

Targeting of the neddylation pathway in intestinal epithelial cells inhibits NF-κB, resulting in unabated apoptosis. These studies provide new insight into neddylation pathways within the mucosal epithelium and highlight an important role for NF-κB in the resolution of ongoing inflammation.
Abstract
Recent work has revealed a central role for neddylation (the conjugation of a Nedd8 moiety to Cullin proteins) in the fine-tuning of the NF-κB response (via Cullin-1). In the present study, we investigated the contribution of Cullin-1 neddylation and NF-κB signaling to mucosal inflammatory responses in vitro and in vivo. Initial in vitro studies using cultured intestinal epithelial cells revealed that the neddylation inhibitor MLN4924 prominently induces the deneddylation of Cullin-1. Parallel Western blot, luciferase reporter, and gene target assays identified MLN4924 as a potent inhibitor of intestinal epithelial NF-κB. Subsequent studies revealed that MLN4924 potently induces epithelial apoptosis but only in the presence of additional inflammatory stimuli. In vivo administration of MLN4924 (3 mg/kg per day) in a TNBS-induced colitis model significantly accentuated disease severity. Indeed, MLN4924 resulted in worsened clinical scores and increased mortality early in the inflammatory response. Histologic analysis of the colon revealed that neddylation inhibition results in increased tissue damage and significantly increased mucosal apoptosis as determined by TUNEL and cleaved caspase-3 staining, which was particularly prominent within the epithelium. Extensions of these studies revealed that ongoing inflammation is associated with significant loss of deneddylase-1 (SENP8) expression. These studies reveal that intact Cullin-1 neddylation is central to resolution of acute inflammation.
INTRODUCTION
Posttranslational protein modifications (PPMs) play an important role in the regulation of protein function, allowing for rapid responses to external stimuli (Song et al., 2010). One of these PPMs, neddylation—the addition of a neural precursor expressed, developmentally down-regulated (Nedd8) moiety to proteins (Bruning et al., 2011)—has recently gained significant attention in a variety of diseases. Neddylation is involved in the regulation of two key transcription factors, facilitating activation of nuclear factor κB (NF-κB; Amir et al., 2002; Ehrentraut et al., 2013) and inhibition of hypoxia-inducible factor (HIF) during inflammatory processes (Cernada et al., 2011; Curtis et al., 2015). Their respective regulation relies on the intricate interplay between NF-κB and HIF, their regulator proteins, IκB, and von Hippel–Lindau protein and a group of scaffolding proteins called Cullin proteins (Pan et al., 2004; Merlet et al., 2009). The ability of these Cullin proteins to mark their target proteins for ubiquitin-dependent proteasomal degradation relies on their neddylation status (Pan et al., 2004).
The ability of proteins to be neddylated is dependent on the availability of free Nedd8, which is bound by the Nedd8-activating enzyme (NAE; also called UBA3-APPB1-E1-ligase; Wada et al., 1998; Mendoza et al., 2003; Huang et al., 2004). Subsequently the Nedd8 moiety is transferred to its E2-ligase (Jones et al., 2002) and then the target Cullin-E3-ligase protein complex, thereby activating it (Parry and Estelle, 2004). Free Nedd8 can be generated through cleavage of conjugated Nedd8 from the Cullin-E3-ligase, a process known as deneddylation. This process depends, at least in part, on the COP9 signalosome, can be positively influenced by commensal bacteria (Kumar et al., 2007; Jones et al., 2015) and extracellular adenosine (Khoury et al., 2007), and offers a potentially protective mechanism during inflammatory processes. In addition, loss of the isopeptidase sentrin-specific protease 8 (SENP8)/deneddylase-1 leads to a loss of neddylatory function and an inability to activate NF-κB (Ehrentraut et al., 2013).
Studies of neddylation and Cullin pathways in vivo have been hampered by the embryonic lethality of gene-targeted mice (Tateishi et al., 2001). Recent advances in the development of small-molecule therapeutics identified the adenosine monophosphate (AMP) homologue MLN4924 (Soucy et al., 2009). This compound disrupts the Cullin-E3-RING-ligase complex neddylation by inhibiting NAE. Given our previous observations that adenosine deneddylates Cullin proteins during ongoing inflammation (Khoury et al., 2007), we investigated the effect of MLN4924 on inflammatory responses and demonstrated that administration of this compound inhibits acute lipopolysaccharide (LPS)–induced endotoxemic shock (Ehrentraut et al., 2013). Other studies showed that low-dose MLN4924 activated HIF through Cullin-2 (Angus, 2011; Curtis et al., 2015). Here we sought to investigate the contribution of protein neddylation to mucosal inflammation using cultured epithelial cells and experimental colitis in mice as model systems.
RESULTS
Cytokine-induced Cullin-1 neddylation is abrogated by MLN4924
In the present study, we examined how neddylation affects epithelial NF-κB responses and mucosal inflammation endpoints. As shown in Figure 1A, Caco-2 intestinal epithelial cell exposure to tumor necrosis factor-α (TNF-α; 10 ng/ml, 1 h) induced p65 nuclear translocation. Coincubation of cells with the NAE inhibitor MLN4924 inhibited both p65 nuclear translocation and Cullin-1 (Cul-1) neddylation in a concentration-dependent manner. At doses as low as 100 nM MLN4924, the neddylated fraction of Cul-1 was significantly decreased (p < 0.05). Parallel studies using NF-κB reporter assays revealed concentration-dependent inhibition of NF-κB activity with MLN4924, with >60% loss of activity at 3 μM MLN4924 (Figure 1B). When Caco-2 and T84 intestinal epithelial cells were exposed to the combination of MLN4924 (3 μM) and TNF-α/interleukin-1β (IL-1β; 10 ng/ml each), we observed a 50–70% decrease in the induction of the NF-κB target genes IL-8 and ICAM-1 (Figure 1C; p < 0.025). These results indicate that MLN4924 is a potent NF-κB inhibitor and that loss of Cul-1 neddylation significantly inhibits NF-κB target gene induction.
FIGURE 1:
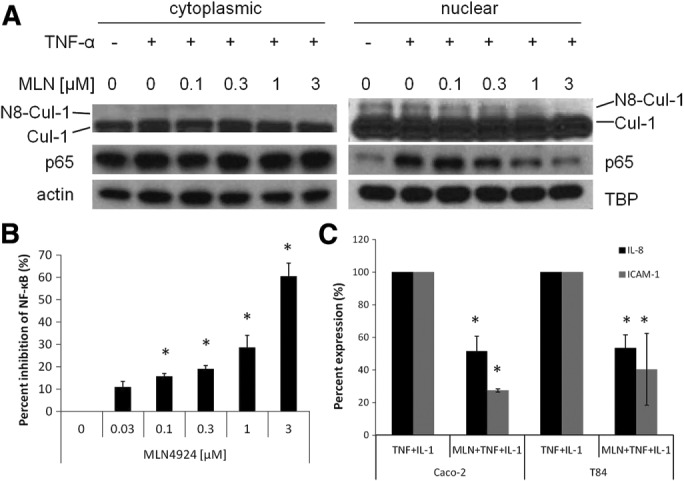
The neddylation inhibitor MLN4924 inhibits NF-κB signaling in intestinal epithelial cells. (A) Western blot of nuclear/cytoplasmic fractionation of Caco-2 cells treated with TNF-α (10 ng/μl) and increasing concentrations of MLN4924 for 1 h leads to deneddylation of Cul-1 and decreased p65 NF-κB in the nucleus, with actin and TATA-binding protein (TBP) as loading controls (n = 3). (B) Luciferase assay in Caco-2 cells transfected with an NF-κB luciferase reporter plasmid and treated with TNF-α and IL-1β (10 ng/μl each) and increasing concentrations of MLN4924 leads to increased inhibition of NF-κB signaling (n = 3). (C) mRNA expression of NF-κB target genes 2 h after treatment with TNF-α and IL-1β (10 ng/ml each) is inhibited by pretreatment (30 min) with 3 μM MLN4924 treatment in the presence of (n = 3), *p < 0.05.
Epithelial barrier function and neddylation
One of the hallmarks of mucosal diseases, including inflammatory bowel disease (IBD), is epithelial barrier dysfunction (Koch and Nusrat, 2012), allowing for translocation of luminal contents into the serosa. Here we examined the influence of neddylation on epithelial barrier function in the presence and absence of inflammatory stimuli. Epithelial barrier function has been widely modeled in vitro through measurement of transepithelial electrical resistance (TER).
For these purposes, T84 intestinal epithelial cells were cultured on polycarbonate inserts and grown to confluence (>1000 Ω.cm2). Cells were exposed to medium alone (control), cytomix (10 ng/ml each of TNF-α, IL-1β, and interferon-γ), MLN4924 (1 μM) alone, or the combination of cytomix and MLN4924. As shown in Figure 2A, exposure of confluent epithelia to MLN4924 alone did not influence baseline epithelial barrier compared with medium alone (p = 0.54), suggesting that neddylation per se is not necessary to maintain epithelial barrier function. Treatment of epithelia with cytomix led to a significant (p < 0.05) decrease in epithelial resistance over 24 h, indicative of a loss of tight junctional integrity (Figure 2A), which has been previously demonstrated (Bruewer et al., 2003). This cytomix-dependent decrease in barrier was markedly enhanced in combination with MLN4924 (Figure 2A; p < 0.01 by analysis of variance [ANOVA]), indicated by an earlier and more severe reduction in TER measurements.
FIGURE 2:

The combination of proinflammatory cytokines with the neddylation inhibitor MLN4924 leads to increased barrier disruption. (A) TER of T84 cells on Transwell inserts during a 24-h time course with control, cytomix (10 ng/μl each of TNF-α, IL-1β, and IFN-γ), 1 μM MLN4924, or cytomix plus MLN4924 (n = 3). (B) FITC-flux assay of T84 cells on Transwell inserts after a 24-h time course with control, cytomix, 1 μM MLN4924, or cytomix plus MLN4924 (n = 3). (C) Luciferase assay detecting activation of caspase-3/7 of T84 cells treated with control, cytomix, 1 μM MLN4924, or cytomix plus MLN4924 for 24 h, *p < 0.05.
TER measurements reflect changes in electrical conductivity from both transcellular and paracellular paths. To verify whether the observed changes were attributable to the paracellular path (i.e., tight junction permeability), we performed paracellular flux assays using 3-kDa fluorescein isothiocyanate (FITC)-dextran as a tracer. As shown in Figure 2B, similar to TER measurements, MLN4924 alone did not increase paracellular flux compared with control (p = 0.10), whereas cytomix treatment increased paracellular flux by a small (∼10%) but significant amount (p < 0.05). The combination of MLN4924 and cytomix, however, increased transepithelial flux by nearly 50-fold (p < 0.01) compared with other treatment groups, clearly indicating that the loss of neddylation in combination with inflammatory stimuli results in a marked loss of tight junction integrity.
Previous studies showed that the disruption of epithelial junctions in response to inflammatory cytokines is related at least in part to an increase in apoptosis through the activation of caspase-3 (Nava et al., 2010). Once activated, caspase-3 initiates the nonreversible apoptotic cascade leading to DNA fragmentation and ultimately cell death (Porter and Janicke, 1999). To determine whether increased apoptosis was responsible for our observed neddylation-dependent loss of barrier, we characterized the influence of the combination of cytokine activation and MLN4924 on caspase-3 activity. As depicted in Figure 2C, MLN4924 alone did not significantly influence caspase-3 activity (p = 0.12). Although cytomix alone trended toward increased caspase-3, the combination of neddylation inhibition and cytomix increased caspase-3 activity by nearly ninefold compared with control and MLN4924 alone (p < 0.01). To define the functional significance of this observation, we used a general caspase inhibitor peptide to block caspase-3 activation. As shown in Figure 3A, the decrease in TER caused by cytomix in combination with MLN4924 was significantly attenuated by the caspase inhibitor peptide compared with cells treated with a negative control peptide over a 24-h period (p < 0.05 by ANOVA). This result was generally specific for caspase-3 because a caspase-3 inhibitor also displayed similar results over a 16-h time course (Figure 3B). Additional experiments with the necroptosis inhibitor necrostatin-1 (100 μM) showed nonsignificant inhibition of TER decreases (p = 0.6; Figure 3C) and flux increases (p = 0.8; unpublished data). These findings indicate that the loss of neddylation in combination with inflammatory stimuli significantly enhances the caspase-3–dependent apoptotic response, negatively affecting barrier integrity.
FIGURE 3:

Proinflammatory cytokines with the neddylation inhibitor MLN4924 lead to increased apoptosis, and NF-κB inhibition leads to increased barrier disruption. (A) TER of T84 cells on Transwell inserts during a 24-h time course with cytomix plus MLN4924 in the presence of 30 μM general caspase inhibitor (Gen cas in) peptide or a negative control (Neg) peptide (n = 3). (B) TER of T84 cells on Transwell inserts during a 16-h time course with cytomix plus MLN4924 in the presence of 30 μM caspase-3 inhibitor (Cas 3 in) peptide or a negative control (Neg) peptide (n = 3). (C) Influence of necroptosis inhibitor necrostatin-1 on permeability of T84 cells to a combination of cytomix and MLN4924 (n = 3). (D) TER of T84 cells on Transwell inserts during a 24-h time course with control, cytomix (10 ng/μl each of TNF-α, IL-1β, and IFN-γ), 1 μM MLN4924, 30 μM Bay 11-7085, or the combination of either cytomix plus MLN4924 or cytomix plus Bay 11-7085 (n = 3). *p < 0.05.
To evaluate whether the apoptotic response seen with MLN4924 treatment arises from NF-κB inhibition, we used Bay 11-7085, a pharmacological inhibitor of NF-κB that irreversibly blocks TNF-α–stimulated IκBα phosphorylation (Pierce et al., 1997). TER measurements of confluent T84 cells exposed to Bay 11-7085 alone (30 μM) showed no loss of barrier function. Cytomix treatment alone again led to a significant decrease (p < 0.05) in epithelial resistance after 24 h. Concomitant treatment of Bay 11-7085 with cytomix also resulted in an earlier and more severe reduction in TER measurements, enhancing the cytomix-dependent decrease in barrier function, similar to the effects seen with MLN4924 (Figure 3D). These findings demonstrate that the inhibition of NF-κB contributes to reduced barrier function and partially recapitulates the response of MLN4924 treatment with cytomix.
Neddylation and intestinal inflammation in vivo
Having defined the importance of neddylation for epithelial barrier integrity in vitro, we extended these results to define the relative importance of neddylation in colonic inflammation in vivo, using a murine 2,4,6-trinitrobenzene sulfonic acid (TNBS) colitis model. This is a model characterized by disruption of the epithelial barrier in vivo (Karhausen et al., 2004). After TNBS instillation, body weight was monitored twice per day. In accordance with previous observations, TNBS treatment led to increased mortality (33% after 3 d, n = 9, compared with 0% death in control group, n = 5, treated with ethanol only; Figure 4A). The earliest time point of animal death was 2 d into the trial period. Treatment with MLN4924 alone (3 mg/kg per day) did not alter this ratio, and all of the animals survived the 3-d trial period (n = 5). Daily subcutaneous injections of MLN4924 combined with TNBS significantly increased mortality (60% of animals by day 3.5, n = 10, p < 0.025 compared with vehicle treatment; Figure 4A).
FIGURE 4:

Neddylation inhibition increases disease severity in a TNBS model of colitis. (A) Treatment of C57/BL6 mice treated with 2.5% TNBS and MLN4924 s.c. at 3 mg/kg per day shows decreased survival in mice receiving TNBS plus MLN4924 (29 total mice). (B) Decreased colon length in mice receiving TNBS plus MLN4924 compared with TNBS alone. (C) Histological colonic sections of mice treated with or without TNBS and with or without MLN4924. (D) TUNEL staining in mouse colon sections from TNBS plus MLN4924 experiment. (E) Cleaved caspase 3 IF staining in mouse colon sections from TNBS plus MLN4924 experiment. (F) Analysis of tissue injury index in mice receiving a combination of TNBS plus MLN4924 in comparison to TNBS alone. (G) Increased IL-1 and IL-6 protein expression as detected by MesoScale in mice receiving TNBS plus MLN4924 compared with TNBS alone.
Colon shortening, a hallmark feature of murine colitis (Karhausen et al., 2004), was not different between vehicle and MLN4924-alone exposures (Figure 4B). Colitis induced with TNBS showed a nonsignificant trend toward shorter colons at the time of killing, which was consistent with previous data for this model (Robinson et al., 2008). However, animals receiving repetitive doses of MLN4924 along with the induction of colitis showed a significant reduction of colon length compared with their littermates receiving only TNBS (Figure 4B).
TNBS colitis has been shown to lead to apoptotic cell death (Crespo et al., 2012; Hjerpe et al., 2012). TNBS-induced inflammatory response was characterized by a loss of crypt architecture and infiltration of large numbers of inflammatory cells with mucosal and submucosal injury (Figure 4C, hematoxylin and eosin staining). MLN4924 treatment alone resulted in no observable change to colonic architecture compared with vehicle control. The combined use of MLN4924 and TNBS, however, significantly increased tissue destruction, leading to total loss of crypt structure, massive inflammatory cell infiltration, and transmural mucosal denudation (Figure 4C, bottom left). As a result, the tissue injury index in mice receiving MLN4924 with TNBS was significantly increased compared with those receiving TNBS alone (Figure 4F).
In addition, colonic tissue of animals treated with only MLN4924 showed no increase in the amount of fragmented DNA (a sign of apoptotic cell death) compared with vehicle-treated controls (Figure 4D; terminal deoxynucleotidyl transferase dUTP nick end labeling [TUNEL] staining, top right and left). TNBS-induced colitis increased the detectable amount of fragmented DNA, indicative of some degree of cell death. This was increased in the colonic epithelium of animals receiving both TNBS and MLN4924 (Figure 4D, bottom). The activation of caspase-3 is another keystone along the apoptotic cell death pathway. In our study, MLN4924 alone did not alter the amounts of detectable caspase-3 in the colonic epithelium. The induction of cell death via TNBS colitis increased the activation of caspase-3 (Figure 4E, bottom left). Again, this proapoptotic response was significantly enhanced in animals receiving both MLN4924 and TNBS, as reflected by cleaved caspase-3 (Figure 4E, bottom right).
A loss of barrier function and cell breakdown is accompanied by the detection of colonic contents (e.g., bacteria) through the innate immune system. One of the major signaling pathways, the Toll-like-receptor pathway, elicits the induction of proinflammatory cytokines such as IL-1β and IL-6. The induction of these cytokines was significantly increased in the colonic tissue of animals undergoing TNBS colitis while concomitantly being dosed with MLN4924 (approximately sixfold compared with normalized control, p < 0.01; Figure 4G).
We previously showed that human deneddylase-1 (SENP8) is crucial for enabling NF-κB–mediated inflammation (Ehrentraut et al., 2013). Whether SENP8 itself is regulated during chronic inflammation is unknown. For this purpose, we investigated the expression of SENP8 in our model systems (cultured epithelia, murine tissue), as well as human tissue from healthy and IBD subjects. As shown in Figure 5, there was a striking similarity between the three models that revealed a correlation between disease and the loss of SENP8 mRNA expression. Exposure of T84 cells to cytomix (24 h) resulted in a nearly 70% decrease in SENP8 mRNA expression (p < 0.01; Figure 5A). Epithelial isolates from animals undergoing TNBS colitis at days 1 and 3 after induction were examined and compared with ethanol-only controls. TNBS-colitic animals tended to express less SENP8 mRNA transcript at day 1 (n = 3 per group, p = 0.07; Figure 5B) and day 3 after colitis induction (p < 0.05, n = 5 per group; Figure 5B). The same response was observed in human tissue samples from patients with active IBD. Regardless of the type of colitis (i.e., ulcerative colitis vs. Crohn’s disease), mRNA levels of SENP8 were significantly lower than in samples from healthy individuals (p < 0.001; four nonactive controls, 19 individual samples per disease cohort; Figure 5C). Collectively these results suggest that intact neddylation is disease protective and that loss of SENP8 expression correlates with the development of mucosal inflammatory disease in both mice and humans.
FIGURE 5:
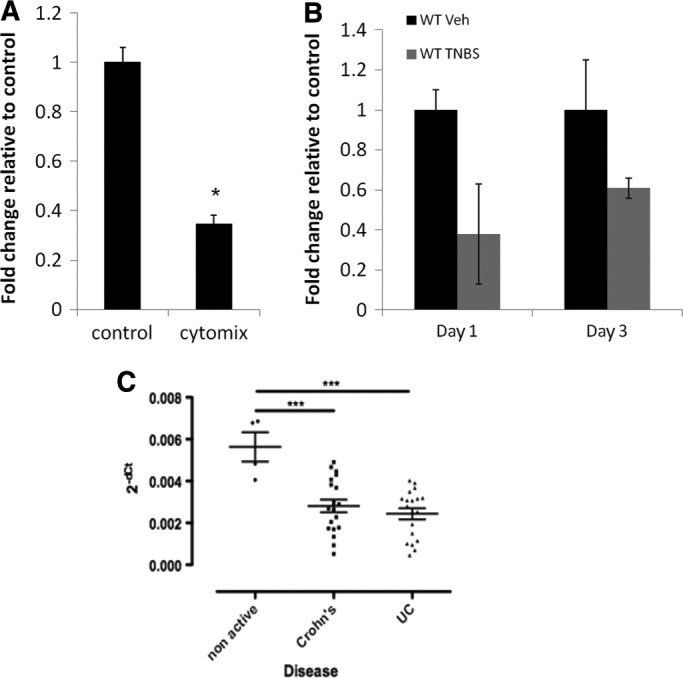
Decreased expression of SENP8 in colitis model and in IBD patients. (A) mRNA expression of SENP8 is decreased in T84 cells after treatment with cytomix (10 ng/μl each of TNF-α, IL-1β, and IFN-γ) for 24 h (n = 3). (B) mRNA expression of SENP8 is decreased in a TNBS model of colitis on days 1 and 3 after treatment (14 total mice). (C) Expression of SENP8 in colon tissue is decreased in patients with active Crohn’s disease and ulcerative colitis compared with nonactive healthy controls, *p < 0.05, ***p < 0.01.
DISCUSSION
Posttranslational modifications of signaling proteins are critical to productive inflammatory responses and resolution of disease (Ehrentraut and Colgan, 2012). NF-κB is the quintessential signaling hub during acute inflammation, and its regulation is fine-tuned by multiple posttranslational modifications, including neddylation (Amir et al., 2002). Cullin proteins, as components of ubiquitin E3 ligases, are neddylated for the polyubiquitination of effectors (e.g., IκB). This neddylation response is regulated, in part, by the deneddylase SENP8 as a mechanism to control E3 ligase activity. An intact Cullin-E3-ligase neddylation process is necessary for eliciting a coordinated inflammatory response to external inflammatory stimuli such as LPS (Ehrentraut et al., 2013), and this response is directly linked to the functional expression of human deneddylase-1/SENP8. We demonstrate here that pharmacological targeting of neddylation using the AMP homologue MLN4924 potently blocks NF-κB activation/signaling and through these actions functions as a preapoptotic sensitizer (Figure 6).
FIGURE 6:
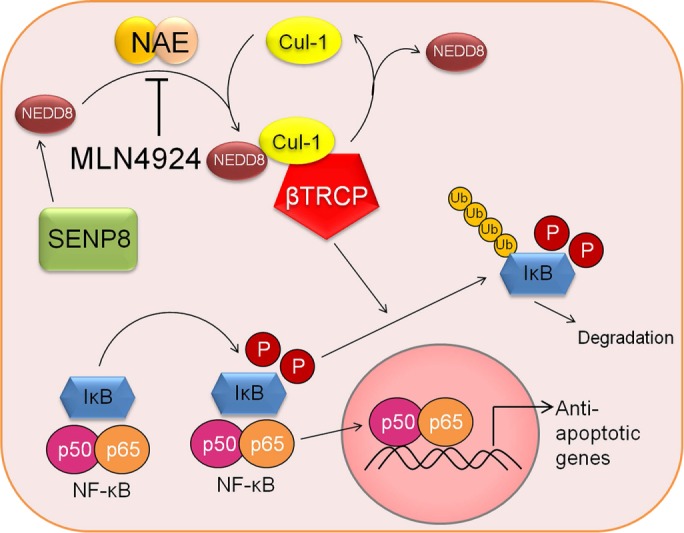
Mechanistic model of the effect of MLN4924 on the NF-κB pathway and downstream apoptotic responses. Under normal conditions, IκB is unphosphorylated and binds NF-κB subunits, sequestering them in the cytoplasm. An inflammatory stimulus leads to the phosphorylation of IκB, which is recognized by the Cul-1-Nedd8-transducin repeat–containing E3 ubiquitin protein ligase (TRCP) complex, targeting IκB for polyubiquitination and degradation by the proteosome. NF-κB can then translocate into the nucleus, leading to the transcription of target genes, including antiapoptotic cascades. Pharmacological inhibition of Cul-1 neddylation using MLN4924 stabilizes cellular IκB levels, keeping NF-κB out the nucleus and leading to decreased transcription of antiapoptotic target genes and increased apoptosis.
The present studies revealed that MLN4924 is an effective (EC50 ≈ 1 μM) Cul-1 deneddylating agent and equally potent inhibitor of NF-κB in intestinal epithelia. Presumably through its actions on Cul-1 activity and subsequent inhibition of E3 ubiquitin ligase activity, MLN4924 appears to function as a “priming” agent of the inflammatory response. Consistent with this premise, in vitro or in vivo exposure to MLN4924 alone resulted in surprisingly little activity within the mucosa. By stark contrast, the addition of activators of the NF-κB pathway profoundly enhanced the inflammatory response. Other model systems show similar actions of MLN4924. For example, studies of T-cell activation revealed that whereas MLN4924 alone does not activate T-cells, neddylation inhibition significantly lowers the threshold for anti-CD3–stimulated cytokine production (Friend et al., 2013). Other studies in T-cells show that MLN4924 may have neddylation targets beyond the Cullins, including proteins in the Ras/Erk pathway and other adaptor proteins such as Shc (Jin et al., 2013). Godbersen et al. (2015) also showed that targeting neddylation with MLN4924 abrogates NF-κB activation in leukemic B-cells and, in the process, regulates a diverse set of target genes. Thus the “priming” activity elicited by MLN4924 within the mucosa likely represents a complex response to neddylation targets beyond that of Cullins.
In intestinal epithelia, MLN4924 more potently inhibits Cul-2 than Cul-1. Our previous studies, in fact, revealed EC50 ≈ 5 nM for MLN4924 actions on Cul-2 and HIF stabilization (Curtis et al., 2015). In these studies, lower concentrations of MLN4924 (0.1 mg/kg, compared with 3 mg/kg used in the present studies) activated HIF in vivo and were protective for dextran sulfate sodium (DSS) colitis at multiple levels. There are distinct differences in our findings here, that is, aggravation of inflammatory tissue damage after MLN4924 plus TNBS–induced inflammatory disease and previously reported findings from our group in DSS colitis. These differences may reflect the different pathogenic mechanisms of both colitis models. DSS colitis is believed to occur independent of adaptive immune cells, whereas TNBS colitis is directly T-cell dependent (Neurath et al., 1996). MLN4924 was initially discovered for the treatment of NF-κB–dependent B-cell lymphoma (Milhollen et al., 2010). Hence use of MLN4924 in a disease model dependent on adaptive immune cells might explain the observed differences. Together these findings suggest that both in vitro and in vivo, Cul-2 responses are significantly more sensitive to MLN4924.
Given the central role of NF-κB in inflammation, it is not surprising that numerous studies have revealed that inhibition of NF-κB is antiinflammatory (Kanarek and Ben-Neriah, 2012). The intestinal mucosa—specifically, epithelial cells—appears to be somewhat unique in this regard (Karrasch and Jobin, 2008). For example, genetic deletion of NF-κB components (e.g., Ikkb) within the intestinal epithelium results in significantly exacerbated pathogen-induced intestinal inflammation (Zaph et al., 2007). These studies revealed increased apoptotic responses with the loss of NF-κB signaling, resulting in a loss of epithelial barrier and ultimately septicemia. Our studies here indicate similar results with targeting neddylation in vivo and that as a preapoptotic sensitizer, the combination of deneddylation in the presence of inflammatory stimuli significantly enhances the inflammatory response. It is noteworthy that throughout these studies, the inhibition of neddylation using MLN4924 had little to no detectable influence on basal epithelial function (i.e., in the absence of additional inflammatory stimuli). Barrier function, for example, was not changed by the inhibition of neddylation responses, even at relatively high concentrations of MLN4924. At multiple levels, these studies revealed a primed inflammatory response that correlated with the loss of neddylation and diminished NF-κB activity in vivo. Direct inhibition of NF-κB via Bay 11-7085 in conjunction with inflammatory stimuli partially recapitulated the reduction in TER measurements and thus barrier function observed with MLN4924.
A central component of the active deneddylation response is SENP8, an isopeptidase capable of directly deneddylating Cullin proteins (Mendoza et al., 2003; Wu et al., 2003), offering a cleavage pathway beyond the COP9 signalosome (Lyapina et al., 2001; Cope and Deshaies, 2003). It was shown, for example, that knockdown of SENP8 prevented LPS-induced NF-κB activation and systemic cytokine release (Ehrentraut et al., 2013). We extended these results to define the expression of SENP8 in murine and human colitis. Across each model tested, including human IBD tissue, inflammation was associated with a loss of SENP8 expression. Such observations suggest that down-regulation of SENP8 in murine/human colitis serves as a compensatory mechanism to quench the ongoing inflammatory response. Mechanisms of such regulation await further studies.
In conclusion, we demonstrate the contribution of epithelial NF-κB and Cul-1 neddylation to inflammatory responses in the intestinal mucosa. In particular, these studies identify MLN4924 as an inhibitor of Cul-1 neddylation, leading to loss of NF-κB signaling. These findings support a role for Cul-1 deneddylation as an apoptotic presensitizer during inflammation, which in turn enhances intestinal inflammatory responses.
MATERIALS AND METHODS
Cell culture
Human T84 and Caco-2 intestinal epithelial cells were cultured in 95% air with 5% CO2 at 37°C in DMEM and DMEM:F12, respectively, supplemented with 10% calf serum (Fisher Scientific, Waltham, MA) and penicillin/streptomycin (100 U/ml, 100 μg/ml; Invitrogen, Carlsbad, CA).
Transcriptional analysis
TRIzol reagent (Invitrogen) was used to isolate RNA from Caco-2 or T84 cells. cDNA was reverse transcribed using the iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA). PCR analysis was performed using SYBR Green (Applied Biosystems, Carlsbad, CA) and the following primer sequences: IL-8, forward 5′-CTGGCCGTGGCTCTCTTG-3′, reverse 5′-CCTTGGCAAAACTGCACCTT-3′; ICAM-1, forward, 5′-ATGCCCAGACATCTGTGTCC-3′, reverse 5′-GGGGTCTCTCTATGCCCAACA-3′; and β-actin, forward, 5′-GGAGAAAATCTGGCACCACA-3′, reverse, 5′-AGAGGCGTACAGGGATAGCA-3′. Each experiment was performed in triplicate.
Western blot analysis
The NE-PER extraction kit was used to prepare nuclear and cytoplasmic lysates from Caco-2 cells per the manufacturer’s instructions (Thermo Scientific, Waltham, MA). Western blotting of these lysates was performed using Cul-1 Rb polyclonal antibody (pAb; Invitrogen), p65 Rb pAb (Cell Signaling, Danvers, MA), TATA-binding protein (TATA-BP) Ms monocolonal antibody (mAb; Abcam, Cambridge, United Kingdom), and human β-actin Rb pAb (Abcam). Each experiment was performed in triplicate.
Luciferase assays
An NF-κB luciferase reporter plasmid (500 ng; Ehrentraut et al., 2013) and a Renilla reporter plasmid (1 ng) were transfected into subconfluent Caco-2 cells using Lipofectamine LTX Reagent (Thermo Scientific). Luciferase activity was determined at 16 h using Promega dual luciferase reagents (Promega, Madison, WI), and luminescence was determined using the GloMax-Multi plate reader (Promega). Each experiment was performed in triplicate.
Barrier integrity, permeability assays, and apoptosis assays
T84 cells were plated on 0.33-cm2, 0.4-μm permeable polyester inserts (Corning, Corning, NY). TER was measured using the EVOM2 voltohmmeter (World Precision Instruments, Sarasota, FL) to monitor barrier formation after treatment of confluent T84 monolayers with MLN49249 (1 μM; Millennium Pharmaceuticals, Cambridge, MA), cytomix (10 ng/μl each of TNF-α, IL-1β, and IFN-γ; eBioscience, San Diego, CA), and Bay 11-7085 (30 μM; Tocris Bioscience, Minneapolis, MN).
Paracellular permeability was assayed using FITC-dextran flux assay described previously (Furuta et al., 2001) on T84 monolayers with MLN49249 (1 μM), cytomix, or a combination of both MLN4924 and cytomix.
To detect caspase-3/7 activity, the Caspase-Glo 3/7 Assay (Promega) was used according to the manufacturer’s instructions. Briefly, 10,000 T84 intestinal epithelial cells per well were plated in 96-well plates and treated with 1 μM MLN4924, cytomix, 30 μM BAY 11-7085, or a combination of either MLN4924 or BAY 11-7085 with cytomix for 24 h. After 24 h, 100 μl of the Caspase-Glo 3/7 reagent was added to each well, and the luminescence was measured using the GloMax-Multi plate reader (Promega).
To inhibit caspase activation, a general caspase inhibitor peptide (Z-VAD-FMK; 30 μM) or a caspase-3 inhibitor peptide (Z-DEVD-FMK; 30 μM) was given as a 30-min pretreatment to T84 cells plated on permeable polyester inserts before treatment with cytomix plus MLN4924 (1 μM; BD PharMingen, San Diego, CA) or cytomix plus Bay 11-7085 (30 μM). A negative control peptide (Z-FA-FMK; 30 μM) was used on additional inserts (BD PharMingen).
Animals
Wild-type C57BL/6 mice (Mus musculus) were obtained from Jackson Laboratories (Bar Harbor, ME). All animal experiments were reviewed and approved by the Institutional Animal Care and Use Committee at the University of Colorado. TNBS colitis was induced as previously described (Louis et al., 2008). Animals were administered either MLN4924 (3 mg/kg) or cyclodextrin (Sigma-Aldrich, St. Louis, MO) via subcutaneous (s.c.) injection at day −1 of TNBS exposure, and this was continued daily. The groups were as follows: ethanol plus cyclodextrin (n = 5), ethanol plus MLN4924 (n = 5), TNBS plus cyclodextrin (n = 9), and TNBS plus MLN4924 (n = 10).
Histology and immunofluorescence
Histological examination was performed on samples of the distal colon from each group; samples were fixed in 10% Formalin before staining with hematoxylin and eosin. Slides used for immunofluorescence were first blocked with 5% normal goat serum (NGS). An In Situ Cell Death Detection kit (Sigma-Aldrich) was used to visualize apoptotic cell death by TUNEL staining. For cleaved caspase-3 staining, Rb mAb was used (Cell Signaling) at a 1:400 dilution. Secondary antibody was Alexa Fluor 488 goat anti-rabbit (Invitrogen), used at 1:500 dilution in 5% NGS. Immunolabeling was visualized with an AxioCam MR c5 attached to an AxioImager A1 microscope (Zeiss, Oberkochen, Germany). Where indicated, series of confocal fluorescence images were obtained using a Zeiss Axiovert 200 M laser-scanning confocal/multiphoton-excitation fluorescence microscope with a Meta spectral detection system (Zeiss NLO 510 with META; Zeiss, Thornwood, NY). Representative hematoxylin and eosin and immunofluorescence sections are presented.
Histology
Histological examination was performed on three samples of the distal colon. Samples were fixed in 10% Formalin before staining with hematoxylin and eosin. All histological quantitation was performed in a blinded manner, using a previously described scoring system (Dieleman et al., 1998). The three independent parameters measured were severity of inflammation (0–3: none, slight, moderate, severe), extent of injury (0–3: none, mucosal, mucosal and submucosal, transmural), and crypt damage (0–4: none, basal one-third damaged, basal two-thirds damaged, only surface epithelium intake, entire crypt and epithelium lost). The score of each parameter was multiplied by a factor reflecting the percentage of tissue involvement (×1: 0–25%; ×2: 26–50%; ×3: 51–75%; ×4: 76–100%), and all numbers were summed. Maximum possible score was 40.
Human tissue
Deidentified human intestinal tissue cDNA was obtained from the TissueScan Crohn’s/ColitisII array (OriGene Technologies, Rockville, MD). Complete patient/sample characteristics can be accessed from the supplier’s website (www.origene.com).
Statistical analysis
Data are expressed as mean values ± SEM. Data were analyzed with Student’s t test between two groups or ANOVA coupled with post hoc Bonferroni test for multiple pairwise comparisons. p < 0.05 was considered to be statistically significant.
Acknowledgments
This work was supported by a Deutsche Forschungsgemeinschaft research fellowship (DFG EH371/1-1) and a BONFOR starting grant to S.F.E., by National Institutes of Health Grants DK103639, DK50189, and DK95491 to S.P.C., and by funding from the Crohn’s and Colitis Foundation of America.
Abbreviations used:
- SENP8
sentrin-specific protease 8/deneddylase-1
- TER
transepithelial electrical resistance.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E16-05-0273) on September 28, 2016.
REFERENCES
- Amir RE, Iwai K, Ciechanover A. The NEDD8 pathway is essential for SCF(beta -TrCP)-mediated ubiquitination and processing of the NF-kappa B precursor p105. J Biol Chem. 2002;277:23253–23259. doi: 10.1074/jbc.M200967200. [DOI] [PubMed] [Google Scholar]
- Angus DC. The search for effective therapy for sepsis: back to the drawing board? J Am Med Assoc. 2011;306:2614–2615. doi: 10.1001/jama.2011.1853. [DOI] [PubMed] [Google Scholar]
- Bruewer M, Luegering A, Kucharzik T, Parkos CA, Madara JL, Hopkins AM, Nusrat A. Proinflammatory cytokines disrupt epithelial barrier function by apoptosis-independent mechanisms. J Immunol. 2003;171:6164–6172. doi: 10.4049/jimmunol.171.11.6164. [DOI] [PubMed] [Google Scholar]
- Bruning U, Fitzpatrick SF, Frank T, Birtwistle M, Taylor CT, Cheong A. NFkappaB and HIF display synergistic behaviour during hypoxic inflammation. Cell Mol Life Sci. 2011;69:1319–1329. doi: 10.1007/s00018-011-0876-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cernada M, Badia N, Modesto V, Alonso R, Mejias A, Golombek S, Vento M. Cord blood interleukin-6 as a predictor of early-onset neonatal sepsis. Acta Paediatr. 2011;101:e203–e207. doi: 10.1111/j.1651-2227.2011.02577.x. [DOI] [PubMed] [Google Scholar]
- Cope GA, Deshaies RJ. COP9 signalosome: a multifunctional regulator of SCF and other cullin-based ubiquitin ligases. Cell. 2003;114:663–671. doi: 10.1016/s0092-8674(03)00722-0. [DOI] [PubMed] [Google Scholar]
- Crespo I, San-Miguel B, Prause C, Marroni N, Cuevas MJ, Gonzalez-Gallego J, Tunon MJ. Glutamine treatment attenuates endoplasmic reticulum stress and apoptosis in TNBS-induced colitis. PLoS One. 2012;7:e50407. doi: 10.1371/journal.pone.0050407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis VF, Ehrentraut SF, Campbell EL, Glover LE, Bayless A, Kelly CJ, Kominsky DJ, Colgan SP. Stabilization of HIF through inhibition of Cullin-2 neddylation is protective in mucosal inflammatory responses. FASEB J. 2015;29:208–215. doi: 10.1096/fj.14-259663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieleman LA, Palmen MJ, Akol H, Bloemena E, Pena AS, Meuwissen SG, Van Rees EP. Chronic experimental colitis induced by dextran sulphate sodium (DSS) is characterized by Th1 and Th2 cytokines. Clin Exp Immunol. 1998;114:385–391. doi: 10.1046/j.1365-2249.1998.00728.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrentraut SF, Colgan SP. Implications of protein post-translational modifications in IBD. Inflamm Bowel Dis. 2012;18:1378–1388. doi: 10.1002/ibd.22859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrentraut SF, Kominsky DJ, Glover LE, Campbell EL, Kelly CJ, Bowers BE, Bayless AJ, Colgan SP. Central role for endothelial human deneddylase-1/SENP8 in fine-tuning the vascular inflammatory response. J Immunol. 2013;190:392–400. doi: 10.4049/jimmunol.1202041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friend SF, Peterson LK, Treacy E, Stefanski AL, Sosinowski T, Pennock ND, Berger AJ, Winn VD, Dragone LL. The discovery of a reciprocal relationship between tyrosine-kinase signaling and cullin neddylation. PLoS One. 2013;8:e75200. doi: 10.1371/journal.pone.0075200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuta GT, Turner JR, Taylor CT, Hershberg RM, Comerford K, Narravula S, Podolsky DK, Colgan SP. Hypoxia-inducible factor 1-dependent induction of intestinal trefoil factor protects barrier function during hypoxia. J Exp Med. 2001;193:1027–1034. doi: 10.1084/jem.193.9.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godbersen JC, Paiva C, Danilova OV, Berger A, Brown JR, Danilov AV. Targeting neddylation effectively antagonizes nuclear factor-kappaB in chronic lymphocytic leukemia B-cells. Leuk Lymphoma. 2015;56:1566–1569. doi: 10.3109/10428194.2014.990901. [DOI] [PubMed] [Google Scholar]
- Hjerpe R, Thomas Y, Chen J, Zemla A, Curran S, Shpiro N, Dick LR, Kurz T. Changes in the ratio of free NEDD8 to ubiquitin triggers NEDDylation by ubiquitin enzymes. Biochem J. 2012;441:927–936. doi: 10.1042/BJ20111671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DT, Miller DW, Mathew R, Cassell R, Holton JM, Roussel MF, Schulman BA. A unique E1-E2 interaction required for optimal conjugation of the ubiquitin-like protein NEDD8. Nat Struct Mol Biol. 2004;11:927–935. doi: 10.1038/nsmb826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin HS, Liao L, Park Y, Liu YC. Neddylation pathway regulates T-cell function by targeting an adaptor protein Shc and a protein kinase Erk signaling. Proc Natl Acad Sci USA. 2013;110:624–629. doi: 10.1073/pnas.1213819110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones D, Crowe E, Stevens TA, Candido EP. Functional and phylogenetic analysis of the ubiquitylation system in Caenorhabditis elegans: ubiquitin-conjugating enzymes, ubiquitin-activating enzymes, and ubiquitin-like proteins. Genome Biol. 2002;3:RESEARCH0002. doi: 10.1186/gb-2001-3-1-research0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones RM, Desai C, Darby TM, Luo L, Wolfarth AA, Scharer CD, Ardita CS, Reedy AR, Keebaugh ES, Neish AS. Lactobacilli modulate epithelial cytoprotection through the Nrf2 pathway. Cell Rep. 2015;12:1217–1225. doi: 10.1016/j.celrep.2015.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanarek N, Ben-Neriah Y. Regulation of NF-kappaB by ubiquitination and degradation of the IkappaBs. Immunol Rev. 2012;246:77–94. doi: 10.1111/j.1600-065X.2012.01098.x. [DOI] [PubMed] [Google Scholar]
- Karhausen J, Furuta GT, Tomaszewski JE, Johnson RS, Colgan SP, Haase VH. Epithelial hypoxia-inducible factor-1 is protective in murine experimental colitis. J Clin Invest. 2004;114:1098–1106. doi: 10.1172/JCI21086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karrasch T, Jobin C. NF-kappaB and the intestine: friend or foe? Inflamm Bowel Dis. 2008;14:114–124. doi: 10.1002/ibd.20243. [DOI] [PubMed] [Google Scholar]
- Khoury J, Ibla JC, Neish AS, Colgan SP. Antiinflammatory adaptation to hypoxia through adenosine-mediated cullin-1 deneddylation. J Clin Invest. 2007;117:703–711. doi: 10.1172/JCI30049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch S, Nusrat A. The life and death of epithelia during inflammation: lessons learned from the gut. Annu Rev Pathol. 2012;7:35–60. doi: 10.1146/annurev-pathol-011811-120905. [DOI] [PubMed] [Google Scholar]
- Kumar A, Wu H, Collier-Hyams LS, Hansen JM, Li T, Yamoah K, Pan ZQ, Jones DP, Neish AS. Commensal bacteria modulate cullin-dependent signaling via generation of reactive oxygen species. EMBO J. 2007;26:4457–4466. doi: 10.1038/sj.emboj.7601867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis NA, Robinson AM, Macmanus CF, Karhausen J, Scully M, Colgan SP. Control of IFN-{alpha}A by CD73: Implications for mucosal inflammation. J Immunol. 2008;180:4246–4255. doi: 10.4049/jimmunol.180.6.4246. [DOI] [PubMed] [Google Scholar]
- Lyapina S, Cope G, Shevchenko A, Serino G, Tsuge T, Zhou C, Wolf DA, Wei N, Deshaies RJ. Promotion of NEDD-CUL1 conjugate cleavage by COP9 signalosome. Science. 2001;292:1382–1385. doi: 10.1126/science.1059780. [DOI] [PubMed] [Google Scholar]
- Mendoza HM, Shen LN, Botting C, Lewis A, Chen J, Ink B, Hay RT. NEDP1, a highly conserved cysteine protease that deNEDDylates Cullins. J Biol Chem. 2003;278:25637–25643. doi: 10.1074/jbc.M212948200. [DOI] [PubMed] [Google Scholar]
- Merlet J, Burger J, Gomes JE, Pintard L. Regulation of cullin-RING E3 ubiquitin-ligases by neddylation and dimerization. Cell Mol Life Sci. 2009;66:1924–1938. doi: 10.1007/s00018-009-8712-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milhollen MA, Traore T, Adams-Duffy J, Thomas MP, Berger AJ, Dang L, Dick LR, Garnsey JJ, Koenig E, Langston SP, et al. MLN4924, a NEDD8-activating enzyme inhibitor, is active in diffuse large B-cell lymphoma models: rationale for treatment of NF-{kappa}B-dependent lymphoma. Blood. 2010;116:1515–1523. doi: 10.1182/blood-2010-03-272567. [DOI] [PubMed] [Google Scholar]
- Nava P, Koch S, Laukoetter MG, Lee WY, Kolegraff K, Capaldo CT, Beeman N, Addis C, Gerner-Smidt K, Neumaier I, et al. Interferon-gamma regulates intestinal epithelial homeostasis through converging beta-catenin signaling pathways. Immunity. 2010;32:392–402. doi: 10.1016/j.immuni.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neurath MF, Pettersson S, Meyer zum Buschenfelde KH, Strober W. Local administration of antisense phosphorothioate oligonucleotides to the p65 subunit of NF-kappa B abrogates established experimental colitis in mice. Nat Med. 1996;2:998–1004. doi: 10.1038/nm0996-998. [DOI] [PubMed] [Google Scholar]
- Pan ZQ, Kentsis A, Dias DC, Yamoah K, Wu K. Nedd8 on cullin: building an expressway to protein destruction. Oncogene. 2004;23:1985–1997. doi: 10.1038/sj.onc.1207414. [DOI] [PubMed] [Google Scholar]
- Parry G, Estelle M. Regulation of cullin-based ubiquitin ligases by the Nedd8/RUB ubiquitin-like proteins. Semin Cell Dev Biol. 2004;15:221–229. doi: 10.1016/j.semcdb.2003.12.003. [DOI] [PubMed] [Google Scholar]
- Pierce JW, Schoenleber R, Jesmok G, Best J, Moore SA, Collins T, Gerritsen ME. Novel inhibitors of cytokine-induced IkappaBalpha phosphorylation and endothelial cell adhesion molecule expression show anti-inflammatory effects in vivo. J Biol Chem. 1997;272:21096–21103. doi: 10.1074/jbc.272.34.21096. [DOI] [PubMed] [Google Scholar]
- Porter AG, Janicke RU. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 1999;6:99–104. doi: 10.1038/sj.cdd.4400476. [DOI] [PubMed] [Google Scholar]
- Robinson A, Keely S, Karhausen J, Gerich ME, Furuta GT, Colgan SP. Mucosal protection by hypoxia-inducible factor prolyl hydroxylase inhibition. Gastroenterology. 2008;134:145–155. doi: 10.1053/j.gastro.2007.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song YR, You SJ, Lee YM, Chin HJ, Chae DW, Oh YK, Joo KW, Han JS, Na KY. Activation of hypoxia-inducible factor attenuates renal injury in rat remnant kidney. Nephrol Dial Transplant. 2010;25:77–85. doi: 10.1093/ndt/gfp454. [DOI] [PubMed] [Google Scholar]
- Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S, Brownell JE, Burke KE, Cardin DP, Critchley S, et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature. 2009;458:732–736. doi: 10.1038/nature07884. [DOI] [PubMed] [Google Scholar]
- Tateishi K, Omata M, Tanaka K, Chiba T. The NEDD8 system is essential for cell cycle progression and morphogenetic pathway in mice. J Cell Biol. 2001;155:571–579. doi: 10.1083/jcb.200104035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada H, Kito K, Caskey LS, Yeh ET, Kamitani T. Cleavage of the C-terminus of NEDD8 by UCH-L3. Biochem Biophys Res Commun. 1998;251:688–692. doi: 10.1006/bbrc.1998.9532. [DOI] [PubMed] [Google Scholar]
- Wu K, Yamoah K, Dolios G, Gan-Erdene T, Tan P, Chen A, Lee CG, Wei N, Wilkinson KD, Wang R, et al. DEN1 is a dual function protease capable of processing the C terminus of Nedd8 and deconjugating hyper-neddylated CUL1. J Biol Chem. 2003;278:28882–28891. doi: 10.1074/jbc.M302888200. [DOI] [PubMed] [Google Scholar]
- Zaph C, Troy AE, Taylor BC, Berman-Booty LD, Guild KJ, Du Y, Yost EA, Gruber AD, May MJ, Greten FR, et al. Epithelial-cell-intrinsic IKK-beta expression regulates intestinal immune homeostasis. Nature. 2007;446:552–556. doi: 10.1038/nature05590. [DOI] [PubMed] [Google Scholar]