

PARP1 and its effector, the ATP-dependent chromatin remodeler Alc1/Chd1L, are identified as key players during the rapid chromatin relaxation at DNA damage sites.
Abstract
Chromatin relaxation is one of the earliest cellular responses to DNA damage. However, what determines these structural changes, including their ATP requirement, is not well understood. Using live-cell imaging and laser microirradiation to induce DNA lesions, we show that the local chromatin relaxation at DNA damage sites is regulated by PARP1 enzymatic activity. We also report that H1 is mobilized at DNA damage sites, but, since this mobilization is largely independent of poly(ADP-ribosyl)ation, it cannot solely explain the chromatin relaxation. Finally, we demonstrate the involvement of Alc1, a poly(ADP-ribose)- and ATP-dependent remodeler, in the chromatin-relaxation process. Deletion of Alc1 impairs chromatin relaxation after DNA damage, while its overexpression strongly enhances relaxation. Altogether our results identify Alc1 as an important player in the fast kinetics of the NAD+- and ATP-dependent chromatin relaxation upon DNA damage in vivo.
INTRODUCTION
The complex multiscale architecture of chromatin poses a formidable challenge for the DNA repair machinery, which requires regulated access to DNA lesions. Early steps of the DNA damage response involve chromatin remodeling, leading to an increased sensitivity of chromatin to nucleases (Smerdon and Lieberman, 1978). Experiments in living cells have shown that DNA damage induced by laser microirradiation leads to an ATP-dependent but ataxia telangiectasia mutated (ATM)-independent chromatin relaxation at sites of DNA damage (Kruhlak et al., 2006). While the dense packing of chromatin may hinder the efficiency of DNA repair (Schuster-Böckler and Lehner, 2012), recent reports also show that chromatin overcompaction at DNA lesions may also be important to inhibit transcription during repair and to keep the broken DNA ends in close proximity (Ayrapetov et al., 2014; Burgess et al., 2014).
One of the earliest events upon DNA damage is the recruitment and activation of poly(ADP-ribose) polymerase 1 (PARP1), a key regulator of chromatin structure during DNA repair and transcription (Lebeaupin et al., 2015). It is activated by DNA lesions and attaches poly(ADP-ribose) (PAR) to itself and other chromatin factors, including histones. The binding of PARP1 to chromatin modifies its compaction state through multiple, sometimes opposite, mechanisms. Inactive PARP1 competes with the linker histone H1, leading to the formation of compact and transcriptionally repressed genomic regions (Kim et al., 2004). In contrast, PARylated polynucleosomes appear as a loose, beads-on-a-string fiber, on electron micrographs (Poirier et al., 1982). It was suggested that the PARylation of chromatinized H1 could counteract its ability to condense chromatin (Huletsky et al., 1989). Additionally, PARylation is also involved in the recruitment and the regulation of several chromatin-remodeling enzymes whose ATP-dependent activity could promote chromatin relaxation (Chou et al., 2010; Polo et al., 2010; Smeenk et al., 2013).
In the present work, we sought to address the impact of PARP1 on chromatin structure and dynamics following DNA damage. Using photoactivated histones, live-cell imaging, and laser microirradiation in human cells, we analyzed the contributions of PARylation, linker histone H1, ATP, and the nucleosome remodeler Alc1 during the transient chromatin relaxation observed upon DNA damage.
RESULTS AND DISCUSSION
DNA damage induction by laser microirradiation induces a rapid chromatin relaxation at the DNA lesions
To assess large-scale chromatin reorganization at sites of DNA damage in living cells, we established an assay using human U2OS cells expressing the core histone H2B labeled with the photoconvertible dyes PAGFP or PATagRFP. By irradiating a predefined nuclear area with a 405 nm laser, we simultaneously photoconvert the tagged histones and, if cells have been Hoechst-presensitized, induce DNA lesions, allowing us to compare chromatin dynamics in the presence or absence of DNA damage (Figure 1A).
FIGURE 1:

DNA damage induced by laser microirradiation induces transient chromatin relaxation. (A) Recruitment of PARP1 at the microirradiated area in cells coexpressing PARP1-mCherry and H2B-PAGFP. Scale bar: 4 μm. In cells not presensitized with Hoechst, the 405 nm irradiation induces local photoactivation of the H2B-PAGFP but no recruitment of PARP1-mCherry. In contrast, in the case of Hoechst presensitization, the 405 nm irradiation induces both photoactivation of the H2B-PAGFP and a marked recruitment of PARP1-mCherry, indicating the presence of DNA lesions. Similarly, we observed the recruitment of 53BP1 only in cells presensitized with Hoechst (unpublished data). (B) Confocal image sequence of a human U2OS nucleus expressing H2B-PAGFP. Scale bar: 4 μm. The automatic segmentation of the histone H2B channel is shown in red below the raw images. The average thickness of the segmented line can be plotted as a function of time after irradiation, as shown in C for cells presensitized (n = 17) or not (n = 23) with Hoechst (mean ± SEM). Based on this analysis, the ratio between the thicknesses of the photoconverted line at time = 60 s and time = 0 s can be calculated to estimate the relative relaxation of the irradiated region. (D) Confocal image sequence of a U2OS cell expressing H2B-PATagRFP (red) and labeled with fluorescent nucleotides dUTP-ATTO633 (green). Scale bar: 4 μm. (E) Enlarged view of the region overlaid in yellow on the previous panel. The segmentation of the photoconverted chromatin area (red outline) and trajectories of individual foci labeled with fluorescent nucleotides (green) are shown. For this experiment, the power of the 405 nm laser used for simultaneous photoactivation and microirradiation was set to 250 μW at the sample level, instead of 125 μW, to induce an enhanced chromatin relaxation, allowing an easier identification of the phase of directed motion for the dUTP-labeled foci. (F) Comparison between the speed at which the width of the H2B-labeled region is growing and the speed of the dUTP-labeled foci perpendicular to the irradiation line. We show the average speed for the 30 s subsequent to laser microirradiation. p Values were calculated by paired t test. (G) Dynamics of the chromatin compaction state at DNA damage sites over long timescales measured in wild-type U2OS cell expressing H2B-PATagRFP (mean ± SEM, n = 16).
On microirradiation at 405 nm of cells expressing photoactivatable H2B and presensitized with Hoechst, we observed a rapid increase of the size of the photoconverted chromatin area (Figure 1, B and C), indicating chromatin relaxation at DNA damage sites, as previously reported (Kruhlak et al., 2006). However, an alternative interpretation could be the local release of photoconverted H2B through nucleosome remodeling induced upon DNA damage (Polo, 2015). To distinguish between these two possibilities, we labeled DNA by incorporating fluorescent nucleotides (Schermelleh et al., 2001). On irradiation, we observed the directional movement of fluorescent spots away from the irradiated line (Figure 1, D and E, and Supplemental Movie 1), with a speed similar to the one characterizing the expansion of the H2B photoconverted area (Figure 1F). These results indicate that the changes in the size of the photoactivated H2B area upon DNA damage reflect the relaxation of chromatinized DNA, rather than the local release of photoactivated H2B. This fast initial chromatin relaxation upon DNA damage is followed by a slow recondensation with chromatin recovering a compaction state close to its predamage level in ∼20 min (Figure 1G).
Chromatin relaxation at DNA damage sites is controlled by PARP1 activation
In agreement with recent reports (Khurana et al., 2014; Strickfaden et al., 2016), we observed that chromatin relaxation at DNA lesions is PARylation dependent (Figures 2A and Supplemental Figure S1, A–D). Interestingly, inhibiting PARylation not only abolished chromatin relaxation at DNA damage sites but also induced a small but significant chromatin overcompaction upon laser microirradiation (Figure 2A). The human PARP enzyme family has multiple members, and we found that PARP1, PARP2, and PARP3 are all recruited at DNA damage sites (Supplemental Figure S1E). PARP1 is responsible for ∼85% of PARylation activity (Woodhouse and Dianov, 2008). Therefore, to address the specific role of PARP1 in chromatin relaxation, we generated PARP1 knockout (KO) U2OS cell lines. While PARP1 was absent from these cells, we could detect similar amounts of PARP2 and PARP3 as compared with wild type (Supplemental Figure S1F). Chromatin relaxation at DNA lesions was dramatically reduced in PARP1 KO cells (Figure 2, A and B), a phenotype that could be partially rescued by reexpressing wild-type PARP1 (Figure 2C). Remarkably, laser irradiation in the PARP1 KOs did not lead to chromatin overcompaction, even after inhibition of PARylation (Figure 2A). Instead, we observed a residual chromatin relaxation independent of PARylation activity, suggesting that it was not triggered by the activity of other PARPs, such as PARP2 or PARP3. Altogether, since PARP inhibitors do not block the recruitment of PARP1 to DNA damage (Timinszky et al., 2009), our data suggest that chromatin overcompaction when inhibiting PARylation is due to PARP1 binding to DNA lesions, whereas its product, PAR, is responsible for chromatin relaxation. These findings reconcile oppositely reported effects of PARP1 on chromatin structure (Poirier et al., 1982; Kim et al., 2004).
FIGURE 2:
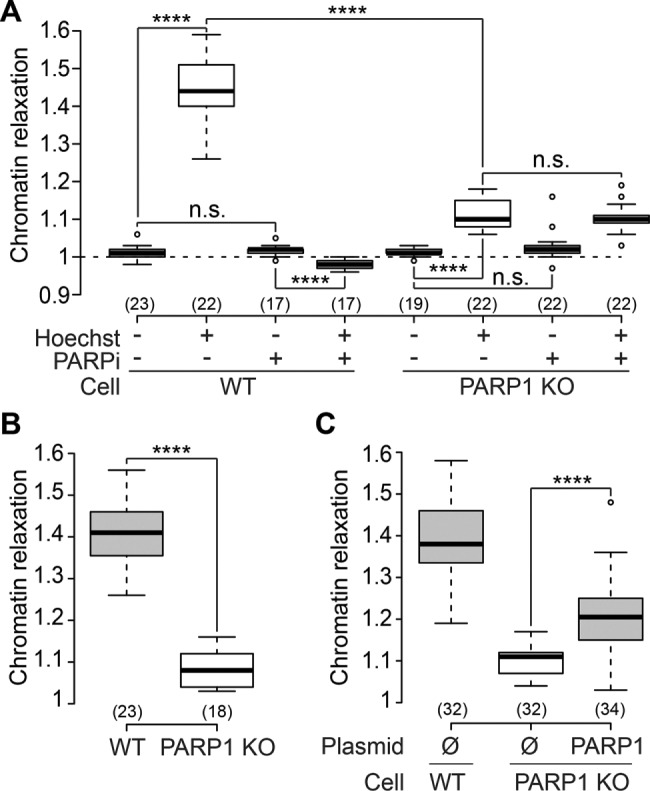
PARP1 activity controls chromatin relaxation at DNA damage sites. (A) Relative chromatin relaxation at 60 s after laser microirradiation in wild-type and PARP1 KO cells (clone C8) transfected with H2B-PAGFP and treated or not with the PARP inhibitor AG14361 (30 μM, 1 h; PARPi). (B) Similar results were obtained with a second PARP1 KO cell clone (clone C12). (C) Partial rescue of the impairment of chromatin relaxation in the PARP1 KO cells (clone C8) by reexpression of wild-type PARP1 fused to mCherry.
Chromatin relaxation at DNA lesions is not directly triggered by the mobilization of linker histone H1
In vitro studies identified the linker histone H1 to be crucial for the formation of compact chromatin (Thoma et al., 1979). Because H1 is a substrate of PARP1, PARylation of H1 could trigger its dissociation from chromatin, as shown for regulated transcription (Ju et al., 2006), and lead to chromatin relaxation. To test this hypothesis, we analyzed H1 (H1.1 variant) dynamics at DNA lesions in cells coexpressing H2B-PATagRFP and H1-PAGFP, allowing us to simultaneously label the damaged chromatin area and follow the dynamics of the H1 proteins localized within this area at the time of irradiation (Figure 3A and Supplemental Movie 2). For quantification of the redistribution of photoactivated H1 from the irradiated area independently of the co-occurring chromatin-relaxation process, the integrated fluorescence signal for H1 was measured within the irradiated area defined by the segmentation of the H2B channel (Figure 3B).
FIGURE 3:

The linker histone H1 is mobilized at DNA lesions. (A) Confocal image sequence of a U2OS nucleus coexpressing H2B-PATagRFP and H1.1-PAGFP. For the H1 channel, the image contrast was enhanced to allow the visualization of H1 redistribution over the entire nucleus following laser microirradiation. This led to an apparent saturation of the image at time = 0 s. Scale bar: 4 μm. (B) Kinetics of the release of the H1 proteins localized at the DNA lesions at the time of laser microirradiation in wild-type cells coexpressing H2B-PATagRFP and H1.1-PAGFP, presensitized or not with Hoechst and treated or not with the PARP1 inhibitor AG14361 (30 μM, 1 h; PARPi) (mean ± SEM, for each condition, 17 < n < 28). (C) Characteristic release time for H1, measured at half fluorescence decay, in wild-type and PARP1 KO cells.
We found that H1 initially localized within the irradiated area is released faster in the presence of DNA damage (Figure 3, B and C). Knowing that most H1 molecules are bound to chromatin at any given time (Beaudouin et al., 2006), this can only reflect impaired binding to chromatin. Once the photoconverted H1 proteins are redistributed over the entire nucleus, the DNA damage area appears to be depleted for H1 (Supplemental Figure S1G). This depletion progressively disappears as chromatin slowly recondenses. At the same time, we observed no significant release of the core histone H2B from the irradiated region (Supplemental Figure S1H).
Inhibiting PARylation reduced H1 dynamics both in the presence and absence of DNA damage, while deleting PARP1 only slowed H1 dynamics in the presence of DNA damage (Figure 3C). These data are consistent with the observation that the PARylation of H1 increases its dynamics (Ju et al., 2006). Nevertheless, even in the presence of PARP inhibitor or in the PARP1 KO cells, H1 dynamics were always much faster after DNA damage as compared with the dynamics observed when no damage was induced (Figure 3C). Because chromatin relaxation was abolished in cells treated with PARP inhibitors and strongly reduced in the PARP1 KOs (Figure 2A), this indicates that chromatin loosening at DNA lesions is not the direct consequence of PAR-driven H1 mobilization at DNA lesions. This result contrasts with a recent report that correlates H1 eviction and PAR-dependent chromatin relaxation at DNA lesions (Strickfaden et al., 2016). The discrepancy with our findings may arise from the difference in laser-irradiation methods, which could lead to different DNA damage (Kong et al., 2009). Nevertheless, we cannot exclude that the PARylation-dependent chromatin relaxation requires concomitant H1 mobilization, which is always observed upon DNA damage independent of PARP1 activation. Furthermore, it is possible that the DNA damage–induced H1 mobilization accounts for the observed residual chromatin relaxation observed in the PARP1 KOs.
Contribution of ATP-dependent processes in chromatin relaxation at DNA lesions
In vitro PARP1 activation results in chromatin loosening in the absence of ATP (Poirier et al., 1982), whereas ATP depletion abolishes chromatin relaxation at DNA lesions in live cells (Kruhlak et al., 2006; Luijsterburg et al., 2012). To establish the role of ATP in our assays, we quantified chromatin relaxation and PARylation levels upon laser microirradiation in cells depleted for ATP. We found that ATP depletion significantly impaired chromatin relaxation upon DNA damage (Figure 4A) while not affecting the level of PARylation at the lesions, as shown by the similar accumulation of the PAR-binder WWE domain of RNF146 at DNA damage sites (Figure 4B) (Wang et al., 2012). Nevertheless, ATP depletion did not fully abolish chromatin relaxation, its amplitude corresponding to approximately half of the control situation. This result suggests that PARylation may act on chromatin in both ATP-dependent and ATP-independent ways but it may also be due to only partial depletion of ATP. A confounding effect of ATP inhibition is chromatin hypercondensation (Figure 4C). To test whether chromatin hypercondensation could explain the inhibition of chromatin relaxation seen upon ATP depletion, we induced chromatin tightening in another way. Cells were bathed in hypertonic medium to induce a shrinking of the nuclear volume (Figure 4, D and E), which in turn leads to chromatin hypercondensation. The chromatin patterns in hypertonic cells visually resembled those obtained after ATP depletion (Figure 4C). The hypertonic treatment itself does not activate the PARylation signaling pathway (Figure 4, F and G). In hypertonic cells, chromatin loosening upon DNA damage was slightly increased compared with isotonic controls (Figure 4H), while PARylation at the site of damage was unchanged (Figure 4I). Thus the reduction of chromatin relaxation at DNA lesions observed upon ATP depletion does not seem to be the mere consequence of a tighter chromatin packing before damage induction.
FIGURE 4:
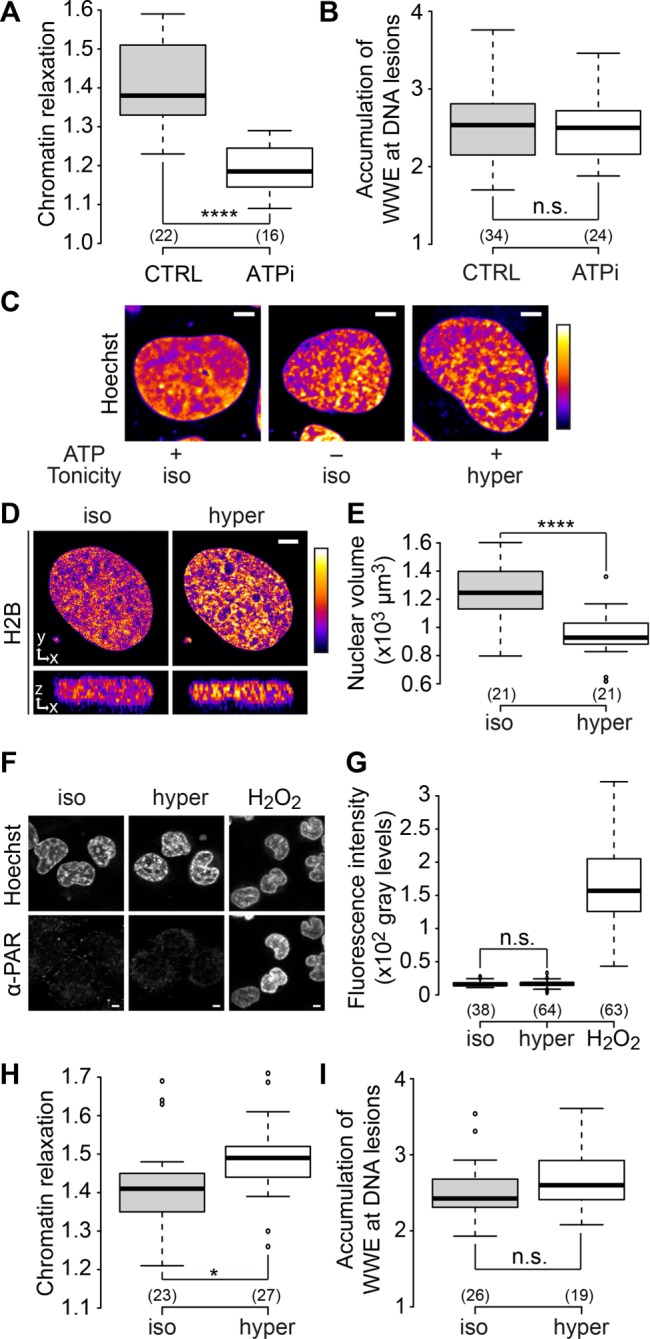
Chromatin relaxation at DNA damage sites partially depends on ATP. (A) Relative chromatin relaxation at 60 s after laser microirradiation in wild-type cells expressing H2B-PAGFP and depleted or not for ATP. (B) Accumulation of the WWE domain of RNF146 at the DNA lesions estimated 60 s after laser microirradiation in wild-type cells expressing an EGFP-tagged version of WWE and depleted or not for ATP. (C) Confocal image of U2OS cell nuclei stained with Hoechst and left untreated, depleted for ATP, or bathed with hypertonic medium. Scale bar: 4 μm. (D) Middle x,y and x,z sections of raw confocal image stacks of a U2OS cell expressing H2B-EGFP before and after the change of the bathing medium from isotonic to hypertonic. Scale bar: 4 μm. For C and D, fluorescence signals are pseudocolored using the lookup table shown on the right of the images. (E) Change in nuclear volumes of U2OS cells expressing H2B-EGFP upon hypertonic treatment. The nuclear volumes were estimated by automatic segmentation of confocal image stacks. (F, G) Images and quantification of immunofluorescence staining with anti-PAR (10H) antibody performed in U2OS cells left untreated, subjected to hypertonic shock, or treated with H2O2 (1 mM in PBS for 10 min). (H) Relative chromatin relaxation at 60 s after laser microirradiation in wild-type cells expressing H2B-PAGFP and bathed in isotonic or hypertonic media. (I) Accumulation of the WWE domain of RNF146 at the DNA lesions estimated 60 s after laser microirradiation in wild-type cells bathed in isotonic or hypertonic medium.
The ATP-dependent remodeler Alc1 contributes to chromatin relaxation at DNA damage sites
Several ATP-dependent chromatin-remodeling enzymes have been shown to be regulated by PARP activation (Chou et al., 2010; Polo et al., 2010; Smeenk et al., 2013). However, the only chromatin-remodeling enzyme with an ADP-ribose–binding domain that actively remodels nucleosomes upon PARP1 activation is Alc1 (Ahel et al., 2009; Gottschalk et al., 2009; Pines et al., 2012). To address the role of Alc1, also known as CHD1L, in chromatin relaxation, we generated an Alc1 KO U2OS cell line (Supplemental Figure S2A). By coexpressing a fluorescently tagged version of Alc1 together with H2B-PAGFP in these cells, we followed the recruitment of this protein at DNA damage sites together with the relaxation process (Figure 5, A and B). The fast accumulation of Alc1 observed at the site of DNA damage, with a maximum recruitment a few seconds after laser microirradiation, is compatible with a role for Alc1 in chromatin relaxation at DNA breaks, a process that lasts approximately 60 s. Moreover, the recruitment of Alc1 at DNA damage sites was abolished by PARP inhibitor treatment or for Alc1 lacking the PAR-binding macrodomain (Supplemental Figure S2, B and C), indicating that Alc1 recruitment, similar to chromatin relaxation, is fully controlled by PARP1 activation at DNA lesions.
FIGURE 5:

The chromatin remodeler Alc1 contributes to chromatin relaxation upon DNA damage. (A) Confocal image sequence of a U2OS nucleus coexpressing H2B-PAGFP and Alc1-mCherry. Scale bar: 4 μm. (B) Relative kinetics of Alc1 recruitment and chromatin relaxation at the DNA lesions measured in Alc1 KO cells coexpressing H2B-PAGFP and Alc1-mCherry (mean ± SEM, n = 20). (C) Confocal images of wild-type and Alc KO U2OS cells labeled with Hoechst. Scale bar: 4 μm. Fluorescence signals are pseudocolored using the lookup table shown on the right of the images. (D) Relative chromatin relaxation at 60 s after laser microirradiation for wild-type vs. Alc1 KO cells cotransfected with H2B-PAGFP and an empty plasmid (Ø), wild-type Alc1 or the catalytic-dead mutant Alc1 E175Q, both fused to mCherry. Cells with comparable expression levels of the wild-type or mutant Alc1 constructs were chosen, as assessed by similar fluorescence signals in the mCherry channel. (E) Relative chromatin relaxation at 60 s after laser microirradiation in Alc1 KO cells expressing H2B-PAGFP and depleted or not for ATP. (F) Relative chromatin relaxation at 60 s after laser microirradiation in Alc1 KO cells expressing H2B-PAGFP and treated or not with the PARP1 inhibitor AG14361 (30 μM, 1 h). (G) Relative chromatin relaxation at 60 s after laser microirradiation for wild-type cells expressing H2B-PAGFP and transfected with uncoupled mCherry, wild-type Alc1 fused to mCherry, or the catalytic-dead mutant Alc1 E175Q fused to mCherry. Cells with comparable expression levels of the transfected constructs were chosen, as assessed by similar fluorescence signals in the mCherry channel. (H) Clonogenic survival after different doses of X-ray irradiation for wild-type U2OS cells, KOs for Alc1 and PARP1, and wild-type cells overexpressing Alc1 fused to YFP.
The loss of Alc1 had no detectable effect on chromatin architecture in the absence of DNA damage (Figure 5C and Supplemental Figure S2, D and E) but led to impaired chromatin relaxation upon laser irradiation (Figure 5D and Supplemental Figure S2, F and G). Expression of wild-type Alc1 in Alc1 KOs fully restored chromatin relaxation at DNA lesions in contrast to the expression of ATPase-dead Alc1 mutants (Alc1-E175Q or Alc1-K77R) despite their efficient recruitment at DNA damage sites (Supplemental Figure S2, B and H). Cells depleted for Alc1 using RNA interference (RNAi) behaved in a similar manner (Supplemental Figure S2, I and J). While ATP depletion only slightly reduced chromatin relaxation in the Alc1 KO cells (Figure 5E), the inhibition of PARylation completely suppressed the relaxation process (Figure 5F), suggesting that the remaining chromatin relaxation observed at DNA damage sites in the absence of Alc1 is mediated mainly by the ATP-independent loosening effect of PARylation. Importantly, the overexpression of Alc1 in wild-type cells strongly increased chromatin relaxation at DNA lesions, while overexpressing the ATPase-dead Alc1-E175Q had no effect (Figure 5G). Altogether these results identify Alc1 as a mediator of PARylation-dependent chromatin relaxation through its ATP-dependent remodeling activity. A recent publication also reported the role of the remodeler CHD2 in the chromatin relaxation at DNA lesions (Luijsterburg et al., 2016). Because CHD2 appears to be recruited at the DNA damage sites slightly later than Alc1, the two remodelers may act sequentially to allow chromatin loosening. Further work is required to understand how the activities of these two remodelers are coordinated.
In conclusion, our present work extends our understanding of the contribution of the PARylation signaling pathway in the early chromatin remodeling at DNA lesions. We demonstrate the dual impact of PARP1 on the chromatin structure. In line with in vitro observations (Kim et al., 2004), our data in living cells indicate that PARP1 binding to DNA breaks leads to chromatin overcompaction while the formation of PAR chains due to PARP1 activity triggers its relaxation, in agreement with a recent report (Strickfaden et al., 2016). Moreover, our data show for the first time the direct contribution of the ATP-dependent chromatin-remodeling activity of the remodeler Alc1 in the rapid chromatin relaxation observed upon DNA damage induction. We also found that the absence of either PARP1 or Alc1 reduces cell survival capacity upon X-ray irradiation (Figure 5H). This result, which corroborates other reports showing that several members of the PARylation signaling pathway are important for efficient DNA repair (Khurana et al., 2014; Nagy et al., 2016), argues for a key regulatory role of the PARylation-dependent modulation of the chromatin compaction state during the DNA damage response. In addition, we propose that the dramatic increase in chromatin relaxation together with the cell hypersensitivity to X-ray irradiation observed in the case of Alc1 overexpression (Figure 5, G and H) might underlie the oncogenic potential of this remodeler, which has been shown to promote cancer progression and metastasis (Cheng et al., 2013).
MATERIALS AND METHODS
Plasmids
The core histone H2B, subcloned from the pH2B-mCherry vector was a gift from J. Ellenberg, European Molecular Biology Laboratory, Heidelberg, Germany (Neumann et al., 2010; Euroscarf accession number P30632), was cloned into pPATagRFP-N1 using NdeI and BamHI restriction sites. pPATagRFP-N1 was a gift from V. Verkhusha, Albert Einstein College of Medicine, Bronx, NY (Subach et al., 2010; Addgene plasmid #31941). The histone H2B-PAGFP and histone H1.1-PAGFP were gifts from J. Ellenberg (Beaudouin et al., 2006; Euroscarf accession numbers P30499 and P30503, respectively). Another construct of H1.1-PAGFP was produced with the PAGFP tag on the other side of the protein to ensure that similar results could be obtained with both constructs (Hutchinson et al., 2015). H1.1 was PCR amplified from the H1.1-PAGFP plasmid and subcloned into pmEGFP-N1 using BglII and ApaI to obtain the H1.1-EGFP construct. Wild-type Alc1 and E175Q Alc1 mutant fused to the C-terminus of enhanced green fluorescent protein (EGFP) or mCherry were obtained by exchanging yellow fluorescent protein (YFP) for the respective fluorescent protein in the constructs described previously (Gottschalk et al., 2009). The Alc1-Δmacro mutant fused to YFP was described previously (Gottschalk et al., 2009).The Alc1-K77R construct fused to mCherry was obtained by first mutating a wild-type ALC1 construct fused to YFP (Gottschalk et al., 2009), using QuikChange in vitro mutagenesis (Agilent, Santa Clara, CA), and then exchanging YFP for mCherry. The WWE domain of RNF146 (amino acids 99–183) was cloned into pmEGFP-C1 using BglII and EcoRI by PCR amplifying it from a cDNA library. PARP1-mCherry was described previously (Timinszky et al., 2009). This construct was also used to generate PARP1-EGFP by exchanging mCherry with EGFP. PARP2-EGFP was generated by PCR amplification of PARP2, digestion with NheI/SmaI, and ligation into pmEGFPC1. PARP3-EGFP (short isoform) was a gift from C. Prigent, Institut de Génétique et Développement de Rennes, CNRS, France (Rouleau et al., 2007). Mammalian expression was under the control of cytomegalovirus (CMV) promoter. All constructs were sequence verified.
Cell culture, inhibitor treatments, and osmotic shocks
Cells used for this work were wild-type U2OS cells or KO cells made from parental U2OS cells. Cells were routinely cultured in DMEM (with 4.5 g/l glucose) supplemented with 10% fetal bovine serum (FBS), 2 mM glutamine, 100 μg/ml penicillin, and 100 U/ml streptomycin in 5% CO2 at 37°C. For microscopy, cells were plated on Lab-Tek II chambered coverglass (Thermo Fisher Scientific, Waltham, MA). Presensitization was achieved by bathing cells for 1 h in culture medium containing 0.3 μg/ml Hoechst 33342 (Life Technologies, Carlsbad, CA). Immediately before imaging, the growth medium was replaced by Leibovitz’s L-15 medium (Life Technologies) supplemented with 20% FBS, 2 mM glutamine, 100 μg/ml penicillin, and 100 U/ml streptomycin. The PARP1 inhibitors AG14361 and Olaparib (Euromedex, Souffelweyersheim, France) were used at 30 and 50 μM, respectively. ATP depletion was achieved as previously described (Platani et al., 2002). The osmotic shock procedure was as previously described (Walter et al., 2013). All experiments presented in this work were performed on unsynchronized cells.
Live-cell DNA labeling with fluorescent nucleotides
U2OS cells expressing H2B-PATagRFP were synchronized at the G1/S phase transition by treating the cells with aphidicolin (Sigma-Aldrich, St. Louis, MO) at 5 μg/ml for 18 h. After aphidicolin release, the cell layer, bathed with growing medium containing 10 μM of dUTP-ATTO633 (Jena Bioscience, Jena, Germany), was scraped using a silicon stick to allow nucleotide loading and integration to the DNA during replication (Schermelleh et al., 2001).
Transfections and generation of stable and KO cell lines
Transient transfections were performed 12–24 h after cells were plated, using XtremeGENE HP (Roche, Basel, Switzerland) or JetPRIME (Polyplus Transfection, Illkirch, France) according to the manufacturers’ instructions. Cells were imaged 48–72 h after transfection.
To establish cell lines stably expressing H2B-PATagRFP or Alc1-YFP (construct described in Gottschalk et al., 2009), wild-type U2OS cells were transfected with the appropriate DNA construct and grown in culture medium containing Geneticin (PAA Laboratories, Pasching, Austria) for selection. Clones with stably integrated H2B-PATagRFP or Alc1-YFP were picked after 2 wk of Geneticin selection. Once selected, these cells were cultured in normal medium supplemented with 500 μg/ml Geneticin.
The KO cell lines were made according to the protocol described by the Zhang lab (Ran et al., 2013). The target sequence for ALC1 (5′-GACTTCCCTCAAGTACGTTAG-3′) and PARP1 (5′-GTCCAACAGAAGTACGTGCAA-3′) was chosen according to the Web-based CRISPR design tool from the Zhang lab (www.genome-engineering.org). The sgRNA oligos were introduced into pX458 expressing Cas9 nuclease fused to green fluorescent protein (GFP; Addgene #48138). pSpCas9(BB)-2A-GFP (PX458) was a gift from Feng Zhang (Broad Institute of MIT and Harvard, Cambridge, MA; Addgene plasmid #48138). We transfected the plasmids using the transfection reagent XtremeGENE HP (Roche) according to the manufacturer’s protocol. Single GFP-positive cells were sorted into 96-well plates using FACS. The KO cell lines grown from the single cells were identified by Western blot using specific antibodies against PARP1 or Alc1.
Small interfering RNA (siRNA) knockdown
For RNAi-mediated knockdown, we used SilencerSelect Negative Control No. 2 (ref. 4390846) and siRNA against Alc1 (CHD1L; ref. s18358) from Ambion (Thermo Fisher Scientific). Cells grown in normal culture medium were transfected with 500 nM siRNA using Oligofectamine (Life Technologies) according to the manufacturer’s instructions. After 48 h, cells were used for imaging or harvested for protein analysis.
Western blot
Cell lysates were separated using SDS–PAGE, transferred to nitrocellulose membranes (GE Healthcare, Little Chalfont, UK), and blocked in 5% (wt/vol) milk powder in 0.05% (vol/vol) phosphate-buffered saline (PBS)-Tween 20 at room temperature. The primary antibodies were diluted in 5% (wt/vol) milk powder in 0.05% (vol/vol) PBS-Tween 20 and used at the following concentrations: affinity-purified anti-Alc1 rabbit polyclonal, 1:2500; anti-actin (Sigma-Aldrich; A5060), 1:1000; anti-PARP1 rabbit polyclonal, 1:10,000; anti-PARP2 polyclonal rabbit antibody (Active Motif, Carlsbad, CA; #39743), 1:1000; anti-PARP3 polyclonal rabbit (Thermo Fisher Scientific; PA5-21478), 2 μg/ml; and the mouse monoclonal (DM1A) anti-tubulin (Sigma-Aldrich; T9026), 1:20,000. Horseradish peroxidase (HRP)-conjugated secondary antibodies were used to detect primary antibodies. The HRP-conjugated anti-rabbit immunoglobulin G (IgG) and anti-mouse IgG antibodies (BioRad, Hercules, CA) were used at 1:10,000, and the blot was developed using the ELC reagent (Merck Millipore, Billerica, MA).
Immunofluorescence staining
Cells were washed once in PBS and incubated in serum-free DMEM containing either dimethyl sulfoxide, Olaparib (50 μM), or AG14631 (30 μM) for 1 h. Cells were then exposed to H2O2 (0.5 mM) in serum-free DMEM for 10 min and fixed in ice-cold methanol:acetone (1:1) for 10 min. After being washed once with PBS and then blocked for 1 h (5% milk in PBS + 0.05% Tween-20), cells were incubated with anti–poly-ADPr mouse monoclonal 10H antibody (ascites) diluted (1:800) in blocking buffer overnight at 4°C. They were then washed three times with PBS + 0.1% Triton X-100 before being incubated with Alexa Fluor 488 anti-mouse IgG (4 μg/ml; Life Technologies) in blocking buffer for 1 h, after which they were washed three times with PBS + 0.1% Triton X-100, and nuclei were stained using Hoescht (1 μg/ml) for 10 min. Cells were washed twice with PBS + 0.1% Triton X-100 before imaging.
Quantification of cell viability upon X-ray irradiation
Cells were seeded at a density of 500 cells per well in a 12-well plate. Plates were immediately treated with X-ray irradiation (1 or 2 Gy) (Faxitron, Tucson, AZ) and returned to the incubator for 11 d to allow colony formation. Cells were washed once with PBS before being fixed and stained for 30 min with a 4% paraformaldehyde and 0.5% crystal violet solution. Staining solution was removed, and plates were immersed in water to remove excess stain. The colony area percentage was calculated using the ColonyArea plug-in for ImageJ according to Guzmán et al. (2014). Average colony area percentage was normalized to an untreated control.
Live-cell imaging and laser microirradiation
Live-cell imaging was performed on an inverted confocal spinning disk (imaging scan head CSU-X1 from Yokogawa [Tokyo, Japan] and microscope body Ti-E from Nikon [Tokyo, Japan]) equipped with a single-point scanning head to allow laser microirradiation and local photoactivation using a 405 nm laser. We used a Plan APO 63×, oil-immersion objective lens (numerical aperture [N.A.] 1.4) and a sCMOS ORCA Flash 4.0 camera (Hamamatsu, Hamamatsu, Japan) for imaging the cells. The pixel resolution at the object plane was 108 nm. The fluorescence of EGFP and the activated form of PAGFP was excited with a laser at 488 nm, and the fluorescence of mCherry and the activated form of PATagRFP was excited with a laser at 561 nm. For fluorescence detection, we used band-pass filters adapted to the fluorophores. Laser powers were adjusted to minimize bleaching during the time-lapse acquisitions. Photoactivation and DNA damage were induced simultaneously using a 405 nm laser. The power of the 405 nm laser used for photoactivation and, for cells presensitized with Hoechst, induction of DNA lesions, was set to 125 μW at the sample level, unless stated otherwise. Cells were irradiated along a 16-μm-long line crossing the nucleus. The microscope was equipped with a heating chamber to maintain cells at 37°C during the imaging experiments. When long time-lapse experiments of 30–60 min to study chromatin remodeling in response to DNA damage were performed, premature cell death that would indicate a phototoxic effect due to imaging was never observed.
Images shown in Supplemental Figure S1, A and C, were taken on an inverted AxioObserver Z1 confocal spinning-disk microscope (Zeiss, Oberkochen, Germany) equipped with a single-point scanning head for laser microirradiation and local photoactivation using a 405 nm laser (Rapp OptoElectronic). We used a C-Apo 63×, water-immersion objective lens (N.A. 1.2), and the images were acquired on a AxioCam HRm CCD camera (Zeiss). The pixel resolution at the object plane was 171 nm. The fluorescence of EGFP and YFP was excited with a laser at 488 nm, and the fluorescence of the activated form of PATagRFP was excited with a laser at 561 nm. For fluorescence detection, we used band-pass filters adapted to the fluorophores. The microirradiation conditions at 405 nm were adjusted to obtain amplitudes of the chromatin relaxation at DNA lesions that were similar to those obtained with the system described above. A heating chamber was used to maintain the cells at 37°C.
Image analysis
The time-lapse sequences were analyzed automatically using custom-made routines written in Matlab (Mathworks, Natick, MA) to quantify chromatin relaxation at DNA lesions. The chromatin area microirradiated at 405 nm and tagged with photoactivatable H2B was segmented by k-means segmentation. An ellipsoid was fitted to the segmented area, and its minor axis length was used to estimate the width of the microirradiated chromatin area and thus assess changes in the chromatin compaction level.
To analyze the release of the photoactivatable H1 proteins from the area irradiated at 405 nm, we measured the H1 integrated intensity inside the segmented microirradiated chromatin area in cells coexpressing H1 and H2B tagged with two different photoactivatable dyes. This intensity was divided by the H1 intensity integrated over the whole nucleus to correct for bleaching and small focus drifts. For this step, whole nuclei were segmented using the low fluorescence signal coming from the nonactivated tagged H2B proteins. The same approach was used to analyze the release of the H2B proteins from DNA lesions and to characterize Alc1 recruitment kinetics.
When necessary, nuclei movements occurring during the time-lapse experiments were corrected using the ImageJ plug-in StackReg (Thévenaz et al., 1998).
The accumulation of the fluorescently tagged WWE domain of RNF146 at the DNA lesions was quantified as follows. The mean fluorescence intensity in three areas was estimated by manual segmentation: at the site of DNA damage (Id), in a region of the nucleus not subjected to laser irradiation (Ind), and outside the cells (Ibg). The accumulation of the WWE domain at the DNA lesions AWWE was then calculated as
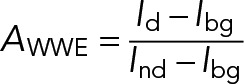 |
For quantifying the immunofluorescence staining with anti-PAR (10H) antibody, the nuclei were segmented using Hoechst staining, and the mean fluorescence intensity for the anti-PAR antibody was measured inside each nucleus after background subtraction.
For chromatin texture analysis, wild-type and Alc1 KO U2OS cells were plated on Lab-Tek II chambered coverglasses, fixed with 4% paraformaldehyde for 10 min at room temperature, and stained with Hoechst 33342 (1 μg/ml) for 1 h. Confocal images were captured on a Leica SP8 confocal microscope using a Plan APO 63×, oil-immersion objective lens (N.A. 1.4). Hoechst staining was excited with a 405-nm laser, and the emission band was chosen to optimize fluorescence collection. The pinhole was set to one Airy unit, and we used a pixel size of 60 nm. The GLCM ImageJ texture plug-in written by Julio E. Cabrera was used to analyze chromatin texture. The correlation and contrast parameters were chosen to characterize chromatin texture using a pixel-to-pixel distance of 7 pixels, which allowed maximizing the differences measured between the cells bathed with the isotonic medium and those subjected to osmotic stress.
In cells labeled with fluorescent nucleotides, the chromatin dynamics was assessed by tracking the fluorescently labeled DNA replication foci using the plug-in Particle Tracker from ImageJ (Sbalzarini and Koumoutsakos, 2005).
Statistics
In the figure legends, n refers to the number of cells used for a given experimental condition. Box plots were generated using a Web tool developed by the Tyers and Rappsilber labs (http://boxplot.tyerslab.com). The box limits correspond to the 25th and 75th percentiles, and the bold line indicates the median value. The whiskers extend 1.5 times the interquartile range, and outliers are shown by dots. The numbers in parentheses refers to the number of cells for each condition. Unless stated otherwise, p values were calculated using an unpaired Student’s t test, assuming unequal variances. On the box plots: *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001; n.s., not significant.
Supplementary Material
Acknowledgments
We thank the Microscopy Rennes Imaging Center (BIOSIT, Université Rennes 1) for technical assistance. This work was supported by the Agence National de la Recherche (JCJC-SVSE2-2011, ChromaTranscript project to S.H.), the Ligue contre le Cancer du Grand-Ouest (commitees 35 and 72 to S.H.), the European Union (FP7-PEOPLE-2011-CIG, ChromaTranscript project to S.H.; and Marie Curie Initial Training Network, Nucleosome4D to A.G.L.), the Deutsche Forschungsgemeinschaft (TI 817/2-1 to G.T.; and SFB collaborative research center 1064 to A.G.L.), and Worldwide Cancer Research (14-1315 to G.T.). H.S.’s PhD fellowship was funded by the Centre National de la Recherche Scientifique and the Région Bretagne. Our collaboration benefited from funding from the Hubert Curien partnership/German Academic Exchange Service–DAAD (28486ZD to S.H.; 55934632 to G.T.) and the Deutsche Forschungsgemeinschaft CIPSM and SyNergy excellence clusters (to A.G.L.).
Abbreviations used:
- GFP
green fluorescent protein
- HRP
horseradish peroxidase
- IgG
immunoglobulin G
- KO
knockout
- PAR
poly(ADP-ribose)
- PARP1
poly(ADP-ribose) polymerase 1
- PBS
phosphate-buffered saline
- RNAi
RNA interference
- siRNA
small interfering RNA
- YFP
yellow fluorescent protein.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E16-05-0269) on October 12, 2016.
REFERENCES
- Ahel D, Horejsí Z, Wiechens N, Polo SE, Garcia-Wilson E, Ahel I, Flynn H, Skehel M, West SC, Jackson SP, et al. Poly(ADP-ribose)-dependent regulation of DNA repair by the chromatin remodeling enzyme ALC1. Science. 2009;325:1240–1243. doi: 10.1126/science.1177321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayrapetov MK, Gursoy-Yuzugullu O, Xu C, Xu Y, Price BD. DNA double-strand breaks promote methylation of histone H3 on lysine 9 and transient formation of repressive chromatin. Proc Natl Acad Sci USA. 2014;111:9169–9174. doi: 10.1073/pnas.1403565111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaudouin J, Mora-Bermúdez F, Klee T, Daigle N, Ellenberg J. Dissecting the contribution of diffusion and interactions to the mobility of nuclear proteins. Biophys J. 2006;90:1878–1894. doi: 10.1529/biophysj.105.071241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess RC, Burman B, Kruhlak MJ, Misteli T. Activation of DNA damage response signaling by condensed chromatin. Cell Rep. 2014;9:1703–1717. doi: 10.1016/j.celrep.2014.10.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng W, Su Y, Xu F. CHD1L: a novel oncogene. Mol Cancer. 2013;12:170. doi: 10.1186/1476-4598-12-170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou DM, Adamson B, Dephoure NE, Tan X, Nottke AC, Hurov KE, Gygi SP, Colaiácovo MP, Elledge SJ. A chromatin localization screen reveals poly (ADP ribose)-regulated recruitment of the repressive polycomb and NuRD complexes to sites of DNA damage. Proc Natl Acad Sci USA. 2010;107:18475–18480. doi: 10.1073/pnas.1012946107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottschalk AJ, Timinszky G, Kong SE, Jin J, Cai Y, Swanson SK, Washburn MP, Florens L, Ladurner AG, Conaway JW, et al. Poly(ADP-ribosyl)ation directs recruitment and activation of an ATP-dependent chromatin remodeler. Proc Natl Acad Sci USA. 2009;106:13770–13774. doi: 10.1073/pnas.0906920106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzmán C, Bagga M, Kaur A, Westermarck J, Abankwa D. ColonyArea: an ImageJ plugin to automatically quantify colony formation in clonogenic assays. PLoS One. 2014;9:e92444. doi: 10.1371/journal.pone.0092444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huletsky A, de Murcia G, Muller S, Hengartner M, Ménard L, Lamarre D, Poirier GG. The effect of poly(ADP-ribosyl)ation on native and H1-depleted chromatin. A role of poly(ADP-ribosyl)ation on core nucleosome structure. J Biol Chem. 1989;264:8878–8886. [PubMed] [Google Scholar]
- Hutchinson JB, Cheema MS, Wang J, Missiaen K, Finn R, Gonzalez Romero R, Th’ng JPH, Hendzel M, Ausió J. Interaction of chromatin with a histone H1 containing swapped N- and C-terminal domains. Biosci Rep. 2015;35:e00209. doi: 10.1042/BSR20150087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju B-G, Lunyak VV, Perissi V, Garcia-Bassets I, Rose DW, Glass CK, Rosenfeld MG. A topoisomerase IIβ-mediated dsDNA break required for regulated transcription. Science. 2006;312:1798–1802. doi: 10.1126/science.1127196. [DOI] [PubMed] [Google Scholar]
- Khurana S, Kruhlak MJ, Kim J, Tran AD, Liu J, Nyswaner K, Shi L, Jailwala P, Sung MH, Hakim O, et al. A macrohistone variant links dynamic chromatin compaction to BRCA1-dependent genome maintenance. Cell Rep. 2014;8:1049–1062. doi: 10.1016/j.celrep.2014.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MY, Mauro S, Gévry N, Lis JT, Kraus WL. NAD+-dependent modulation of chromatin structure and transcription by nucleosome binding properties of PARP-1. Cell. 2004;119:803–814. doi: 10.1016/j.cell.2004.11.002. [DOI] [PubMed] [Google Scholar]
- Kong X, Mohanty SK, Stephens J, Heale JTGomez-Godinez V, Shi LZ, Kim J-S, Yokomori K, Berns MW. Comparative analysis of different laser systems to study cellular responses to DNA damage in mammalian cells. Nucleic Acids Res. 2009;37:e68. doi: 10.1093/nar/gkp221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruhlak MJ, Celeste A, Dellaire G, Fernandez-Capetillo O, Müller WG, McNally JG, Bazett-Jones DP, Nussenzweig A. Changes in chromatin structure and mobility in living cells at sites of DNA double-strand breaks. J Cell Biol. 2006;172:823–834. doi: 10.1083/jcb.200510015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebeaupin T, Sellou H, Timinszky G, Huet S. Chromatin dynamics at DNA breaks: what, how and why. AIMS Biophys. 2015;2:458–475. [Google Scholar]
- Luijsterburg MS, Lindh M, Acs K, Vrouwe MG, Pines A, van Attikum H, Mullenders LH, Dantuma NP. DDB2 promotes chromatin decondensation at UV-induced DNA damage. J Cell Biol. 2012;197:267–281. doi: 10.1083/jcb.201106074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luijsterburg MS, de Krijger I, Wiegant WW, Shah RG, Smeenk G, de Groot AJ, Pines A, Vertegaal AC, Jacobs JJ, Shah GM, et al. PARP1 links CHD2-mediated chromatin expansion and H3.3 deposition to DNA repair by non-homologous end-joining. Mol Cell. 2016;61:547–562. doi: 10.1016/j.molcel.2016.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy Z, Kalousi A, Furst A, Koch M, Fischer B, Soutoglou E. Tankyrases promote homologous recombination and check point activation in response to DSBs. PLoS Genetics. 2016;12:e1005791. doi: 10.1371/journal.pgen.1005791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann B, Walter T, Hériché JK, Bulkescher J, Erfle H, Conrad C, Rogers P, Poser I, Held M, Liebel U, et al. Phenotypic profiling of the human genome by time-lapse microscopy reveals cell division genes. Nature. 2010;464:721–727. doi: 10.1038/nature08869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pines A, Vrouwe MG, Marteijn JA, Typas D, Luijsterburg MS, Cansoy M, Hensbergen P, Deelder A, de Groot A, Matsumoto S, et al. PARP1 promotes nucleotide excision repair through DDB2 stabilization and recruitment of ALC1. J Cell Biol. 2012;199:235–249. doi: 10.1083/jcb.201112132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platani M, Goldberg I, Lamond AI, Swedlow JR. Cajal body dynamics and association with chromatin are ATP-dependent. Nat Cell Biol. 2002;4:502–508. doi: 10.1038/ncb809. [DOI] [PubMed] [Google Scholar]
- Polo SE. Reshaping chromatin after DNA damage: the choreography of histone proteins. J Mol Biol. 2015;427:626–636. doi: 10.1016/j.jmb.2014.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polo SE, Kaidi A, Baskcomb L, Galanty Y, Jackson SP. Regulation of DNA-damage responses and cell-cycle progression by the chromatin remodelling factor CHD4. EMBO J. 2010;29:3130–3139. doi: 10.1038/emboj.2010.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poirier GG, de Murcia G, Jongstra-Bilen J, Niedergang C, Mandel P. Poly(ADP-ribosyl)ation of polynucleosomes causes relaxation of chromatin structure. Proc Natl Acad Sci USA. 1982;79:3423–3427. doi: 10.1073/pnas.79.11.3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouleau M, McDonald D, Gagné P, Ouellet M-E, Droit A, Hunter JM, Dutertre S, Prigent C, Hendzel MJ, Poirier GG. PARP-3 associates with polycomb group bodies and with components of the DNA damage repair machinery. J Cell Biochem. 2007;100:385–401. doi: 10.1002/jcb.21051. [DOI] [PubMed] [Google Scholar]
- Sbalzarini IF, Koumoutsakos P. Feature point tracking and trajectory analysis for video imaging in cell biology. J Struct Biol. 2005;151:182–195. doi: 10.1016/j.jsb.2005.06.002. [DOI] [PubMed] [Google Scholar]
- Schermelleh L, Solovei I, Zink D, Cremer T. Two-color fluorescence labeling of early and mid-to-late replicating chromatin in living cells. Chromosome Res. 2001;9:77–80. doi: 10.1023/a:1026799818566. [DOI] [PubMed] [Google Scholar]
- Schuster-Böckler B, Lehner B. Chromatin organization is a major influence on regional mutation rates in human cancer cells. Nature. 2012;488:504–507. doi: 10.1038/nature11273. [DOI] [PubMed] [Google Scholar]
- Smeenk G, Wiegant WW, Marteijn JA, Luijsterburg MS, Sroczynski N, Costelloe T, Romeijn RJ, Pastink A, Mailand N, Vermeulen W, et al. Poly(ADP-ribosyl)ation links the chromatin remodeler SMARCA5/SNF2H to RNF168-dependent DNA damage signaling. J Cell Sci. 2013;126:889–903. doi: 10.1242/jcs.109413. [DOI] [PubMed] [Google Scholar]
- Smerdon MJ, Lieberman MW. Nucleosome rearrangement in human chromatin during UV-induced DNA-repair synthesis. Proc Natl Acad Sci USA. 1978;75:4238–4241. doi: 10.1073/pnas.75.9.4238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strickfaden H, McDonald D, Kruhlak MJ, Haince JF, Th’ng JP, Rouleau M, Ishibashi T, Corry GN, Ausio J, Underhill DA, et al. Poly(ADP-ribosyl)ation-dependent transient chromatin decondensation and histone displacement following laser micro-irradiation. J Biol Chem. 2016;291:1789-802. doi: 10.1074/jbc.M115.694992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subach FV, Patterson GH, Renz M, Lippincott-Schwartz J, Verkhusha VV. Bright monomeric photoactivatable red fluorescent protein for two-color super-resolution sptPALM of live cells. J Am Chem Soc. 2010;132:6481–6491. doi: 10.1021/ja100906g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thévenaz P, Ruttimann UE, Unser M. A pyramid approach to subpixel registration based on intensity. IEEE Trans Image Process. 1998;7:27–41. doi: 10.1109/83.650848. [DOI] [PubMed] [Google Scholar]
- Thoma F, Koller T, Klug A. Involvement of histone H1 in the organization of the nucleosome and of the salt-dependent superstructures of chromatin. J Cell Biol. 1979;83:403–427. doi: 10.1083/jcb.83.2.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timinszky G, Till S, Hassa PO, Hothorn M, Kustatscher G, Nijmeijer B, Colombelli J, Altmeyer M, Stelzer EH, Scheffzek K, et al. A macrodomain-containing histone rearranges chromatin upon sensing PARP1 activation. Nat Struct Mol Biol. 2009;16:923–929. doi: 10.1038/nsmb.1664. [DOI] [PubMed] [Google Scholar]
- Walter A, Chapuis C, Huet S, Ellenberg J. Crowded chromatin is not sufficient for heterochromatin formation and not required for its maintenance. J Struct Biol. 2013;184:445–53. doi: 10.1016/j.jsb.2013.10.004. [DOI] [PubMed] [Google Scholar]
- Wang Z, Michaud GA, Cheng Z, Zhang Y, Hinds TR, Fan E, Cong F, Xu W. Recognition of the iso-ADP-ribose moiety in poly(ADP-ribose) by WWE domains suggests a general mechanism for poly(ADP-ribosyl)ation-dependent ubiquitination. Genes Dev. 2012;26:235–240. doi: 10.1101/gad.182618.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodhouse BC, Dianov GL. Poly ADP-ribose polymerase-1: an international molecule of mystery. DNA Repair (Amst) 2008;7:1077–1086. doi: 10.1016/j.dnarep.2008.03.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.