

Abstract
Purpose
Human organic anion transporter 4 (hOAT4) belongs to a family of organic anion transporters that play critical roles in the body disposition of clinically important drugs. We have previously shown that the activity of hOAT4 was down-regulated by activation of PKC and up-regulated by PDZ protein NHERF-1. Here, we investigated the mechanisms underlying such regulations.
Methods
COS-7 cells expressing hOAT4 were treated with PKC activator phorbol 12-myristate 13-acetate (PMA) or transfected with dominant negative mutants of dynamin-2 or Eps15 or transfected with NHERF-1. The internalization and the function of hOAT4 were then determined.
Results
We showed that hOAT4 constitutively internalized from and recycled back to plasma membrane. Transfection of dominant negative mutants of dynamin-2 or Eps15 into the cells, all of which block clathrin-dependent endocytotic pathway, significantly blocked hOAT4 internalization. Treatment of cells with PMA accelerated hOAT4 internalization, whereas transfection of cells with NHERF-1 attenuated hOAT4 internalization.
Conclusion
Our studies demonstrated that i) hOAT4 undergoes constitutive trafficking between cell surface and intracellular compartments, ii) hOAT4 internalization partly occurs through clathrin-dependent pathway, iii) the down-regulation of hOAT4 activity by activation of PKC and the up-regulation of hOAT4 activity by NHERF-1 are mediated through alteration of hOAT4 internalization.
Keywords: drug transporter, membrane trafficking, NHERF-1, protein kinase C, regulation
INTRODUCTION
Human organic anion transporter 4 (hOAT4) belongs to a family of organic anion transporters of 10 members (OAT1-10), which play critical roles in the body disposition of clinically important drugs, including anti-human–immunodeficiency–virus therapeutics, anti-tumor drugs, antibiotics, antihypertensives, and anti-inflammatories (1–4). hOAT4 is abundantly expressed in the kidney and placenta (5,6). In the kidney, hOAT4 localizes at the apical membrane of the proximal tubule, and functions as an organic anion exchanger for exchanging organic anions across the apical membrane. In the placenta, hOAT4 is localized to the basolateral membrane of syncytiotrophoblasts (7). It is believed that estrogen biosynthesis in the placenta uses dehydroepiandrosterone sulfate (DHEAS), a precursor produced in large amount by the fetal adrenals. Accumulation of excess DHEAS is associated with intrauterine growth retardation (8). DHEAS is an OAT4 substrate. Therefore, OAT4 may play an important role in efficient uptake of DHEAS by the placenta for the production of estrogens and for the protection of fetus from the cytotoxicity of DHEAS.
Given such an important role, understanding the regulation of hOAT4 has profound clinical significance. We and others have previously shown that hOAT4 activity was down-regulated by activation of protein kinase C (PKC) (9) and up-regulated by PDZ protein NHERF-1 (10,11). The up-regulation of hOAT4 activity by NHERF-1 is through the interaction between PDZ protein NHERF-1 and PDZ consensus-binding site(s) at the carboxyl terminus of hOAT4 (10,11). A relatively large number of PDZ proteins has been identified. These PDZ proteins interact with the PDZ consensus-binding site of its target protein and modulate its function. We further showed (9,10) that both down-regulation of hOAT4 activity by activation of PKC and up-regulation of hOAT4 activity by NHERF-1 regulation occur through altering the cell surface expression of the transporter: with activation of PKC, a decreased surface expression of hOAT4 was observed, whereas with NHERF-1, an increased cell surface expression of hOAT4 was observed. However, the mechanisms underlying such regulations were not known. The current study was carried out to address this issue.
MATERIALS AND METHODS
Materials
[3H] estrone sulfate was purchased from Perkin-Elmer Life and Analytical Sciences (Boston, MA). Dynamin-2/K44A mutant were purchased from American Type Culture Collection (Manssas, VA). Eps15 mutant (D95/295) was generously provided by Dr. Jennifer Lippincott-Schwartz from National Institute of Child Health and Human Development, National Institutes of Health (Bethesda, MD). NHS-SS-biotin and streptavidin-agarose beads were purchased from Pierce Chemical (Rockford, IL). All other reagents were from Sigma-Aldrich (St. Louis, MO).
Expression in COS-7 Cells
COS-7 cells were grown in 48-well plates (50–80% confluency) at 37°C and 5% CO2 in Dulbecco’s modified Eagle’s medium (Cellgro, USA) supplemented with 10% fetal bovine serum (Sigma), 100 units/ml penicillin, and 100 mg/ml streptomycin. Cells were transfected with either pcDNA3.1-hOAT4-myc, or pcDNA3.1 vector alone (2) using LipofectAmine 2000™ reagent (Life Technologies, Inc.) following the manufacturer’s instruction. The epitope myc was tagged to the carboxyl terminus of hOAT4 for immunodetection (9,10). The cells were used for studies described below 48 h after transfection.
Transport Measurement
Cells were plated in 48-well plates. For each well, uptake solution was added. The uptake solution consisted of phosphate-buffered saline (PBS)/Ca2+/Mg2+ (137 mM NaCl, 2.7 mM KCl, 4.3 mM Na2HPO4, 1.4 mM KH2PO4, 1 mM CaCl2, and 1 mM MgCl2, pH 7.4) and [3H]estrone sulfate. Four min after adding the uptake solution, the uptake was stopped by aspirating off the uptake solution and rapidly washing the well with ice-cold PBS. The cells were then solubilized in 0.2 N NaOH, neutralized in 0.2 N HCl, and aliquotted for liquid scintillation counting. The uptake count was standardized by the amount of protein in each well. Values are means ± SE (n=3).
Cell Surface Biotinylation
Cell surface expression levels of hOAT4 were examined using the membrane-impermeant biotinylation reagent NHS-SS-biotin (Pierce Chemical, USA). The cells were seeded onto six-well plates at 5×105 cells per well. After 24 h, the medium was removed, and the cells were washed twice with 3 ml of ice-cold PBS, pH 8.0. The plates were kept on ice, and all solutions were kept ice-cold for the rest of the procedure. Each well of cells was incubated with 1 ml of freshly made NHS-SS-biotin (0.5 mg/ml in PBS, pH 8.0) in two successive 20-min incubations on ice with very gentle shaking. Biotinylation was quenched by first briefly washing each well with 3 ml of 100 mM glycine and followed by incubation with 100 mM glycine on ice for 20 min. The cells were then dissolved on ice for 40 min in 400 μl of lysis buffer (10 mM Tris, 150 mM NaCl, 1 mM EDTA, 0.1% SDS, 1% Triton X-100, and protease inhibitor cocktail (Sigma, St. Louis, MO), pH 7.4). The unlysed cells were removed by centrifugation at 13,000 rpm at 4°C. Streptavidin-agarose beads (50 μl; Pierce Chemical, USA) were then added to the supernatant to isolate cell membrane proteins. hOAT4 was detected in the pool of surface proteins by polyacrylamide gel electrophoresis and immunoblotting using an anti-myc antibody (1:100) (Mount Sinai School of Medicine, NY, USA).
Internalization Assay
We followed the procedure established previously in our lab (12). hOAT4-expressing cells underwent biotinylation with 0.5 mg/ml sulfo-NHS-SS-biotin as we described above. Following biotinylation, one set of cells was washed with PBS and kept at 4°C to determine the total initial surface hOAT4 and stripping efficiencies. To initiate internalization, cells in the duplicate plate were washed repeatedly with prewarmed (37°C) PBS containing either 1 μM of PMA or PBS only and incubated with the same solutions for indicated time at 37°C. Residual cell surface biotin was stripped by incubating cells three times for 20 min with freshly prepared 50 mM MesNa in NT buffer (150 mM NaCl, 1 mM EDTA, 0.2% bovine serum albumin, 20 mM Tris, pH 8.6). Stripping efficiency was determined for each experiment on biotinylated cells kept in parallel at 4°C. Cells were lysed in lysis buffer with protease inhibitor cocktail (Sigma-Aldrich, USA). Biotinylated proteins were separated from non-biotinylated proteins by streptavidin-agarose resin (Thermo Scientific, USA) similarly as we described above. Samples were then eluted from the beads by adding sample buffer and resolved by SDS-PAGE and analyzed by Western blotting with anti-myc antibody. Relative hOAT4 internalized was calculated as % of the total initial cell surface hOAT4 pool.
Recycling Assay
We followed the procedure previously established in our lab (12). hOAT4-expressing cells underwent biotinylation with 1.0 mg/ml sulfo-NHS-SS-biotin at 4°C for 30 min to label cell surface pool of hOAT4. Subsequently, one set of cells was continuously biotinylated at 4°C. Cells in the duplicate plate were warmed to 37°C and continuously biotinylated at 37°C. At the time points indicated in the figures, biotinylation was stopped, and biotin-labeled hOAT4 was analyzed by SDS-PAGE and Western blotting as described above. Relative hOAT4 recycled was calculated as the difference between hOAT4 biotin-labeled at 37°C and hOAT4 biotin-labeled at 4°C.
Electrophoresis and Western Blotting
Protein concentration was determined by Bradford method. Protein sample (100 μg) was loaded on each lane and was resolved on 7.5% SDS-PAGE minigels and electroblotted onto polyvinylidene difluoride membranes. The blots were blocked for 1 h with 5% nonfat dry milk in PBS-0.05% Tween, and incubated overnight at 4°C with anti-myc antibody (1:100). The membranes were washed and then incubated with goat anti-mouse IgG (Thermo Scientific, USA) conjugated to horseradish peroxidase (1:5,000), and signals were detected using a SuperSignal West Dura extended duration substrate kit (Thermo Scientific, USA). Images were captured by Fluorchem® 8800 system (Alpha Innotech, USA). Density of bands was analyzed by Quantity One software (Bio-Rad, USA).
Immunofluorescence Analysis
hOAT4-expressing COS-7 cells were grown on coverslips (22 mm) for 48 h, washed three times in PBS, and then fixed with 3% paraformaldehyde for 20 min at room temperature, permeabilized with 0.01% Triton x-100 for 5 min three times, and incubated with 5% dry milk at room temperature for 1 h. Afterwards, the cells were incubated with rabbit anti-c-Myc antibody (Sigma, 1:300) to label c-myc hOAT4 and with mouse anti-EEA1 antibody (BD Biosciences,1:250) at 4°C overnight. The coverslips were then incubated with Alexa Fluor® 488 goat anti-mouse IgG (H+L) (Molecular Probes, 1:500) or Alexa Fluor® 555 goat anti-rabbit IgG (H+L) (Molecular Probes, 1:1000) at room temperature for 2 h. After washing, the coverslips were mounted on slides for image acquisition and analysis. Samples were visualized with a Zeiss LSM-510 laser-scanning microscope (Carl Zeiss Inc., Thornwood, NY).
Data Analysis
Statistical analysis was conducted using Student’s paired t test for comparing two treatments. A one-way ANOVA followed by a Dunnett’s post hoc test was used for comparing among more than two treatments. A P value <0.05 was considered significant.
RESULTS
Constitutive hOAT4 Trafficking
We have recently shown that hOAT1 constitutively internalizes from and recycles back to the plasma membrane (12). We then asked whether hOAT4 undergoes similar constitutive trafficking. To address this issue, we took a biotinylation-based strategy. OAT4-expressing COS-7 cells were biotinylated with cell-impermeable biotinylation reagent sulfo-NHS-SS-biotin under trafficking-impermissive condition (4°C). The labeled cells were then rewarmed back to trafficking-permissive condition (37°C) to allow internalization to occur. At indicated time points after initiation of internalization, biotin from biotinylated proteins remaining on the surface was removed by treatment with MesNa, a nonpermeant reducing agent that cleaves disulfide bond and liberates biotin from biotinylated proteins at the cell surface. The amount of biotinylated proteins resistant (inaccessible) to MesNa treatment was defined as “the amount of protein internalized.” Our result (Fig. 1, a and b) showed that 0 min after initiation of internalization, there was no surface-labeled hOAT4 internalized. However, 5, 10, or 15 min after initiation of internalization, approximately 20%, 40%, or 50% of surface-labeled hOAT4 was detectable in the intracellular compartments. Therefore, hOAT4 undergoes constitutive internalization in COS-7 cells. We next investigated whether hOAT4 constitutively recycles back to the cell surface (Fig. 2). hOAT4-expressing cells first underwent biotinylation with 1.0 mg/ml sulfo-NHS-SS-biotin at 4°C for 30 min to completely label cell surface pool of hOAT4. Subsequently, one set of cells was continuously biotinylated at 4°C. Cells in the duplicate plate were warmed to 37°C and continuously biotinylated at 37°C (12). The rationale was that if hOAT4 constitutively traffics between the cell surface and the intracellular compartments, then biotinylation under trafficking-permissive conditions (37°C) should significantly increase the amount of biotinylated hOAT4 as compared with biotinylation performed under trafficking-restrictive conditions (4°C). The experimental conditions were first tested with a positive control protein, transferrin receptor, which is known to participate in membrane recycling. As shown in Fig. 2a, the amount of transferrin receptor biotin-labeled 30 min after the initiation of recycling (37°C) was much greater than that before the initiation of recycling (4°C). Under the same experimental conditions, we further showed that the amount of hOAT4 biotin-labeled at 37°C was much greater than that biotin-labeled at 4°C (Fig. 2b, and c). These results demonstrated hOAT4 constitutively recycles back to the cell surface.
Fig. 1.

Biotinylation analysis of constitutive hOAT4 internalization in COS-7 cells. a. hOAT4 internalization was analyzed as described in “Materials and Methods” section followed by Western blotting using anti-myc antibody (1:100). b. Densitometry plot of results from Fig. 1a as well as from other experiments. Internalized hOAT4 was expressed as % of total initial cell surface hOAT4 pool. Values are mean ± S.E. (n=3).
Fig. 2.

Biotinylation analysis of constitutive hOAT4 recycling. a. 30-min recycling of transferrin receptor (TfR) was analyzed as described in “Materials and Methods” section followed by Western blotting using anti-TfR antibody (1:100). b. hOAT4 recycling was analyzed at a 5-min time point followed by Western blotting using anti-myc antibody (1:100). c. Densitometry plot of results from Fig. 2b as well as from other experiments. Total biotin-labeled hOAT4 was expressed as % of hOAT4 biotinylated at 4°C (before initiation of recycling). Values are mean ± S.E. (n=3).
Clathrin-Dependent hOAT4 Internalization
Transporter internalization critically depends on their interaction with the cellular internalization machinery. So far, three different internalization pathways have been described for other transporters: (i) clathrin-mediated internalization, (ii) caveolae-mediated internalization, and (iii) clathrin- and caveolae-independent internalization. To determine whether hOAT4 internalizes through a calthrin-dependent pathway, we measured hOAT4 internalization in cells transfected with or without dominant negative mutants of dynamin-2 and Eps15, both of which block clathrin-dependent pathway (12). Our results showed that the amount of surface-labeled hOAT4 internalized decreased from ~50% in the absence of dominant negative dynamin-2 (Fig. 3a) to ~20% in the presence of dominant negative dynamin-2 (Fig. 3b)
Fig. 3.

Dominant negative mutant of dynamin 2 blocked constitutive hOAT4 internalization. a. Top panel: cells were transfected with control plasmid (pcDNA vector). 48 h later, hOAT4 internalization (15 min) was analyzed as described in “Materials and Methods” section followed by Western blotting using anti-myc antibody (1:100). Bottom panel: Densitometry analysis of results from top panel as well as other experiments. Internalized hOAT4 was expressed as % of total initial cell surface hOAT4 pool. Values are mean ± S.E. (n=3). b. Top panel: Cells were transfected with cDNA encoding dominant negative mutant of dynamin-2 (Dyn-2 mutant). 48 h later, hOAT4 internalization (15 min) was analyzed as described in “Materials and Methods” section followed by Western blotting using anti-myc antibody (1:100). Bottom panel: Densitometry analysis of results from top panel as well as other experiments. Internalized hOAT4 was expressed as % of total initial cell surface hOAT4 pool. Values are mean ± S.E. (n=3). c. Top panel: Steady-state surface expression of hOAT4 in cells transfected with or without Dyn-2 mutant was determined by biotinylation analyses. Bottom panel: Densitometry analysis of results from top panel as well as other experiments. Surface expression of hOAT4 in cell transfected with Dyn-2 mutant was expressed as % of total cell surface hOAT4 pool in control cells. Values are mean ± S.E. (n=3). d. 4-min uptake of [3H] estrone sulfate (100 nM) into cells transfected with Dyn-2 mutant. Uptake activity was expressed as a percentage of the uptake measured in control cells. Values are mean ± S.E. (n=3).
The effect of reduced hOAT4 internalization on hOAT4 expression and function in cells transfected with dominant negative mutants of dynamin-2 was further analyzed. A reduced hOAT4 internalization resulted in an increase in steady-state cell surface expression of hOAT4 (Fig. 3c). Consequently, hOAT4 transport activity was increased (Fig. 3d). Similar results were obtained with cells transfected with Eps15 (Fig. 4).
Fig. 4.

Dominant negative mutant of Eps15 blocked constitutive hOAT4 internalization. a. Top panel: cells were transfected with control plasmid (pcDNAvector). 48 h later, hOAT4 internalization (15 min) was analyzed as described in “Materials and Methods” section followed by Western blotting using anti-myc antibody (1:100). Bottom panel: Densitometry analysis of results from top panel as well as other experiments. Internalized hOAT4 was expressed as % of total initial cell surface hOAT4 pool. Values are mean ± S.E. (n=3). b. Top panel: Cells were transfected with cDNA encoding dominant negative mutant of Eps15. 48 h later, hOAT4 internalization (15 min) was analyzed as described in “Materials and Methods” section followed by Western blotting using anti-myc antibody (1:100). Bottom panel: Densitometry analysis of results from top panel as well as other experiments. Internalized hOAT4 was expressed as % of total initial cell surface hOAT4 pool. Values are mean ± S.E. (n=3). c. Top panel: Steady-state surface expression of hOAT4 in cells transfected with or without Eps15 mutant was determined by biotinylation analyses. Bottom panel: Densitometry analysis of results from top panel as well as other experiments. Surface expression of hOAT4 in cell transfected with Eps15 mutant was expressed as % of total cell surface hOAT4 pool in control cells. Values are mean ± S.E. (n=3). d. 4-min uptake of [3H] estrone sulfate (100 nM) into cells transfected with Eps15 mutant. Uptake activity was expressed as a percentage of the uptake measured in control cells. Values are mean ± S.E. (n=3).
Immunolocalization of hOAT4 and EEA1
The cellular distribution of hOAT4 was examined by immunofluorescence microscopy. We previously showed that hOAT1 constitutively traffics between plasma membrane and recycling endosomes (12). To determine whether hOAT4 traffics through the same route, we immunolocalized hOAT4 and EEA1, a recycling endosome marker. The fluorescence images (Fig. 5) showed that hOAT4 partially colocalized with EEA1-positive recycling endosomes.
Fig. 5.

Immuno-localization of hOAT4 and EEA1. The cells were immunostained for hOAT4, and early endosome marker EEA1. Fluorescence images were taken for hOAT4 (red), and EEA1 (green). The merged image of hOAT4 and EEA1 was shown as orange/yellow. Bar=~10 μm.
PKC Activation Enhances hOAT4 Internalization
We previously showed that activation of PKC by PMA inhibited hOAT4 activity by reducing its cell surface expression (9). To further investigate the mechanism underlying such regulation, we examined whether PMA modulates hOAT4 internalization. Using similar biotinylation approach as we used to investigate constitutive hOAT4 internalization, we observed (Fig. 6a) that the amount of surface-labeled hOAT4 internalized in the presence of 1 μM PMA (Fig. 6a lane 3) was much greater than that in the absence of PMA (Fig. 6a, lane 2), suggesting that activation of PKC by PMA accelerated hOAT4 internalization into the intracellular compartments. As a result, the steady-state cell surface expression of hOAT4 was reduced (Fig. 6b).
Fig. 6.
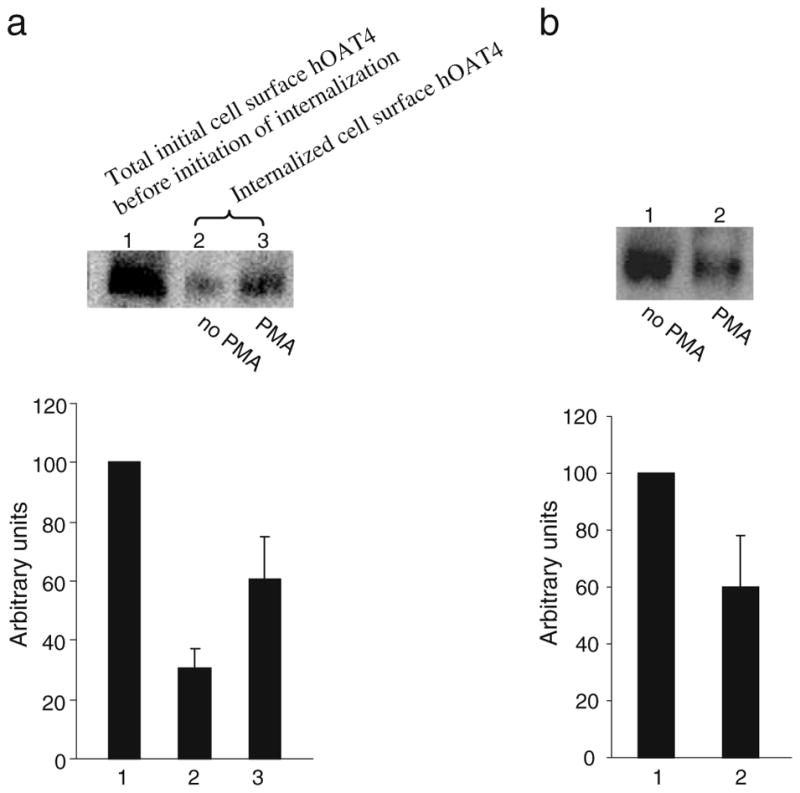
Biotinylation analysis of PKC-modulated hOAT4 internalization and steady-state cell surface expression. a. Top panel: hOAT4 internalization (15 min) was analyzed as described in “Materials and Methods” section in the presence and the absence of 1 μM PMA followed by Western blotting using anti-myc antibody (1:100). Bottom panel: Densitometry analyses of results from top panel as well as from other experiments. Internalized hOAT4 was expressed as % of total initial cell surface hOAT4 pool. Values are mean ± S.E. (n=3). b. Top panel: The effect of PMA on the steady-state expression of hOAT4 at cell surface was analyzed by biotinylation approach. Bottom panel: Densitometry analyses of results from top panel as well as from other experiments. The cell surface expression of hOAT4 in the presence of PMA was expressed as % of the cell surface hOAT4 in the absence of PMA. Values are mean ± S.E. (n=3).
NHERF-1 decreases hOAT4 internalization
We previously showed that NHERF-1 had an opposite effect on hOAT4 to that of PMA. NHERF-1 enhances hOAT4 activity by increasing its cell surface expression (10). By analyzing hOAT4 internalization in the presence and absence of NHERF-1, we observed that the amount of surface-labeled hOAT4 internalized in the presence of NHERF-1 (Fig. 7b) was much less than that in the absence of NHERF-1 (Fig. 7a), suggesting that NHERF-1 decreases hOAT4 internalization into the intracellular compartments. As a result, the steady-state cell surface expression of hOAT4 was increased (Fig. 7c).
Fig. 7.

Biotinylation analysis of NHERF-1-modulated hOAT4 internalization and steady-state cell surface expression. a. Top panel: Cells were transfected with control plasmid (pcDNA vector). 48 h later, hOAT4 internalization (15 min) was analyzed as described in “Materials and Methods” section followed by Western blotting using anti-myc antibody (1:100). Bottom panel: Densitometry analysis of results from top panel as well as other experiments. Internalized hOAT4 was expressed as % of total initial cell surface hOAT4 pool. Values are mean ± S.E. (n=3). b. Top panel: Cells were transfected with cDNA encoding NHERF-1. 48 h later, hOAT4 internalization (15 min) was analyzed as described in “Materials and Methods” section followed by Western blotting using anti-myc antibody (1:100). Bottom panel: Densitometry analysis of results from top panel as well as other experiments. Internalized hOAT4 was expressed as % of total initial cell surface hOAT4 pool. Values are mean ± S.E. (n=3). c. Top panel: Steady-state surface expression of hOAT4 in cells transfected with or without NHERF-1 mutant was determined by biotinylation analyses. Bottom panel: Densitometry analysis of results from top panel as well as other experiments. Surface expression of hOAT4 in cell transfected with NHERF-1 mutant was expressed as % of total cell surface hOAT4 pool in control cells. Values are mean ± S.E. (n=3).
DISCUSSION
Compared to other major members of OAT family, OAT4 has its unique properties. First, OAT4 locates at the apical membrane of the kidney proximal tubule cells, whereas OAT1, OAT2 and OAT3 locate at the basolateral membrane of the kidney proximal tubule cells. Secondly, OAT4 has distinct substrate specificity from other OATs. Due to these differences, it is important to compare the regulation of hOAT4 with other members of OAT family.
We previously showed that hOAT4 activity can be down-regulated by activation of PKC and up-regulated by PDZ protein NHERF-1. However, the mechanisms underlying such regulation are not known. In the current study, we addressed such issue, and the major findings from the current study are that i) hOAT4 undergoes constitutive trafficking between cell surface and intracellular compartments, ii) hOAT4 internalization partly occurs through clathrin-dependent pathway, iii) the down-regulation of hOAT4 activity by activation of PKC and the up-regulation of hOAT4 activity by NHERF-1 are mediated through alteration of hOAT4 internalization.
Several reports indicate that many transporters, such as dopamine transporter (13), are not static at the cell surface; rather, they dynamically cycles to and from the plasma membrane under basal conditions. Recently, our lab provided evidence that hOAT1, another member of OAT family, indeed undergoes such constitutive trafficking (12). The direct evidence on constitutive hOAT4 trafficking came from our biotinylation study. We showed that under basal condition, hOAT4 robustly internalized from (Fig. 1) and recycled back (Fig. 2) to the cell surface. The rates for both internalization and recycling were ~5%/min. This trafficking rate is comparable to that observed form hOAT1 described in our previous study (12).
The pathway through which hOAT4 internalizes was next investigated (Fig. 3 and Fig. 4). We observed that transfection of dominant negative mutants of dynamin-2 and Eps 15 into the cells, all of which blocked clathrin-dependent pathway (12), significantly reduced hOAT4 internalization, suggesting that hOAT4 internalizes at least in part through a clathrin-dependent pathway. Whether hOAT4 internalizes through other pathways, such as caveolae-dependent pathway and clathrin- and caveolae-independent pathways, will be the focus of future study. Indeed, Lee et al. have shown that hOAT4 coimunoprecipitates with caveolin in human placenta (14). Caveolin is an important component of caveolae complex. The intracellular compartments, to which hOAT4 internalizes, were identified as early recycling endosomes by our immunolocalization study (Fig. 5), showing that hOAT4 colocalized with EEA1, a recycling endosome marker.
We have previously shown that acute PKC activation inhibits hOAT4 activity by decreasing the surface expression of the transporter, and PDZ protein NHERF-1 enhanced hOAT4 activity by increasing the surface expression of the transporter. In the current study, we further explored the reason behind such changes in surface expression and we observed that the decrease in surface expression, of hOAT4 by activation of PKC was achieved by an increase in hOAT4 internalization, and the increase in surface expression of hOAT4 by NHERF-1 was achieved by a decrease in hOAT4 internalization. Indeed, NHERF-1 has been shown previously to regulate trafficking of several G protein-coupled receptors (15,16).
In summary, this is the first study demonstrating that regulation of hOAT4 activity can be attained by altering the already existent trafficking kinetics of the transporter.
Acknowledgments
This work was supported by grants (to Dr. Guofeng You) from the National Institute of Health (R01-DK 60034 and R01-GM 079123).
References
- 1.Srimaroeng C, Perry JL, Pritchard JB. Physiology, structure, and regulation of the cloned organic anion transporters. Xenobiotica. 2008;38:889–935. doi: 10.1080/00498250801927435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rizwan AN, Burckhardt G. Organic anion transporters of the SLC22 family: biopharmaceutical, physiological, and pathological roles. Pharm Res. 2007;24:450–70. doi: 10.1007/s11095-006-9181-4. [DOI] [PubMed] [Google Scholar]
- 3.Zhou F, You G. Molecular insights into the structure-function relationship of organic anion transporters OATs. Pharm Res. 2007;24:28–36. doi: 10.1007/s11095-006-9144-9. [DOI] [PubMed] [Google Scholar]
- 4.Anzai N, Kanai Y, Endou H. Organic anion transporter family: current knowledge. J Pharmacol Sci. 2006;100:411–26. doi: 10.1254/jphs.crj06006x. [DOI] [PubMed] [Google Scholar]
- 5.Ekaratanawong S, Anzai N, Jutabha P, Miyazaki H, Noshiro R, Takeda M, et al. Human organic anion transporter 4 is a renal apical organic anion/dicarboxylate exchanger in the proximal tubules. J Pharmacol Sci. 2004;94:297–304. doi: 10.1254/jphs.94.297. [DOI] [PubMed] [Google Scholar]
- 6.Cha SH, Sekine T, Kusuhara H, Yu E, Kim JY, Kim DK, et al. Molecular cloning and characterization of multispecific organic anion transporter 4 expressed in the placenta. J Biol Chem. 2000;275:4507–12. doi: 10.1074/jbc.275.6.4507. [DOI] [PubMed] [Google Scholar]
- 7.Ugele B, St-Pierre MV, Pihusch M, Bahn A, Hantschmann P. Characterization and identification of steroid sulfate transporters of human placenta. Am J Physiol Endocrinol Metab. 2003;284:E390–8. doi: 10.1152/ajpendo.00257.2002. [DOI] [PubMed] [Google Scholar]
- 8.Rabe T, Hosch R, Runnebaum B. Diagnosis of intrauterine fetal growth retardation (IUGR) and placental insufficiency by a dehydroepiandrosterone sulfate (DHAS) loading test. Biol Res Pregnancy Perinatol. 1983;4:130–6. [PubMed] [Google Scholar]
- 9.Zhou F, Hong M, You G. Regulation of human organic anion transporter 4 by progesterone and protein kinase C in human placental BeWo cells. Am J Physiol Endocrinol Metab. 2007;293:E57–61. doi: 10.1152/ajpendo.00696.2006. [DOI] [PubMed] [Google Scholar]
- 10.Zhou F, Xu W, Tanaka K, You G. Comparison of the interaction of human organic anion transporter hOAT4 with PDZ proteins between kidney cells and placental cells. Pharm Res. 2008;25:475–80. doi: 10.1007/s11095-007-9359-4. [DOI] [PubMed] [Google Scholar]
- 11.Miyazaki H, Anzai N, Ekaratanawong S, Sakata T, Shin HJ, Jutabha P, et al. Modulation of renal apical organic anion transporter 4 function by two PDZ domain-containing proteins. J Am Soc Nephrol. 2005;16:3498–506. doi: 10.1681/ASN.2005030306. [DOI] [PubMed] [Google Scholar]
- 12.Zhang Q, Hong M, Duan P, Pan Z, Ma J, You G. Organic anion transporter OAT1 undergoes constitutive and protein kinase C-regulated trafficking through a dynamin- and clathrin-dependent pathway. J Biol Chem. 2008;283:32570–9. doi: 10.1074/jbc.M800298200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Loder MK, Melikian HE. The dopamine transporter constitutively internalizes and recycles in a protein kinase C-regulated manner in stably transfected PC12 cell lines. J Biol Chem. 2003;278:22168–74. doi: 10.1074/jbc.M301845200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee WK, Choi JK, Cha SH. Co-localization and interaction of human organic anion transporter 4 with caveolin-1 in primary cultured human placental trophoblasts. Exp Mol Med. 2008;40:505–13. doi: 10.3858/emm.2008.40.5.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wheeler D, Sneddon WB, Wang B, Friedman PA, Romero G. NHERF-1 and the cytoskeleton regulate the traffic and membrane dynamics of G protein-coupled receptors. J Biol Chem. 2007;282:25076–87. doi: 10.1074/jbc.M701544200. [DOI] [PubMed] [Google Scholar]
- 16.Wang B, Bisello A, Yang Y, Romero GG, Friedman PA. NHERF1 regulates parathyroid hormone receptor membrane retention without affecting recycling. J Biol Chem. 2007;282:36214–22. doi: 10.1074/jbc.M707263200. [DOI] [PubMed] [Google Scholar]