine sediments accurately predicts bladder cancer recurrence. Clin Cancer Res. 2014;20:1978–89. doi: 10.1158/1078-0432.CCR-13-2637. [DOI] [PubMed] [Google Scholar]
- 68.Kim J, Akbani R, Creighton CJ, Lerner SP, Weinstein JN, Getz G, et al. Invasive bladder cancer: genomic insights and therapeutic promise. Clin Cancer Res. 2015;21:4514–24. doi: 10.1158/1078-0432.CCR-14-1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kim EJ, Kim YJ, Jeong P, Ha YS, Bae SC, Kim WJ, et al. Methylation of the RUNX3 promoter as a potential prognostic marker for bladder tumor. J Urol. 2008;180:1141–5. doi: 10.1016/j.juro.2008.05.002. [DOI] [PubMed] [Google Scholar]
- 70.Maruyama R, Toyooka S, Toyooka KO, Harada K, Virmani AK, Zöchbauer-Müller S, et al. Aberrant promoter methylation profile of bladder cancer and its relationship to clinicopathological features. Cancer Res. 2001;61:8659–63. [PubMed] [Google Scholar]
- 71.Marsit CJ, Karagas MR, Andrew A, Liu M, Danaee H, Schned AR, et al. Epigenetic inactivation of SFRP genes and TP53 alteration act jointly as markers of invasive bladder cancer. Cancer Res. 2005;65:7081–5. doi: 10.1158/0008-5472.CAN-05-0267. [DOI] [PubMed] [Google Scholar]
- 72.Catto JW, Azzouzi AR, Rehman I, Feeley KM, Cross SS, Amira N, et al. Promoter hypermethylation is associated with tumor location, stage, and subsequent progression in transitional cell carcinoma. J Clin Oncol. 2005;23:2903–10. doi: 10.1200/JCO.2005.03.163. [DOI] [PubMed] [Google Scholar]
- 73.He YF, Li BZ, Li Z, Liu P, Wang Y, Tang Q, et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science. 2011;333:1303–7. doi: 10.1126/science.1210944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kohli RM, Zhang Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature. 2013;502:472–9. doi: 10.1038/nature12750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–92. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Munari E, Chaux A, Vaghasia AM, Taheri D, Karram S, Bezerra SM, et al. Global 5-hydroxymethylcytosine levels are profoundly reduced in multiple genitourinary malignancies. PLoS One. 2016;11:e0146302. doi: 10.1371/journal.pone.0146302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Liu M, Ohtani H, Zhou W, Ørskov AD, Charlet J, Zhang YW, et al. Vitamin C increases viral mimicry induced by 5-aza-2’-deoxycytidine. Proc Natl Acad Sci U S A. 2016;113:10238–44. doi: 10.1073/pnas.1612262113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–80. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 79.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 80.Vallot C, Stransky N, Bernard-Pierrot I, Hérault A, Zucman-Rossi J, Chapeaublanc E, et al. A novel epigenetic phenotype associated with the most aggressive pathway of bladder tumor progression. J Natl Cancer Inst. 2011;103:47–60. doi: 10.1093/jnci/djq470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ellinger J, Bachmann A, Göke F, Behbahani TE, Baumann C, Heukamp LC, et al. Alterations of global histone H3K9 and H3K27 methylation levels in bladder cancer. Urol Int. 2014;93:113–8. doi: 10.1159/000355467. [DOI] [PubMed] [Google Scholar]
- 82.Wu S, Yang Z, Ye R, An D, Li C, Wang Y, et al. Novel variants in MLL confer to bladder cancer recurrence identified by wholeexome sequencing. Oncotarget. 2016;7:2629–45. doi: 10.18632/oncotarget.6380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Charlet J, Duymich CE, Lay FD, Mundbjerg K, Dalsgaard Sørensen K, Liang G, et al. Bivalent regions of cytosine methylation and H3K27 acetylation suggest an active role for DNA methylation at enhancers. Mol Cell. 2016;62:422–31. doi: 10.1016/j.molcel.2016.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yoshino H, Seki N, Itesako T, Chiyomaru T, Nakagawa M, Enokida H, et al. Aberrant expression of microRNAs in bladder cancer. Nat Rev Urol. 2013;10:396–404. doi: 10.1038/nrurol.2013.113. [DOI] [PubMed] [Google Scholar]
- 85.Friedman JM, Liang G, Liu CC, Wolff EM, Tsai YC, Ye W, et al. The putative tumor suppressor microRNA-101 modulates the cancer epigenome by repressing the polycomb group protein EZH2. Cancer Res. 2009;69:2623–9. doi: 10.1158/0008-5472.CAN-08-3114. [DOI] [PubMed] [Google Scholar]
- 86.Saito Y, Liang G, Egger G, Friedman JM, Chuang JC, Coetzee GA, et al. Specific activation of microRNA-127 with downregulation of the proto-oncogene BCL6 by chromatin-modifying drugs in human cancer cells. Cancer Cell. 2006;9:435–43. doi: 10.1016/j.ccr.2006.04.020. [DOI] [PubMed] [Google Scholar]
- 87.Saito Y, Saito H, Liang G, Friedman JM. Epigenetic alterations and microRNA misexpression in cancer and autoimmune diseases: a critical review. Clin Rev Allergy Immunol. 2014;47:128–35. doi: 10.1007/s12016-013-8401-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cheng Y, Deng X, Yang X, Li P, Zhang X, Li P, et al. Urine microRNAs as biomarkers for bladder cancer: a diagnostic meta-analysis. Onco Targets Ther. 2015;8:2089–96. doi: 10.2147/OTT.S86908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lorch Y, LaPointe JW, Kornberg RD. Nucleosomes inhibit the initiation of transcription but allow chain elongation with the displacement of histones. Cell. 1987;49:203–10. doi: 10.1016/0092-8674(87)90561-7. [DOI] [PubMed] [Google Scholar]
- 90.Lee CK, Shibata Y, Rao B, Strahl BD, Lieb JD. Evidence for nucleosome depletion at active regulatory regions genome-wide. Nat Genet. 2004;36:900–5. doi: 10.1038/ng1400. [DOI] [PubMed] [Google Scholar]
- 91.Lay FD, Liu Y, Kelly TK, Witt H, Farnham PJ, Jones PA, et al. The role of DNA methylation in directing the functional organization of the cancer epigenome. Genome Res. 2015;25:467–77. doi: 10.1101/gr.183368.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ghosh M, Brancato SJ, Agarwal PK, Apolo AB. Targeted therapies in urothelial carcinoma. Curr Opin Oncol. 2014;26:305–20. doi: 10.1097/CCO.0000000000000064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mooso BA, Vinall RL, Mudryj M, Yap SA, deVere White RW, Ghosh PM. The role of EGFR family inhibitors in muscle invasive bladder cancer: a review of clinical data and molecular evidence. J Urol. 2015;193:19–29. doi: 10.1016/j.juro.2014.07.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.You JS, Jones PA. Cancer genetics and epigenetics: two sides of the same coin? Cancer Cell. 2012;22:9–20. doi: 10.1016/j.ccr.2012.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Nervi C, De Marinis E, Codacci-Pisanelli G. Epigenetic treatment of solid tumours: a review of clinical trials. Clin Epigenetics. 2015;7:127. doi: 10.1186/s13148-015-0157-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ding XL, Yang X, Liang G, Wang K. Isoform switching and exon skipping induced by the DNA methylation inhibitor 5-Aza-2’-deoxycytidine. Sci Rep. 2016;6:24545. doi: 10.1038/srep24545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Nebbioso A, Carafa V, Benedetti R, Altucci L. Trials with ‘epigenetic’ drugs: an update. Mol Oncol. 2012;6:657–82. doi: 10.1016/j.molonc.2012.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hurst CD, Platt FM, Taylor CF, Knowles MA. Novel tumor subgroups of urothelial carcinoma of the bladder defined by integrated genomic analysis. Clin Cancer Res. 2012;18:5865–77. doi: 10.1158/1078-0432.CCR-12-1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mayland CR, Bennett MI, Allan K. Vitamin C deficiency in cancer patients. Palliat Med. 2005;19:17–20. doi: 10.1191/0269216305pm970oa. [DOI] [PubMed] [Google Scholar]