

Abstract
Phosphatidylinositol-3-kinase (PI3K) pathway signaling is an established oncogenic signal transduction pathway implicated in multiple malignancies. Therapeutic targeting of PI3K pathway components has improved outcomes in chronic lymphocytic leukemia, kidney cancer, breast cancer, and neuroendocrine tumors. Gastric cancers harbor some of the highest rates of oncogenic alterations in PI3K but attempts to translate this genomic observation have met with limited clinical success and novel approaches are needed. In the following review we discuss PI3K signaling, previous preclinical and clinical investigations in gastric cancer, and discuss future strategies aimed at overcoming resistance and improving efficacy. Identification and refinement of molecular tumor subtypes, development of predictive biomarkers along, and rational drug combination strategies are key to capitalizing on the therapeutic potential of PI3K pathway directed therapies in gastric cancers.
Keywords: PIK3CA portein, Stomach Neoplasms, Molecular Targeted Therapies, AKT oncogene protein, mTOR protein, Clinical Trial
INTRODUCTION
Gastric adenocarcinoma (GC) is the fifth most common cancer in the world, with approximately one million new cases and 730,000 deaths occurring annually [1]. In the United States, approximately 21,600 new cases occurred in 2015 [2]. Therapeutic advances in gastric cancer have been slow and 5-year survival is less than 10% with a median overall survival of 1 year for advanced disease [3,4]. Surgery with or without chemotherapy and/or radiation is potentially curative in resectable disease although metastatic recurrence rates remain high [5-7]. Systemic multiagent chemotherapy remains the mainstay of treatment for advanced disease [8,9]. The phase III ToGA trial (Trastuzumab for Gastric Cancer) was the first study to demonstrate success for a targeted therapy in ERBB2-amplified gastric cancer, but this is only relevant for the 12%–20% of advanced gastric cancer patients who harbor ERBB2 (Her2) amplification, and new therapies are needed [10].
Geographic differences in gastric cancer incidence are well described, and gastric cancers are the second most commonly diagnosed cancer and a leading cause of cancer-related death in Korea [11]. Risk is partly related to geographic and ethnic origins and observations that migration and associated change in environmental risk factors affects gastric cancer risk has led to some questions about underlying biologic differences [12,13]. The incidence of gastric cancer in Asian American populations has remained stable or slightly decreased over time, largely mirroring the larger United States trend [14]. Although data is limited it appears PIK3CA mutations may be less common in east Asian populations whereas phosphatase and tensin homolog (PTEN) deletion more frequent when compared to Caucasian gastric cancer patients [15]. Despite the lack of well characterized genomic differences, large phases II and III trials continue to report outcome differences in Asian and non-Asian populations [16-19].
Molecular characterization of gastric cancers has revealed high rates of recurrent somatic alterations in members of the PI3K/AKT/mTOR pathway, suggesting potential therapeutic targets [20]. The PI3K/AKT/mTOR pathway is an important promoter of cell growth, metabolism, survival, metastasis, and resistance to chemotherapy. Despite the appreciation of frequent PI3K pathway alterations in GC, the ability to translate this genomic observation to improved outcomes has been limited. In the following review we discuss PI3K signaling, preclinical rationale, current clinical data, and future directions.
CANONICAL PI3K SIGNALING
Phosphatidylinositol-3-kinase (PI3K) is a lipid kinase existing as a heterodimer consisting of a regulatory subunit p85 (p85α, p85β, and p55γ) and a catalytic subunit p110 (p110α, p110β, p110γ, and p110δ). Although there are 3 classes of PI3K based on the structure, distribution, and mechanism of activation, class IA PI3K is mostly associated with malignancy [21,22]. Under physiologic conditions PI3K is activated by multiple receptor tyrosine kinases (RTKs) and/or G-protein-coupled receptors located at the cell surface (Fig. 1). Upon RTK activation PI3K phosphorylates phosphatidylinositol 4,5 bisphosphate (PI(4,5) P2) at the 3’ OH position to become phosphatidylinositol 3,4,5 trisphosphate (PI(3,4,5)P3), which in turn directly binds to the pleckstrin homology (PH) domains of various signaling proteins, including phosphoinositide-dependent kinase 1 (PDK1) [23]. PDK1 phosphorylates AKT in the kinase domain at threonine 308 while PDK2 phosphorylates AKT at serine 473 domain, leading to full AKT activation [24]. Once activated, AKT phosphorylates multiple downstream targets in the cytoplasm and nucleus to promote cell growth and survival [25]. For instance, AKT inhibits proapoptotic Bcl-2 proteins such as BAX and BAD and antagonizes p-53 mediated apoptosis by phosphorylating Mdm2 [26-28]. AKT also phosphorylates TSC2, which prevents the TSC1/TSC2 complex from inhibiting Rheb. Activated Rheb stimulates the mammalian target of rapamycin (mTOR) complex 1 (mTORC1), resulting in increased activity of eukaryotic initiation factor 4E (4EBP1) and the ribosomal S6 protein (S6K1), leading downstream to cell proliferation [27]. mTORC1 also upregulates other genes involved in cell division and angiogenesis, such as cyclin D and hypoxia-inducible factor-1a (HIF-1a), respectively [29,30]. The major negative regulator of the PI3K pathway is the lipid PTEN, a tumor suppressor gene that encodes a lipid phosphatase that converts PIP3 back to PIP2 [31]. Loss of PTEN results in constitutive activation of Akt and alteration of downstream factors in Akt signaling in multiple preclinical models (Fig. 1).
Fig. 1.
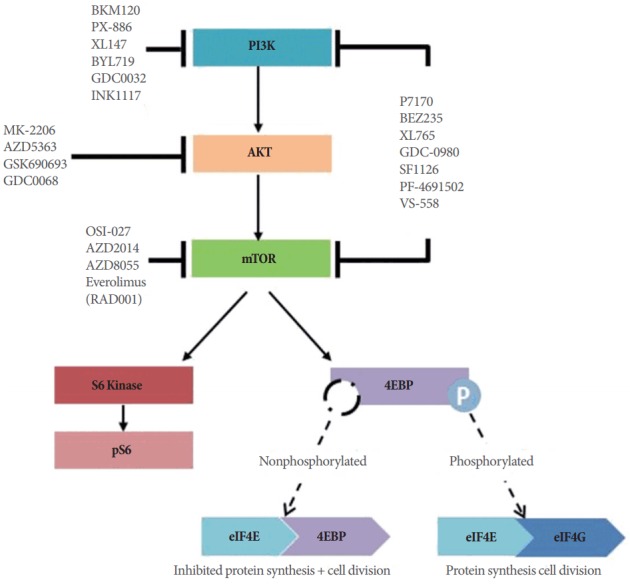
Canonical phosphatidylinositol-3-kinase (PI3K) signaling. Key nodes are represented and examples of small molecule inhibitors acting on key components are shown. mTOR, mammalian target of rapamycin; 4EBP1, eukaryotic initiation factor 4E.
PI3K PATHWAY ALTERATIONS IN GASTRIC CANCER
The PI3K/AKT/mTOR pathway is the second most commonly altered pathway in human cancer after the p53 pathway with the observed frequency of 30%–60% across tumor types [32]. Pathologic PI3K pathway activation is mediated by several mechanisms ranging from upstream RTKs, decreased expression of PTEN, genetic alteration in PIK3CA and AKT and other less frequent events [33]. Within GC PIK3CA is the most commonly mutated PI3K isoform with a mutation and amplification frequency of 18% and 5% respectively (Fig. 2). This finding is consistent with previous studies that reported PI3KCA mutation rates of 4%–25% [34-37]. Approximately 80% of PIK-3CA mutations occur at three recurrent hotspots; E545K and E542K in the helical domain (exon 9) and H1047R in the kinase domain (exon 20) [33]. PIK3CA mutation frequency in GC correlates with stage, seen in 21.4% of pT4 tumors compared to 6.4% in pT2 lesions [37]. In addition to stage there is a strong predilection for PIK3CA mutation in Epstein-Barr virus (EBV) positive gastric cancer with a nonsilent mutation rate up to 80% [20]. In contrast the activating alterations in other p110 isoforms (delta and beta) are rare (Fig. 2).
Fig. 2.
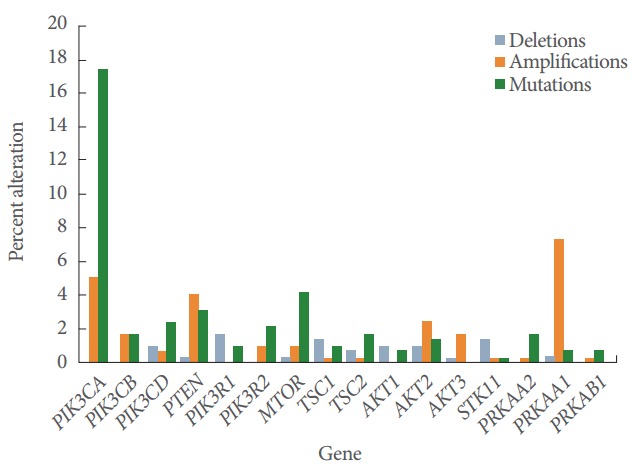
Frequency of alterations in key PI3K-pathway genes in gastric adenocarcinoma. Data derived from TCGA via MSKCC cbio portal (www.cbioportal.org, accessed 2016 Aug). PI3K, phosphatidylinositol-3-kinase.
PTEN is a tumor suppressor gene on chromosome 10q23.3 and the major negative regulator of PI3K activity under physiologic conditions. Within the cancer genome atlas (TCGA) database, the frequency of deletion, mutation and amplification of PTEN in gastric cancer is 0.3%, 3.1%, and 4%, respectively (Fig. 2). There is significant difference in the rate of PIK3CA and PTEN mutations between Asian and Caucasian GC patients. A meta-analysis found that East Asian and Caucasian GC patients differ significantly among the frequencies of PIK3CA Exon 9 and 20 mutations (7% vs. 15%, respectively), PTEN deletion (21% vs. 4%) and PTEN loss (47% vs. 78%) [15]. Another study identified 19% PTEN mutation rate in a Chinese population [38]. The genetic alterations in the study included missense (55.6%), nonsense (33.3%), deletion (7.4%), and a mutation within PTEN intron 6 (3.7%). PTEN missense mutations abrogated or attenuated phosphatase activity up to 90% of the time suggesting functional relevance [39]. PTEN mutation rate is higher in advanced stage and poorly differentiated gastric cancer [40]. PTEN status did not predict response to PI3K/mTOR inhibitors reliably, perhaps because patients with negative and reduced PTEN expression had a higher incidence of simultaneous MAPK (KRAS, NRAS, BRAF) mutations.
The Akt family of genes consists of Akt1, Akt2, and Akt3 genes. Akt1 and 2 are normally expressed in all tissues while Akt 3 is mainly restricted to brain and testes. Although the overexpression of AKT by immunohistochemistry is up to 74% in gastric cancer, genomic alterations are relatively rare at 1%–3% in gastric cancer (Fig. 2) [41]. Differential isoform expression has functional implications and may be relevant for future isoform-specific therapeutic approaches. For example, AKT1 promotes cellular survival and proliferation while AKT2 stimulates cellular invasiveness and mesenchymal transformation [42,43]. Loss of AKT2 expression may decrease metastatic potential while loss of AKT1 paradoxically can increase invasiveness, presumably due to a shunt to AKT2 isoform production [44]. The functional implications of genomic aberrations in downstream signaling nodes MTOR are less well studied although effective inhibitor of mTOR signaling appears important for optimal efficacy. Preclinical studies have demonstrated activity with AKT selective inhibitors in gastric cancer [45].
PI3K-PATHWAY INHIBITORS
There are 4 major classes of drugs that target the PI3K-AKT pathway: PI3K inhibitors, dual PI3K-mTOR inhibitors, mTOR inhibitors, and AKT inhibitors (Table 1). PI3K inhibitors have been developed as either isoform-specific or pan-PI3K inhibitors. Theoretically, isoform-specific inhibitors should have an inherent specificity thereby minimizing on-target side effects results from inhibition of all isoforms. However, potential disadvantage of selective PI3K inhibition is an incomplete blockade of AKT activation in conditions where multiple p110 isoforms exist.
Table 1.
Summary of clinical investigations targeting the PI3K/AKT/mTOR pathway in gastric cancer
| Compound | Target | Study design | Result |
|---|---|---|---|
| Everolimus | mTOR | Phase III | No OS benefit |
| BKM120 | Pan-PI3K | Phase II | N/A |
| PX-886 | Pan-PI3K | Phase I | No DLT |
| SD in 2 of 6 patients | |||
| XL147 | Pan-PI3K | Phase I | DLT = grade 3 rash |
| SD in 6 of 39 patients | |||
| WX-037 | Pan-PI3K | Phase I | N/A |
| BYL719 | p110α PI3K | Phase I | N/A |
| GDC0032 | p110α PI3K | Phase I | DLTs = diarrhea and hyperglycemia |
| INK1117 | p110α PI3K | Phase I | N/A |
| P7170 | PI3K/mTOR | Phase I | N/A |
| BEZ235 | PI3K/mTOR | Phase I/II | DLT = diarrhea |
| XL765 | PI3K/mTOR | Phase I | DLTs = nausea/vomiting, diarrhea, elevated liver enzymes |
| SD in five of 36 patients | |||
| GDC-0980 | PI3K/mTOR | Phase I | DLTs = hyperglycemia and mucositis |
| SF1126 | PI3K/mTOR | Phase I | N/A |
| PF-05212384 | PI3K/mTOR | Phase I | N/A |
| PF-4691502 | PI3K/mTOR | Phase I | DLTs = fatigue and rash |
| SD in 12 of 36 patients | |||
| VS-558 | PI3K/mTOR | Phase I | N/A |
| OSI-027 | mTOR 1/2 | Phase I | N/A |
| AZD2014 | mTOR 1/2 | Phase I | N/A |
| AZD8055 | mTOR 1/2 | Phase I | N/A |
| Temsirolimus | mTOR | Phase I | N/A |
| MK-2206 | Allosteric AKT | Phase I | DLT=rash and mucositis |
| SD in 6 of 19 pts | |||
| AZD5363 | Catalytic AKT | Phase II | N/A |
| AZD5363 | Catalytic AKT | Phase II | N/A |
| GSK690693 | Catalytic AKT | Phase I | DLTs = hyperglycemia and hyperlipidemia |
| GDC0068 | Catalytic AKT | Phase II | N/A |
PI3K, phosphatidylinositol-3-kinase; mTOR, mammalian target of rapamycin; OS, overall survival; N/A, not available; SD, stable disease; DLT, dose limiting toxicity.
Compounds inhibiting the mTOR catalytic site block both mTOR1 and mTOR2, a critical node mediating downstream function. Catalytic site inhibitors largely overcome the insulin receptor substrate 1 (IRS-1)-mediated feedback thought to limit the efficacy of earlier mTOR inhibitors like sirolimus (rapamycin) [46-48]. Patients with AKT1 and AKT2 amplifications may be theoretically more sensitive to AKT inhibitors. Most of these drugs are either ATP mimetics or noncatalytic site allosteric inhibitors. Allosteric inhibitors block the PH domain from binding phosphoinositides at the plasma membrane, preventing Akt phosphorylation [44]. Similar to the theory of PI3K inhibitors, it is possible that AKT inhibitors may eventually be isoform specific for AKT1 or AKT2, to deliver lower effective doses with less side effects. A phase II S1005 study evaluated MK-2206, an allosteric inhibitor of AKT, as second-line therapy for 66 patients with advanced gastric cancer [49]. There were two treatment related deaths (cardiac arrest and respiratory failure). Common toxicities included anemia 15%, anorexia 27%, diarrhea 24%, fatigue 48%, hyperglycemia 29%, nausea 40%, vomiting 20%, maculopapular rash 30%, and acneiform rash 8%. The response rate (RR), progression free survival (PFS), and overall survival (OS) were 2%, 1.8 month, and 5 months, respectively. The study did not meet primary endpoint of OS 6.5 months. A multinational phase II JAGUAR trial comparing the efficacy of mFOLFOX6 plus ipatasertib, an oral AKT inhibitor, versus mFOLFOX6 plus placebo in 120 patients with advanced untreated gastric cancer is currently undergoing [50].
Though PI3K signaling inhibitors are promising, there are some theoretical shortcomings which raise concern for their clinical efficacy. Preclinical studies in breast cancer cells with PI3KCA mutations show in vivo efficacy of PI3K-mTOR inhibitors or Akt inhibitors. However, treatment leads to cytostatic rather than cytotoxic effects when given as monotherapies [51,52]. It remains to be seen whether inhibitors of the PI3K signaling pathway will be efficacious in single-agent therapy, though unlikely. A potential reason for limited efficacy of single-agent therapy is positive feedback loops and activation of coinciding proliferative signaling pathways. The most common alternate pathway involves nodes in the Ras-Raf-MEK-ERK signal transduction pathway. Logically, this observation has led to the development of combination strategies. Murine models of lung cancer with KRAS overexpression showed no efficacy of either a PI3K or MEK inhibitor alone, however, the combination was highly effective [53]. KRAS mutant colorectal cancer mouse model cells that demonstrated resistance to PI3K/mTOR inhibitor had restored sensitivity to dual PI3K/mTOR inhibitor in combination with a pan-ERBB inhibitor [54]. Similarly, the combination of PI3K/mTOR and RAS/ERK pathway inhibitors shows synergy in treating ovarian cancer [55]. Studies such as these suggest that combination therapy may delay resistance and by used in cancers which were initially responsive [56].
EARLY CLINICAL EXPERIENCE
Preclinical studies demonstrated that treatment of gastric cancer cell lines with everolimus and sirolimus lead to G1 cell cycle arrest and growth inhibition [57,58]. In 5-FU-resistant gastric cancer cells, the addition of everolimus to chemotherapy demonstrated synergistic growth inhibition [59]. Administration of rapamycin to mouse xenograft models decreased tumor volume and microvessel density, as well as downstream factors such as S6K1, 4EBP1, HIF-1a and vascular endothelial growth factor [60]. In a phase 2 multicenter trial in 53 advanced heavily pretreated gastric cancer patients who was treated with everolimus, the disease control rate (DCR), PFS, and OS were 56%, 2.7 months, and 10.1 months, respectively [61]. There was no observed partial response (PR) or complete response, and common side effects included stomatitis (73.6%), anorexia (52.8%), fatigue (50.9%), rash (45.3%), nausea (32.1%), peripheral edema (22.6%), diarrhea (20.8%), and pruritus (18.9%). Based on these results, the phase III GRANITE 1 trial evaluated the safety and efficacy of everolimus versus placebo in 656 patients who progressed after the first or second line chemotherapy, randomized to a 2:1 schedule [62]. Although PFS was modestly improved (median: 1.7 months vs. 1.4 months, P<0.001), overall survival, the primary endpoint, was not (median: 5.4 months vs. 4.3 months, P=0.124). However, there were patients who derived durable stable disease and the modest PFS improvement suggested potential benefit in selected patients. Multiple potential predictive biomarkers have been explored, including PIK3CA/PTEN mutation status, AKT activation, pS6Ser240/4 and other members of the AKT/mTOR pathway. One preclinical study demonstrated that tumor cells harboring PIK3CA and/or PTEN mutations were more likely to be rapamycin responsive (P=0.0123) [63]. Additionally, Akt phosphorylation (S473 and T308) was significantly higher in rapamycin sensitive cells (P<0.0001) in this study. Another study demonstrated significant correlations between everolimus sensitivity and a battery of markers consisting of total S6 levels, p235-S6, p240-S6, pEIF4, Rictor, Raptor, total AKT, and pAKT [64]. However, the authors noted that such approach would be infeasible in real clinical practice. The ratio of p235-S6/total-S6 and pAKT alone are able to provide adequate predictive power. The potential role of pS6Ser240/4 as predictive marker was shown in an Asian phase II study evaluating PFS rate at 4 months in 54 advanced gastric cancer pts receiving everolimus [65]. The DCR was 38.9% with 3.7% PR (2 patients). The 4-month PFS rate was 18.4% (less than the hypothesized PFS of 30%) and the median PFS and OS were 1.7 and 8.3 months, respectively. However, high baseline expression of pS6Ser240/4, defined as immunohistochemistry (IHC) staining >2, was significantly associated with higher disease control rate (DCR) (P <0.043) and prolonged PFS (P<0.001). The US study showed numerically inferior outcomes compared to the Asian study with median OS 3.4 months and PFS 1.8 months. However, this study reaffirmed a strong correlation between ≥2+IHC staining for p-S6 in tumor samples with better PFS (P <0.0001) and DCR (P =0.0001) [66]. Anecdotal evidence seems to validate the p-S6 overexpression and concurrent PIK3CA mutation may identify a group of patient able to derive prolonged benefit from PI3K-directed therapy [67]. Further studies are required to validate these biomarkers not only for MTOR inhibitors but also other PI3K pathway inhibitors.
FUTURE DIRECTIONS
The disappointing activity of everolimus in advanced GC highlighted the need for a more biologically informed development process for PI3K pathway inhibitors in gastric cancer. Recently Lei et al. [68] proposed a new gastric cancer classification that was distinct from the well-known 1965 Lauren classification. This classification included mesenchymal, proliferative and metabolic as the 3 distinct histologic subtypes based on the differentially expressed genes. For instance, the gene clusters in the mesenchymal tumors are mostly involved in the epithelial mesenchymal transition pathway with high mTOR pathway activity. The mesenchymal subtype was found to be sensitive to PIK-3CA inhibitors while the proliferative subtype was more sensitive to 5-FU in vitro. Another subgroup that may benefit from PIK3CA inhibitors are those who carry EBV positive tumors which was shown to harbor PIK3CA mutation up to 80% according to the TCGA study [20].
We believe that –omics platforms characterizing gastric cancer and other tumors are a critical step toward refining the role of PI3K compounds. While hotspot mutational analysis is readily available we feel that the genomic context of a PI3K/AKT/mTOR alterations will be important in guiding outcomes. The relatively low response rate even for PIK3CA mutated tumors may be due to intrinsic resistance, acquired resistance mechanism and crosstalk between different pathways. One preclinical study demonstrated that HNSCC cell lines with PIK3CA mutations were universally sensitive to PI3K, mTOR inhibitors, or both. However, PIK3CA amplification, PTEN loss, and basal PIK3CA/AKT/MTOR pathway activity did not predict response [69]. As more clinical experience with PI3K inhibitors becomes available, we will be able to predict which specific PI3K pathway gene dysregulation would be sensitive to specific combination of inhibitors. Whole genome sequencing will also identify genetic alteration in other pathways such as RAS or BRAF, which are known to negate the effect of PI3K inhibitor. One study using the PIK3CA/PTEN/Ras testing protocol prior to PI3K pathway inhibition found that treated patients with wild-type KRAS had a higher PR rate of 31% compared to 6% in patients with simultaneous KRAS mutations (P=0.05) [70]. The PI3K/AKT/mTOR and RAS/RAF/MEK/ERK pathways are known to interact at multiple points, resulting in cross-activation, cross-inhibition, and pathway convergence. Examples include activation of PI3K directly by RAS, suppression of TSC2 by phosphorylated ERK or AKT, initiating mTORC1 complex formation [71].
Multiple resistance mechanisms to PIK3CA inhibitors have been identified and they likely contribute to poor efficacy. The most well-known mechanism is the activation of multiple RTK pathways after PIK3CA inhibition. Inhibition of mTORC1 with rapamycin and catalytic mTOR inhibitors increase insulin receptor substrate 1 (IRS-1) levels and induce AKT phosphorylation and downstream signaling [52,72]. AKT inhibition leads to loss of transcription factor FOXO3 regulation and subsequent increased expression of multiple RTKs [73,74]. Moreover, simultaneous Wnt–β-catenin pathway hyperactivation and PI3K-AKT signaling inhibition promote nuclear accumulation of β-catenin and FOXO3a, and eventual metastasis [75]. Additionally, enhanced HER2 signaling following PI3K inhibition has been reported to lead to ERK activation [76]. Dual blockade with either MEK and PI3K or ERK and PI3K inhibitors lead to higher rate of apoptosis in numerous preclinical studies, prompting the rationale for combination therapy [32,76].
The clinical experience with dual MEK and PI3K inhibitors in treating solid tumors are limited as most trials are still in early phases. The preliminary results from these studies implicate potential efficacy in the combination therapy. There are no known published gastric cancer cases that are sensitivity to dual pathway blockade. In a phase I trial where 49 patients with RAS or BRAF mutant advanced solid tumors were treated with trametinib and BKM120, 3 patients with RAS ovarian cancers achieved PR, 2 with BRAF mutant melanoma achieved stable disease (SD) [77]. The combination was well tolerated with common adverse events including rash, nausea, fatigue, vomiting, decreased appetite, and elevated CPK. Grade 3 DLTs included stomatitis, dysphagia, decreased ejection fraction, CPK elevation, nausea and anorexia. The combination of GDC-0973 and GDC-0941 was evaluated in 78 patients with solid tumors yielded 3 PRs (1 BRAF mutant melanoma, 1 BRAF mutant pancreatic, and 1 KRAS mutant endometrial cancer) and 5 SD [78]. This combination therapy was well tolerated, with comparable toxicities to monotherapy. In a retrospective study, 5 patients with coactivation of PI3K/AKT and RAS/RAF/MEK pathways who received dual inhibition had tumor regression/stabilization from 2%–64% (3 colon, 2 melanoma). Conversely, all 4 colon cancer patients in the single pathway inhibition group developed progressive disease. This finding is consistent with other studies that PI3K inhibition alone is insufficient for tumors with concurrent Ras/BRAF activation. For the unselected population, there was no significant difference in both tumor control rate for single or dual inhibition (52.7% and 64.6%, respectively, P=0.16) [79]. There were more grade III/IV adverse events observed in the dual pathway inhibition group. However, this study was limited by its retrospective nature. The data were derived from different phase I trials that are heterogeneous. The response rates are low and appear mainly restricted to RAS and RAF mutated cancers. The most responsive tumors included BRAF positive melanoma, KRAS positive ovarian cancer, and KRAS positive colon cancer.
CONCLUSIONS
Gastric cancer is a heterogeneous disease and recent genomic characterization has refined our understanding of molecular subtypes [80]. Alterations in the PI3K/AKT/mTOR pathway occur across gastric cancer types but have yet to translate to improved outcomes in prospective trials. Anecdotal reports provide preliminary support for a possible rare subset with multiple PI3K pathway activating alterations and the absence of MAPK alterations which may provide ready resistance. The need to assess multiple genomic alterations in order to identify a narrow subset makes large prospective trials more difficult, however, is likely a key need to maximize the potential to exploit oncogenic PI3K pathway alterations in gastric and other tumors. We will eagerly await additional information from some of the aforementioned ongoing clinical trials.
Footnotes
Conflict of Interest
No potential conflict of interest relevant to this article was reported.
REFERENCES
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65:5–29. doi: 10.3322/caac.21254. [DOI] [PubMed] [Google Scholar]
- 3.Power DG, Kelsen DP, Shah MA. Advanced gastric cancer--slow but steady progress. Cancer Treat Rev. 2010;36:384–92. doi: 10.1016/j.ctrv.2010.01.005. [DOI] [PubMed] [Google Scholar]
- 4.Sastre J, Garcia-Saenz JA, Diaz-Rubio E. Chemotherapy for gastric cancer. World J Gastroenterol. 2006;12:204–13. doi: 10.3748/wjg.v12.i2.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cunningham D, Allum WH, Stenning SP, Thompson JN, Van de Velde CJ, Nicolson M, et al. Perioperative chemotherapy versus surgery alone for resectable gastroesophageal cancer. N Engl J Med. 2006;355:11–20. doi: 10.1056/NEJMoa055531. [DOI] [PubMed] [Google Scholar]
- 6.Stahl M, Walz MK, Stuschke M, Lehmann N, Meyer HJ, RieraKnorrenschild J, et al. Phase III comparison of preoperative chemotherapy compared with chemoradiotherapy in patients with locally advanced adenocarcinoma of the esophagogastric junction. J Clin Oncol. 2009;27:851–6. doi: 10.1200/JCO.2008.17.0506. [DOI] [PubMed] [Google Scholar]
- 7.Ychou M, Boige V, Pignon JP, Conroy T, Bouché O, Lebreton G, et al. Perioperative chemotherapy compared with surgery alone for resectable gastroesophageal adenocarcinoma: an FNCLCC and FFCD multicenter phase III trial. J Clin Oncol. 2011;29:1715–21. doi: 10.1200/JCO.2010.33.0597. [DOI] [PubMed] [Google Scholar]
- 8.Juergens RA, Forastiere A. Combined modality therapy of esophageal cancer. J Natl Compr Canc Netw. 2008;6:851–60. doi: 10.6004/jnccn.2008.0063. [DOI] [PubMed] [Google Scholar]
- 9.Haghighat P, Bekaii-Saab T. An update on biochemotherapy of advanced gastric and gastroesophageal adenocarcinoma. J Natl Compr Canc Netw. 2008;6:895–900. doi: 10.6004/jnccn.2008.0068. [DOI] [PubMed] [Google Scholar]
- 10.Bang YJ, Van Cutsem E, Feyereislova A, Chung HC, Shen L, Sawaki A, et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet. 2010;376:687–97. doi: 10.1016/S0140-6736(10)61121-X. [DOI] [PubMed] [Google Scholar]
- 11.Jung KW, Won YJ, Kong HJ, Oh CM, Cho H, Lee DH, et al. Cancer statistics in Korea: incidence, mortality, survival, and prevalence in 2012. Cancer Res Treat. 2015;47:127–41. doi: 10.4143/crt.2015.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haenszel W. Variation in incidence of and mortality from stomach cancer, with particular reference to the United States. J Natl Cancer Inst. 1958;21:213–62. [PubMed] [Google Scholar]
- 13.Haenszel W, Kurihara M. Studies of Japanese migrants. I. Mortality from cancer and other diseases among Japanese in the United States. J Natl Cancer Inst. 1968;40:43–68. [PubMed] [Google Scholar]
- 14.Gomez SL, Noone AM, Lichtensztajn DY, Scoppa S, Gibson JT, Liu L, et al. Cancer incidence trends among Asian American populations in the United States, 1990-2008. J Natl Cancer Inst. 2013;105:1096–110. doi: 10.1093/jnci/djt157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chong ML, Loh M, Thakkar B, Pang B, Iacopetta B, Soong R. Phosphatidylinositol-3-kinase pathway aberrations in gastric and colorectal cancer: meta-analysis, co-occurrence and ethnic variation. Int J Cancer. 2014;134:1232–8. doi: 10.1002/ijc.28444. [DOI] [PubMed] [Google Scholar]
- 16.Pavlakis N, Sjoquist KM, Martin AJ, Tsobanis E, Yip S, Kang YK, et al. Regorafenib for the Treatment of Advanced Gastric Cancer (INTEGRATE): A Multinational Placebo-Controlled Phase II Trial. J Clin Oncol. 2016;34:2728–35. doi: 10.1200/JCO.2015.65.1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hecht JR, Bang YJ, Qin SK, Chung HC, Xu JM, Park JO, et al. Lapatinib in combination with capecitabine plus oxaliplatin in human epidermal growth factor receptor 2-positive advanced or metastatic gastric, esophageal, or gastroesophageal adenocarcinoma: TRIO-013/LOGiC--A randomized phase III trial. J Clin Oncol. 2016;34:443–51. doi: 10.1200/JCO.2015.62.6598. [DOI] [PubMed] [Google Scholar]
- 18.Fuchs CS, Tomasek J, Yong CJ, Dumitru F, Passalacqua R, Goswami C, et al. Ramucirumab monotherapy for previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (REGARD): an international, randomised, multicentre, placebo-controlled, phase 3 trial. Lancet. 2014;383:31–9. doi: 10.1016/S0140-6736(13)61719-5. [DOI] [PubMed] [Google Scholar]
- 19.Wilke H, Muro K, Van Cutsem E, Oh SC, Bodoky G, Shimada Y, et al. Ramucirumab plus paclitaxel versus placebo plus paclitaxel in patients with previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (RAINBOW): a double-blind, randomised phase 3 trial. Lancet Oncol. 2014;15:1224–35. doi: 10.1016/S1470-2045(14)70420-6. [DOI] [PubMed] [Google Scholar]
- 20.Cancer Genome Atlas Research Network Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202–9. doi: 10.1038/nature13480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Okumura N, Yoshida H, Kitagishi Y, Murakami M, Nishimura Y, Matsuda S. PI3K/AKT/PTEN signaling as a molecular target in leukemia angiogenesis. Adv Hematol. 2012;2012:843085. doi: 10.1155/2012/843085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yuan TL, Cantley LC. PI3K pathway alterations in cancer: variations on a theme. Oncogene. 2008;27:5497–510. doi: 10.1038/onc.2008.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–7. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 24.Hodgkinson CP, Sale EM, Sale GJ. Characterization of PDK2 activity against protein kinase B gamma. Biochemistry. 2002;41:10351–9. doi: 10.1021/bi026065r. [DOI] [PubMed] [Google Scholar]
- 25.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–74. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Courtney KD, Corcoran RB, Engelman JA. The PI3K pathway as drug target in human cancer. J Clin Oncol. 2010;28:1075–83. doi: 10.1200/JCO.2009.25.3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7:606–19. doi: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]
- 28.Duronio V. The life of a cell: apoptosis regulation by the PI3K/PKB pathway. Biochem J. 2008;415:333–44. doi: 10.1042/BJ20081056. [DOI] [PubMed] [Google Scholar]
- 29.Patel PH, Chadalavada RS, Chaganti RS, Motzer RJ. Targeting von Hippel-Lindau pathway in renal cell carcinoma. Clin Cancer Res. 2006;12:7215–20. doi: 10.1158/1078-0432.CCR-06-2254. [DOI] [PubMed] [Google Scholar]
- 30.Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18:1926–45. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- 31.Ortega-Molina A, Serrano M. PTEN in cancer, metabolism, and aging. Trends Endocrinol Metab. 2013;24:184–9. doi: 10.1016/j.tem.2012.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Klempner SJ, Myers AP, Cantley LC. What a tangled web we weave: emerging resistance mechanisms to inhibition of the phosphoinositide 3-kinase pathway. Cancer Discov. 2013;3:1345–54. doi: 10.1158/2159-8290.CD-13-0063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Markman B, Atzori F, Pérez-García J, Tabernero J, Baselga J. Status of PI3K inhibition and biomarker development in cancer therapeutics. Ann Oncol. 2010;21:683–91. doi: 10.1093/annonc/mdp347. [DOI] [PubMed] [Google Scholar]
- 34.Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304:554. doi: 10.1126/science.1096502. [DOI] [PubMed] [Google Scholar]
- 35.Velho S, Oliveira C, Ferreira A, Ferreira AC, Suriano G, Schwartz S, Jr, et al. The prevalence of PIK3CA mutations in gastric and colon cancer. Eur J Cancer. 2005;41:1649–54. doi: 10.1016/j.ejca.2005.04.022. [DOI] [PubMed] [Google Scholar]
- 36.Barbi S, Cataldo I, De Manzoni G, Bersani S, Lamba S, Mattuzzi S, et al. The analysis of PIK3CA mutations in gastric carcinoma and metanalysis of literature suggest that exon-selectivity is a signature of cancer type. J Exp Clin Cancer Res. 2010;29:32. doi: 10.1186/1756-9966-29-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sukawa Y, Yamamoto H, Nosho K, Ito M, Igarashi H, Naito T, et al. HER2 expression and PI3K-Akt pathway alterations in gastric cancer. Digestion. 2014;89:12–7. doi: 10.1159/000356201. [DOI] [PubMed] [Google Scholar]
- 38.Wen YG, Wang Q, Zhou CZ, Qiu GQ, Peng ZH, Tang HM. Mutation analysis of tumor suppressor gene PTEN in patients with gastric carcinomas and its impact on PI3K/AKT pathway. Oncol Rep. 2010;24:89–95. doi: 10.3892/or_00000832. [DOI] [PubMed] [Google Scholar]
- 39.Han SY, Kato H, Kato S, Suzuki T, Shibata H, Ishii S, et al. Functional evaluation of PTEN missense mutations using in vitro phosphoinositide phosphatase assay. Cancer Res. 2000;60:3147–51. [PubMed] [Google Scholar]
- 40.Carracedo A, Alimonti A, Pandolfi PP. PTEN level in tumor suppression: how much is too little? Cancer Res. 2011;71:629–33. doi: 10.1158/0008-5472.CAN-10-2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nam SY, Lee HS, Jung GA, Choi J, Cho SJ, Kim MK, et al. Akt/PKB activation in gastric carcinomas correlates with clinicopathologic variables and prognosis. APMIS. 2003;111:1105–13. doi: 10.1111/j.1600-0463.2003.apm1111205.x. [DOI] [PubMed] [Google Scholar]
- 42.Maroulakou IG, Oemler W, Naber SP, Tsichlis PN. Akt1 ablation inhibits, whereas Akt2 ablation accelerates, the development of mammary adenocarcinomas in mouse mammary tumor virus (MMTV)-ErbB2/neu and MMTV-polyoma middle T transgenic mice. Cancer Res. 2007;67:167–77. doi: 10.1158/0008-5472.CAN-06-3782. [DOI] [PubMed] [Google Scholar]
- 43.Irie HY, Pearline RV, Grueneberg D, Hsia M, Ravichandran P, Kothari N, et al. Distinct roles of Akt1 and Akt2 in regulating cell migration and epithelial-mesenchymal transition. J Cell Biol. 2005;171:1023–34. doi: 10.1083/jcb.200505087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009;9:550–62. doi: 10.1038/nrc2664. [DOI] [PubMed] [Google Scholar]
- 45.Li J, Davies BR, Han S, Zhou M, Bai Y, Zhang J, et al. The AKT inhibitor AZD5363 is selectively active in PI3KCA mutant gastric cancer, and sensitizes a patient-derived gastric cancer xenograft model with PTEN loss to Taxotere. J Transl Med. 2013;11:241. doi: 10.1186/1479-5876-11-241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thoreen CC, Kang SA, Chang JW, Liu Q, Zhang J, Gao Y, et al. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J Biol Chem. 2009;284:8023–32. doi: 10.1074/jbc.M900301200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shor B, Gibbons JJ, Abraham RT, Yu K. Targeting mTOR globally in cancer: thinking beyond rapamycin. Cell Cycle. 2009;8:3831–7. doi: 10.4161/cc.8.23.10070. [DOI] [PubMed] [Google Scholar]
- 48.O’Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66:1500–8. doi: 10.1158/0008-5472.CAN-05-2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ramanathan RK, McDonough SL, Kennecke HF, Iqbal S, Baranda JC, Seery TE, et al. A phase II study of MK-2206, an allosteric inhibitor of AKT as second-line therapy for advanced gastric and gastroesophageal junction (GEJ) cancer: A SWOG Cooperative Group trial (S1005) J Clin Oncol. 2014;32(5(Suppl)) doi: 10.1002/cncr.29363. Abstract No. 4041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bang YJ, Kang YK, Alsina M, Wainberg ZA, Chau I, Zhu J, et al. JAGUAR: A randomized phase II study of the AKT inhibitor ipatasertib (GDC-0068) versus placebo in combination with mFOLFOX6 chemotherapy in patients (pts) with locally advanced or metastatic HER2-negative gastric (G) or gastroesophageal junction (GEJ) adenocarcinoma. J Clin Oncol. 2014;32(5(Suppl)) Abstract No. TPS4147. [Google Scholar]
- 51.She QB, Chandarlapaty S, Ye Q, Lobo J, Haskell KM, Leander KR, et al. Breast tumor cells with PI3K mutation or HER2 amplification are selectively addicted to Akt signaling. PLoS One. 2008;3:e3065. doi: 10.1371/journal.pone.0003065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Serra V, Markman B, Scaltriti M, Eichhorn PJ, Valero V, Guzman M, et al. NVP-BEZ235, a dual PI3K/mTOR inhibitor, prevents PI3K signaling and inhibits the growth of cancer cells with activating PI3K mutations. Cancer Res. 2008;68:8022–30. doi: 10.1158/0008-5472.CAN-08-1385. [DOI] [PubMed] [Google Scholar]
- 53.Engelman JA, Chen L, Tan X, Crosby K, Guimaraes AR, Upadhyay R, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med. 2008;14:1351–6. doi: 10.1038/nm.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Belmont PJ, Jiang P, McKee TD, Xie T, Isaacson J, Baryla NE, et al. Resistance to dual blockade of the kinases PI3K and mTOR in KRAS-mutant colorectal cancer models results in combined sensitivity to inhibition of the receptor tyrosine kinase EGFR. Sci Signal. 2014;7:ra107. doi: 10.1126/scisignal.2005516. [DOI] [PubMed] [Google Scholar]
- 55.Sheppard KE, Cullinane C, Hannan KM, Wall M, Chan J, Barber F, et al. Synergistic inhibition of ovarian cancer cell growth by combining selective PI3K/mTOR and RAS/ERK pathway inhibitors. Eur J Cancer. 2013;49:3936–44. doi: 10.1016/j.ejca.2013.08.007. [DOI] [PubMed] [Google Scholar]
- 56.Zunder ER, Knight ZA, Houseman BT, Apsel B, Shokat KM. Discovery of drug-resistant and drug-sensitizing mutations in the oncogenic PI3K isoform p110 alpha. Cancer Cell. 2008;14:180–92. doi: 10.1016/j.ccr.2008.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cejka D, Preusser M, Woehrer A, Sieghart W, Strommer S, Werzowa J, et al. Everolimus (RAD001) and anti-angiogenic cyclophosphamide show long-term control of gastric cancer growth in vivo. Cancer Biol Ther. 2008;7:1377–85. doi: 10.4161/cbt.7.9.6416. [DOI] [PubMed] [Google Scholar]
- 58.Fuereder T, Jaeger-Lansky A, Hoeflmayer D, Preusser M, Strommer S, Cejka D, et al. mTOR inhibition by everolimus counteracts VEGF induction by sunitinib and improves anti-tumor activity against gastric cancer in vivo. Cancer Lett. 2010;296:249–56. doi: 10.1016/j.canlet.2010.04.015. [DOI] [PubMed] [Google Scholar]
- 59.Lee KH, Hur HS, Im SA, Lee J, Kim HP, Yoon YK, et al. RAD001 shows activity against gastric cancer cells and overcomes 5-FU resistance by downregulating thymidylate synthase. Cancer Lett. 2010;299:22–8. doi: 10.1016/j.canlet.2010.07.020. [DOI] [PubMed] [Google Scholar]
- 60.Lang SA, Gaumann A, Koehl GE, Seidel U, Bataille F, Klein D, et al. Mammalian target of rapamycin is activated in human gastric cancer and serves as a target for therapy in an experimental model. Int J Cancer. 2007;120:1803–10. doi: 10.1002/ijc.22442. [DOI] [PubMed] [Google Scholar]
- 61.Doi T, Muro K, Boku N, Yamada Y, Nishina T, Takiuchi H, et al. Multicenter phase II study of everolimus in patients with previously treated metastatic gastric cancer. J Clin Oncol. 2010;28:1904–10. doi: 10.1200/JCO.2009.26.2923. [DOI] [PubMed] [Google Scholar]
- 62.Ohtsu A, Ajani JA, Bai YX, Bang YJ, Chung HC, Pan HM, et al. Everolimus for previously treated advanced gastric cancer: results of the randomized, double-blind, phase III GRANITE-1 study. J Clin Oncol. 2013;31:3935–43. doi: 10.1200/JCO.2012.48.3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Meric-Bernstam F, Akcakanat A, Chen H, Do KA, Sangai T, Adkins F, et al. PIK3CA/PTEN mutations and Akt activation as markers of sensitivity to allosteric mTOR inhibitors. Clin Cancer Res. 2012;18:1777–89. doi: 10.1158/1078-0432.CCR-11-2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.O’Reilly T, McSheehy PM. Biomarker development for the clinical activity of the mTOR inhibitor everolimus (RAD001): processes, limitations, and further proposals. Transl Oncol. 2010;3:65–79. doi: 10.1593/tlo.09277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yoon DH, Ryu MH, Park YS, Lee HJ, Lee C, Ryoo BY, et al. Phase II study of everolimus with biomarker exploration in patients with advanced gastric cancer refractory to chemotherapy including fluoropyrimidine and platinum. Br J Cancer. 2012;106:1039–44. doi: 10.1038/bjc.2012.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wainberg ZA, Soares HP, Patel R, DiCarlo B, Park DJ, Liem A, et al. Phase II trial of everolimus in patients with refractory metastatic adenocarcinoma of the esophagus, gastroesophageal junction and stomach: possible role for predictive biomarkers. Cancer Chemother Pharmacol. 2015;76:61–7. doi: 10.1007/s00280-015-2744-5. [DOI] [PubMed] [Google Scholar]
- 67.Park JH, Ryu MH, Park YS, Park SR, Na YS, Rhoo BY, et al. Successful control of heavily pretreated metastatic gastric cancer with the mTOR inhibitor everolimus (RAD001) in a patient with PIK-3CA mutation and pS6 overexpression. BMC Cancer. 2015;15:119. doi: 10.1186/s12885-015-1139-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lei Z, Tan IB, Das K, Deng N, Zouridis H, Pattison S, et al. Identification of molecular subtypes of gastric cancer with different responses to PI3-kinase inhibitors and 5-fluorouracil. Gastroenterology. 2013;145:554–65. doi: 10.1053/j.gastro.2013.05.010. [DOI] [PubMed] [Google Scholar]
- 69.Mazumdar T, Byers LA, Ng PK, Mills GB, Peng S, Diao L, et al. A comprehensive evaluation of biomarkers predictive of response to PI3K inhibitors and of resistance mechanisms in head and neck squamous cell carcinoma. Mol Cancer Ther. 2014;13:2738–50. doi: 10.1158/1535-7163.MCT-13-1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Janku F, Garrido-Laguna I, Wheler JJ, Hong DS, Naing A, Falchook GS, et al. Screening for PIK3CA mutations, PTEN loss, and RAS/RAF mutations in early-phase protocols with PI3K/mTOR pathway inhibitors. J Clin Oncol. 2011;29(Suppl) Abstract No. 10507. [Google Scholar]
- 71.Britten CD. PI3K and MEK inhibitor combinations: examining the evidence in selected tumor types. Cancer Chemother Pharmacol. 2013;71:1395–409. doi: 10.1007/s00280-013-2121-1. [DOI] [PubMed] [Google Scholar]
- 72.Ma L, Chen Z, Erdjument-Bromage H, Tempst P, Pandolfi PP. Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell. 2005;121:179–93. doi: 10.1016/j.cell.2005.02.031. [DOI] [PubMed] [Google Scholar]
- 73.Garrett JT, Olivares MG, Rinehart C, Granja-Ingram ND, Sánchez V, Chakrabarty A, et al. Transcriptional and posttranslational upregulation of HER3 (ErbB3) compensates for inhibition of the HER2 tyrosine kinase. Proc Natl Acad Sci U S A. 2011;108:5021–6. doi: 10.1073/pnas.1016140108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chandarlapaty S, Sawai A, Scaltriti M, Rodrik-Outmezguine V, Grbovic-Huezo O, Serra V, et al. AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell. 2011;19:58–71. doi: 10.1016/j.ccr.2010.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tenbaum SP, Ordóñez-Morán P, Puig I, Chicote I, Arqués O, Landolfi S, et al. β-catenin confers resistance to PI3K and AKT inhibitors and subverts FOXO3a to promote metastasis in colon cancer. Nat Med. 2012;18:892–901. doi: 10.1038/nm.2772. [DOI] [PubMed] [Google Scholar]
- 76.Serra V, Scaltriti M, Prudkin L, Eichhorn PJ, Ibrahim YH, Chandarlapaty S, et al. PI3K inhibition results in enhanced HER signaling and acquired ERK dependency in HER2-overexpressing breast cancer. Oncogene. 2011;30:2547–57. doi: 10.1038/onc.2010.626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bedard P, Tabernero J, Kurzrock R, Britten CD, Stathis A, Perez-Garcia JM, et al. A phase lb, open-label, multicenter, dose-escalation study of the oral pan-PI3K inhibitor BKM120 in combination with the oral MEK1/2 inhibitor GSK1120212 in patients (pts) with selected advanced solid tumors. J Clin Oncol. 2012;30(Suppl) Abstract No. 3003. [Google Scholar]
- 78.LoRusso P, Shapiro G, Pandya SS, Kwak EL, Jones C, Belvin M, et al. A first-in-human phase Ib study to evaluate and c.w.t.p.-P. K.i.G.-. the MEK inhibitor GDC-0973, in patients with advanced solid tumors. J Clin Oncol. 2012;30(Suppl) Abstract No. 2566. [Google Scholar]
- 79.Shimizu T, Tolcher AW, Papadopoulos KP, Beeram M, Rasco DW, Smith LS, et al. The clinical effect of the dual-targeting strategy involving PI3K/AKT/mTOR and RAS/MEK/ERK pathways in patients with advanced cancer. Clin Cancer Res. 2012;18:2316–25. doi: 10.1158/1078-0432.CCR-11-2381. [DOI] [PubMed] [Google Scholar]
- 80.Ali SM, Sanford EM, Klempner SJ, Rubinson DA, Wang K, Palma NA, et al. Prospective comprehensive genomic profiling of advanced gastric carcinoma cases reveals frequent clinically relevant genomic alterations and new routes for targeted therapies. Oncologist. 2015;20:499–507. doi: 10.1634/theoncologist.2014-0378. [DOI] [PMC free article] [PubMed] [Google Scholar]