

The GOLPH3 oncogene functions in Golgi trafficking. GOLPH3 promotes cell migration, which is important in cancer. GOLPH3, by linking the Golgi to F-actin, promotes both Golgi reorientation and forward trafficking, which together drive trafficking to the leading edge. These findings provide insight into how GOLPH3 drives cell migration.
Abstract
The mechanism of directional cell migration remains an important problem, with relevance to cancer invasion and metastasis. GOLPH3 is a common oncogenic driver of human cancers, and is the first oncogene that functions at the Golgi in trafficking to the plasma membrane. Overexpression of GOLPH3 is reported to drive enhanced cell migration. Here we show that the phosphatidylinositol-4-phosphate/GOLPH3/myosin 18A/F-actin pathway that is critical for Golgi–to–plasma membrane trafficking is necessary and limiting for directional cell migration. By linking the Golgi to the actin cytoskeleton, GOLPH3 promotes reorientation of the Golgi toward the leading edge. GOLPH3 also promotes reorientation of lysosomes (but not other organelles) toward the leading edge. However, lysosome function is dispensable for migration and the GOLPH3 dependence of lysosome movement is indirect, via GOLPH3’s effect on the Golgi. By driving reorientation of the Golgi to the leading edge and driving forward trafficking, particularly to the leading edge, overexpression of GOLPH3 drives trafficking to the leading edge of the cell, which is functionally important for directional cell migration. Our identification of a novel pathway for Golgi reorientation controlled by GOLPH3 provides new insight into the mechanism of directional cell migration with important implications for understanding GOLPH3’s role in cancer.
INTRODUCTION
Cell migration is critical to a range of normal biological processes during development and for adaptive and regenerative changes in adult organisms (Locascio and Nieto, 2001; Friedl and Gilmour, 2009). Importantly, cell migration is also at the heart of the pathophysiology of cell invasion and metastasis that render cancers lethal (Friedl and Wolf, 2003). Understanding the cellular mechanisms of cell migration, in particular the components that are limiting and thus susceptible to pathophysiological enhancement and therapeutic intervention, remains an important biological problem. Directional cell migration involves reorganization of the actin cytoskeleton, for example, at lamellipodia at the leading edge of the cell (Insall and Machesky, 2009; Ridley, 2011; Krause and Gautreau, 2014). Interestingly, directional cell migration also involves reorientation of the Golgi toward the leading edge (Kupfer et al., 1982; Millarte and Farhan, 2012). However, the link between actin-mediated migration and Golgi reorientation remains poorly understood.
We previously identified GOLPH3 as an effector of phosphatidylinositol-4-phosphate (PtdIns(4)P) that bridges the Golgi to the actin cytoskeleton and is required for efficient Golgi–to–plasma membrane trafficking (Dippold et al., 2009; Bishé et al., 2012; Ng et al., 2013; Farber-Katz et al., 2014; Buschman et al., 2015b). We showed that GOLPH3 binds to PtdIns(4)P and thus localizes to the PtdIns(4)P-rich trans-Golgi. GOLPH3 also binds to the unconventional myosin, myosin 18A (MYO18A), thus linking the Golgi to F-actin (Dippold et al., 2009; Bishé et al., 2012; Ng et al., 2013; Taft et al., 2013; Farber-Katz et al., 2014). The PtdIns(4)P/GOLPH3/MYO18A/F-actin complex applies a tensile force to Golgi membranes that is essential for efficient vesicle exit for transport to the plasma membrane. Surprisingly, GOLPH3 was discovered to be an oncogene, commonly overexpressed in human cancers (Scott et al., 2009; Buschman et al., 2015a). Thus GOLPH3 is the first example of an oncogene that functions in the secretory pathway at the Golgi, raising hope that it will provide fresh insights into mechanisms of oncogenesis.
Overexpression of GOLPH3 has been reported to enhance cell migration, with obvious implications for understanding GOLPH3’s role in driving cancer mortality that results from cancer invasion and metastasis (Zhou et al., 2013; Isaji et al., 2014; Tokuda et al., 2014; Buschman et al., 2015a; Zhang et al., 2015). Here we examine the consequence of overexpression of GOLPH3, and investigate the mechanism of enhanced cell migration. We demonstrate that the PtdIns(4)P/GOLPH3/MYO18A/F-actin pathway is required for cell migration by enabling reorientation of the Golgi toward the wound edge for directional secretion. Moreover, overexpression of GOLPH3, as observed in many human cancers, is sufficient to drive this pathway to produce increased cell migration. Our study of the GOLPH3 pathway sheds light on the cellular basis of cell migration, providing new insight into cancer biology and potential targets for therapeutic intervention.
RESULTS
GOLPH3 overexpression promotes scratch assay wound healing
To test the consequences of overexpression of GOLPH3, we generated a bicistronic retroviral expression vector to overexpress untagged GOLPH3 together with green fluorescent protein (GFP) or a control vector to express GFP alone. We used these vectors to produce pools of MDA-MB-231 human breast cancer cells that stably overexpress GOLPH3 plus GFP or GFP alone. Overexpression of GOLPH3 was validated by Western blot and immunofluorescence (IF; Figure 1, A and B).
FIGURE 1:

GOLPH3 overexpression promotes scratch wound healing. (A) Western blot of duplicate SDS lysates demonstrates increased expression of GOLPH3 (wild-type [WT] or R90L mutant) compared with parental MDA-MB-231 cells or IRES-GFP control cells. GAPDH provides a control to demonstrate similar loading. Dashed lines indicate omission of irrelevant intervening lanes. (B) IF demonstrates endogenous GOLPH3 (green) in parental and IRES-GFP control cells, increased GOLPH3 at the Golgi in cells overexpressing WT GOLPH3, and increased GOLPH3 throughout the cytosol in cells overexpressing GOLPH3-R90L, a mutant unable to bind to PtdIns(4)P. GM130 marks the Golgi (red) and DAPI marks the nucleus (blue). (C) Representative images of scratch wound healing by MDA-MB-231 cells overexpressing IRES-GFP (control), GOLPH3-IRES-GFP, or GOLPH3-R90L-IRES-GFP. Top, images of random fields at the scratch were taken at time of wounding (t = 0 h), with the scratch area indicated by the white box. Bottom, the same fields after 15 h, fixed and stained with DAPI for cell counting (t = 15 h). (D) Quantification of wound healing from C relative to control. Overexpression of GOLPH3 results in a significant, approximately twofold increase in cell migration into the scratch compared with control or GOLPH3-R90L–expressing cells. Graphed are mean ± SEM. The number of fields measured (n, pooled from four independent experiments) and p values (t test with Holm-Bonferroni correction) are indicated.
GOLPH3 overexpression has been reported to drive increased wound healing, as observed in cell culture scratch assays (Isaji et al., 2014; Tokuda et al., 2014). We sought to recapitulate the published result. We examined wound healing by creating a scratch in a confluent monolayer of MDA-MB-231 cells. First, we used live imaging of cells stained with calcein AM to examine cell migration. This revealed enhanced cell migration into the wound in cells that overexpress GOLPH3 (Supplemental Movie 1). Next we performed endpoint assays to measure the number of cells that migrated into the monolayer wound after 15 h. As shown in the images (Figure 1C) and quantification (Figure 1D), enhanced migration into the wound was observed in cells that overexpress GOLPH3, recapitulating the published reports.
Tokuda et al. (2014) showed that Golgi PtdIns(4)P promotes cell migration via GOLPH3. Similarly, to determine whether the ability of GOLPH3 to drive increased wound healing depends on its function at the Golgi, we made use of a previously described mutant. The R90L mutation in the PtdIns(4)P binding pocket largely abolishes the ability of GOLPH3 to bind to PtdIns(4)P, thus rendering GOLPH3-R90L unable to localize to the Golgi (Dippold et al., 2009). We generated a bicistronic retroviral vector to express GOLPH3-R90L together with GFP and produced pools of MDA-MB-231 cells that stably overexpress GOLPH3-R90L (Figure 1, A and B). When compared in cell culture scratch assays, these cells behave in a manner similar to GFP-expressing control cells, unlike GOLPH3-overexpressing cells (Figure 1, C and D). Therefore we conclude that the ability of GOLPH3 to drive cell culture wound healing is dependent on its ability to localize to the Golgi.
GOLPH3 and MYO18A are required for scratch assay wound healing
Because GOLPH3 is capable of driving enhanced wound healing in scratch assays, we wondered whether it is generally required for scratch wound healing. Specific small interfering RNA (siRNA) to knock down GOLPH3 was compared with a control siRNA. As shown in Figure 2A, knockdown of GOLPH3 significantly impaired scratch assay wound healing by MDA-MB-231 cells. A variety of roles have been proposed for GOLPH3 (Rizzo et al., 2016). To test whether the requirement for GOLPH3 is due to its function in the PtdIns(4)P/GOLPH3/MYO18A/F-actin pathway, we examined the effect of siRNA knockdown of MYO18A. We observed that MYO18A knockdown also significantly impaired wound healing by MDA-MB-231 cells. To determine whether the requirement for GOLPH3 and MYO18A is unique to MDA-MB-231 cells or is more generally true, we also examined wound healing in another, unrelated cell type, NRK (normal rat kidney) cells. Again, GOLPH3 and MYO18A were each required for scratch assay wound healing (Figure 2B). Thus we conclude that the PtdIns(4)P/GOLPH3/MYO18A/F-actin pathway is generally required for scratch assay wound healing.
FIGURE 2:
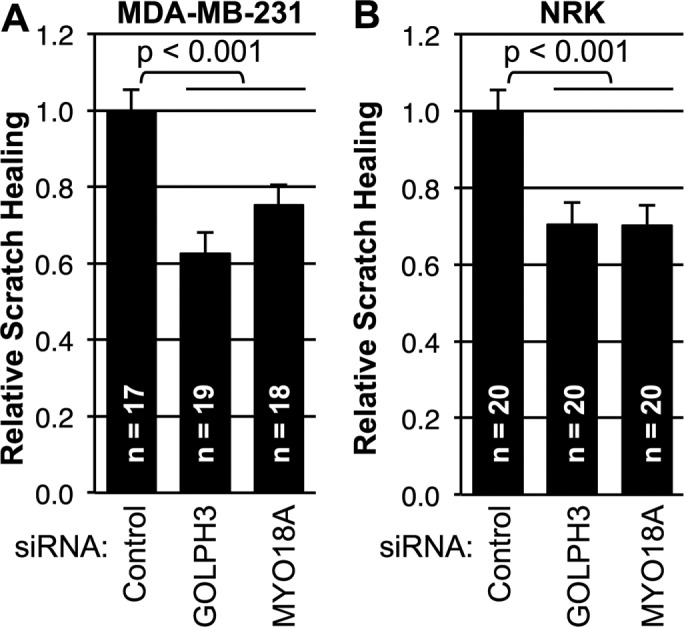
GOLPH3 and MYO18A are required for scratch wound healing. (A, B) Quantification of scratch assay wound healing by MDA-MB-231 cells and NRK cells, respectively. Cells were transfected with control siRNA or siRNA targeting GOLPH3 or MYO18A before monolayer wounding. Effectiveness of knockdown was confirmed by parallel Western blots (not shown; see Supplemental Figure 1 for representative examples). Scratch wound healing is expressed relative to control. Interference with the GOLPH3/MYO18A pathway significantly impairs wound healing in both MDA-MB-231 and NRK cells. Graphed are mean ± SEM pooled from two independent experiments. Number of fields measured (n) and p values (t test with Holm-Bonferroni correction) are indicated.
GOLPH3 does not affect cell proliferation, sensing of loss of contact, or known polarization pathways, but drives cell migration speed
To determine the mechanism by which the GOLPH3 pathway contributes to enhanced cell migration, we considered a range of possibilities. We first examined whether overexpression of GOLPH3 led to increased cell proliferation. We compared the rate of proliferation of GOLPH3 overexpressing MDA-MB-231 cells with the parental, GFP only, and GOLPH3-R90L controls. We found that all four proliferated at essentially identical rates (Figure 3, A and B).
FIGURE 3:

GOLPH3 does not affect MDA-MB-231 proliferation or sensing of loss of contact but enhances wound healing independently from centrosomes or Cdc42 by driving cell migration speed. (A) Rate of proliferation of cell lines was measured by counting on days 1, 3, and 5 following initial seeding of equal numbers of cells and expressed as the binary logarithm of cell count normalized to day 1. (B) Doubling time for each cell line was calculated from (A) and shown in corresponding color. GOLPH3 overexpression has no significant effect on cell proliferation rate. (A, B) Graphs indicate mean ± SEM. For each data point, n = 3. (C) MDA-MB-231 cells overexpressing GFP (control), GOLPH3, or GOLPH3-R90L were grown to confluence and transfected with control siRNA or MYO18A siRNA before wounding. Subcellular YAP distribution of confluent cells and cells at wound edge was measured as the ratio of nuclear YAP vs. cytoplasmic YAP. As expected, monolayer wounding caused loss of confluence and YAP translocation to the nucleus. However, neither GOLPH3 overexpression nor MYO18A knockdown had any effect on YAP distribution. Graphed are mean ± SEM pooled from two independent experiments. Numbers of cells measured (n) and p values (t test) are indicated. MYO18A knockdown was confirmed by parallel Western blots (unpublished data; comparable to Supplemental Figure 1). (D, E) Quantification of wound healing from Supplemental Figure 2, C and D, respectively, relative to control. Graphed are mean ± SEM. The number of fields measured (n, pooled from two independent experiments) and p values (t test with Holm-Bonferroni correction) are indicated. (F) Speed of actively migrating MDA-MB-231 cells overexpressing GFP (control) or GOLPH3 were measured from time-lapse images following wounding (Supplemental Movie 1). Overexpression of GOLPH3 enhances cell migration. Graphed are mean ± SEM, with number of cells (n) and p value (t test) indicated.
Next we investigated whether GOLPH3 affects cell migration by modulating cells’ sensing of loss of contact upon monolayer wounding. Cell confluence is known to regulate the subcellular localization of the transcriptional coactivator YAP between the nucleus and cytoplasm via the Hippo pathway (Zhao et al., 2007). Therefore we could use YAP subcellular localization as a readout of Hippo pathway activity and thus a readout of cellular sensing of confluence. We measured the relative levels of YAP in the nucleus versus cytoplasm using IF in confluent cells compared with cells at the wound edge. We compared control cells with cells overexpressing GOLPH3 and also examined cells following siRNA knockdown of MYO18A versus siRNA control. As expected, we observed significant translocation of YAP from the cytoplasm into the nucleus in cells at the wound edge compared with confluent cells (Figure 3C). However, overexpression of GOLPH3 or knockdown of MYO18A did not significantly alter the subcellular localization of YAP. We conclude that perturbation of the GOLPH3 pathway does not significantly affect the ability of MDA-MB-231 cells to sense a change in cell–cell contact upon wounding.
We considered the possibility that GOLPH3 overexpression may enhance cell migration via pathways known to contribute to cellular polarization in response to cell wounding. The centrosome responds to cell wounding by repositioning toward the leading edge (Kupfer et al., 1982). Signals responsible for cell polarization and directional migration have been reported to act via centrosomes (Etienne-Manneville and Hall, 2001). To test whether enhanced cell migration due to GOLPH3 overexpression depends on centrosomes, we generated cells deficient in centrosomes using centrinone, a Plk4-specific inhibitor (Wong et al., 2015). We grew MDA-MB-231 cells in the presence of 100 nM centrinone for more than 2 wk, resulting in nearly complete loss of centrosomes (Supplemental Figure 2, A and B). We observed scratch wound healing in vehicle (dimethyl sulfoxide [DMSO]) and centrinone-treated cells (Supplemental Figure 2C). Loss of centrosomes resulted in significantly impaired scratch wound healing (Figure 3D). However, overexpression of GOLPH3 still enhanced scratch wound healing in centrosome-deficient cells. We conclude that, while centrosomes contribute to cell migration, GOLPH3’s ability to drive cell migration occurs independent of centrosomes.
Cdc42 has been proposed to be a key signaling molecule that drives cell polarization and cell migration (Nobes and Hall, 1999; Etienne-Manneville and Hall, 2001), although with some disagreement between experiments using dominant-negative, knockout, and small molecule inhibitors in different cell types and culture conditions (Nobes and Hall, 1999; Etienne-Manneville and Hall, 2001; Cau and Hall, 2005; Czuchra et al., 2005; Kiwanuka et al., 2016). We tested the requirement for Cdc42 for GOLPH3-driven cell migration using the specific Cdc42 inhibitor, ML141 (Hong et al., 2013; Kiwanuka et al., 2016). We performed scratch wound healing assays in control (DMSO) or ML141-treated cells (Figure 3E and Supplemental Figure 2D). Inhibition of Cdc42 by ML141 resulted in significant impairment of scratch wound healing. However, overexpression of GOLPH3 still enhanced cell migration, despite inhibition of Cdc42. We conclude that, while Cdc42 contributes to cell migration, GOLPH3’s ability to drive cell migration occurs independently of Cdc42.
Next we considered the possibility that overexpression of GOLPH3 might enhance wound healing by increasing the speed of cell migration. Using time-lapse imaging, we measured the traveling speed of actively migrating cells following wounding. We observed that overexpression of GOLPH3 resulted in a significant increase in the speed of cell migration (Figure 3F).
GOLPH3 overexpression promotes Golgi reorientation in wound healing
Reorientation of the Golgi toward the leading edge is known to be an early, limiting step in cell migration (Kupfer et al., 1982; Nobes and Hall, 1999; Millarte and Farhan, 2012). Because GOLPH3 serves to bridge the Golgi and the actin cytoskeleton, we wondered whether GOLPH3 plays a role in cell migration by facilitating Golgi reorientation in wound healing. Thus we examined Golgi repositioning in MDA-MB-231 cells that overexpress GOLPH3 compared with control cells after monolayer wounding. In control cells, we observed that the Golgi had a random orientation before wounding. At the time of wounding, the Golgi began to polarize toward the wound edge in cells at the edge of the wound and by 5 h was generally pointing toward the wound (Figure 4A). By comparison, cells overexpressing GOLPH3 exhibited a more rapid and robust reorientation of the Golgi toward the wound edge. To quantify Golgi reorientation, we measured for each cell the angle between a line from the nucleus to the wound edge and from the nucleus to the center of the Golgi (Figure 4B). Quantification showed Golgi reorientation toward the wound edge upon wounding is significantly enhanced by overexpression of GOLPH3 (Figure 4C).
FIGURE 4:

GOLPH3 overexpression promotes Golgi reorientation toward the wound edge. (A) Representative IF images of confluent monolayer of MDA-MB-231 cells overexpressing GFP (control), GOLPH3, or GOLPH3-R90L before and 1, 2, and 5 h after wounding are shown, with the Golgi marked by GM130 (red) and the nucleus by DAPI (blue). (B) Illustration of measurement of Golgi orientation angle. For each cell, a first line was drawn from the center of the nucleus to the wound edge and a second line was drawn from the center of the nucleus to the center of the Golgi. The angle formed between the lines was measured. (C) Quantification of Golgi orientation angles from A. The Golgi polarizes toward the wound edge upon monolayer wounding. GOLPH3 overexpression significantly enhances Golgi reorientation. Graphed are mean ± SEM pooled from two independent experiments, with p values (ANOVA with Holm-Bonferroni correction) indicated. (D) Representative IF images of MDA-MB-231 cells overexpressing GFP, GOLPH3, or GOLPH3-R90L 5 h after wounding are shown. Each cell line was transfected with control siRNA or MYO18A siRNA before monolayer wounding. (E) Quantification of Golgi orientation angles from D. GOLPH3-driven enhancement on Golgi reorientation toward the leading edge is abrogated by MYO18A depletion. Graphed are mean ± SEM pooled from two independent experiments. Numbers of cells measured (n) and p values (t test with Holm-Bonferroni correction) are indicated.
To determine the mechanism by which overexpression of GOLPH3 drives reorientation of the Golgi, we first examined whether it is dependent on centrosomes or Cdc42, both of which play a role in generating cell polarity (Kupfer et al., 1982; Nobes and Hall, 1999; Etienne-Manneville and Hall, 2001). Experiments in centrosome-deficient cells (Supplemental Figure 3, A and B) and cells with inhibition of Cdc42 by ML141 (Supplemental Figure 3, C and D) indicate that GOLPH3’s ability to drive Golgi reorientation occurs independent of centrosomes and Cdc42. Next we asked whether GOLPH3 acts through the PtdIns(4)P/GOLPH3/MYO18A/F-actin pathway. To test whether GOLPH3 requires binding to PtdIns(4)P to act, we examined Golgi reorientation in cells overexpressing GOLPH3-R90L. We noted that they behaved in a manner similar to the control cells (Figure 4, A and C), indicating that GOLPH3-driven enhancement of Golgi repositioning depends on GOLPH3’s interaction with PtdIns(4)P. We also examined the requirement for MYO18A in Golgi reorientation by siRNA knockdown of MYO18A in the MDA-MB-231 cell lines before wounding. As shown in Figure 4, D and E, we recapitulated the enhancement of Golgi reorientation due to overexpression of GOLPH3 in cells transfected with control siRNA. However, siRNA depletion of MYO18A significantly impaired Golgi polarization toward the wound and eliminated any effect due to overexpression of GOLPH3. Because the ability of overexpressed GOLPH3 to promote Golgi reorientation in wound healing depends on its ability to bind to PtdIns(4)P and on MYO18A, we conclude that it acts through the PtdIns(4)P/GOLPH3/MYO18A/F-actin pathway.
GOLPH3 and MYO18A are required for Golgi reorientation in wound healing
Having established that overexpression of GOLPH3 is able to drive Golgi reorientation upon wounding, we wondered whether the PtdIns(4)P/GOLPH3/MYO18A/F-actin pathway is generally required for Golgi reorientation. We depleted GOLPH3 and MYO18A in MDA-MB-231 cells using three different siRNA oligos for each (Supplemental Figure 1A). In control siRNA-transfected cells, the Golgi was oriented randomly before wounding and polarized toward the wound edge within 5 h after monolayer wounding, as expected (Figure 5, A and B). However, Golgi reorientation was significantly impaired upon knockdown of either GOLPH3 or MYO18A. To determine whether the requirement for GOLPH3 and MYO18A for Golgi reorientation in wound healing is also true in other cell types, we performed a similar experiment in NRK cells. Here, again, we observed impaired Golgi reorientation after knockdown of either GOLPH3 or MYO18A (Supplemental Figure 1B and Figure 5, C and D). Thus we conclude that the PtdIns(4)P/GOLPH3/MYO18A/F-actin pathway is generally required for efficient Golgi polarization toward the wound edge in response to monolayer wounding.
FIGURE 5:

GOLPH3 and MYO18A are required for normal Golgi reorientation toward the wound edge. (A) MDA-MB-231 cells and (C) NRK cells were transfected with control siRNA, or siRNA oligos targeting GOLPH3 or MYO18A before wounding. Representative IF images of cells before and 5 h after wounding are shown, with the Golgi marked by GM130 (green) and the nucleus marked by DAPI (blue). (B, D) Quantification of Golgi orientation angles from A and C, respectively. The Golgi had a random orientation before wounding and polarized toward the leading edge after wounding. Golgi reorientation is significantly impaired if either GOLPH3 or MYO18A is depleted with different siRNA oligos. Graphed are mean ± SEM. The numbers of cells measured (n) and p values (t test with Holm-Bonferroni correction) are indicated.
Lysosomes reorient with the Golgi in wound healing
To know whether GOLPH3’s effect is restricted to the Golgi or also affects other organelles, we examined the movement of the Golgi and other cellular organelles upon wounding using time-lapse imaging of live cells. We first compared movement of mitochondria with movement of the Golgi (Supplemental Movie 2). At the time of wounding, we observed general rotation of the whole cell, and the movement of mitochondria reflected that rotation. In comparison, the Golgi actively moved around the nucleus toward the wound edge at a faster speed, resulting in faster movement of the Golgi relative to mitochondria and the rest of the cell. To assess the effect of overexpression of GOLPH3 on mitochondrial (vs. Golgi) reorientation, we measured the orientation of mitochondria and the Golgi relative to the direction of the monolayer wound 5 h after wounding (Figure 6A). We found that, while Golgi reorientation was significantly enhanced by overexpression of GOLPH3, reorientation of mitochondria was insensitive to overexpression of GOLPH3 (Figure 6B). Thus the movement of mitochondria and the Golgi do not share the same mechanism.
FIGURE 6:

Comparison of Golgi to mitochondria, nucleus, and lysosomes in response to monolayer wounding. (A) IF example of cell at wound edge with mitochondria marked by ATP synthase (green), Golgi marked by GOLPH3 (red), and the nucleus marked by DAPI (blue). (B) Quantification of Golgi and mitochondria orientation angles from MDA-MB-231 cells overexpressing GFP (control), GOLPH3, or GOLPH3-R90L. Orientation of mitochondria is insensitive to GOLPH3 overexpression, whereas Golgi reorientation is driven by GOLPH3 overexpression. (C) Measurement of Golgi and nuclear rotation speed upon wounding. The Golgi rotates significantly faster than the nucleus. Data are pooled from 10 movies (e.g., Supplemental Movie 3) with p value indicated (t test). (D) Representative IF images of cells at wound edge with Golgi marked by GM130 (green), lysosomes marked by LAMP1 (red), and the nucleus marked by DAPI (blue). (E) Quantification of Golgi and lysosome orientation angles from D, showing GOLPH3-driven reorientation upon wounding for both organelles. (B, E) Mean ± SEM pooled from two independent experiments with number of cells (n) and p values (t test with Holm-Bonferroni correction) indicated.
Next we compared movement of the nucleus with movement of the Golgi after cell monolayer wounding. As shown in Supplemental Movie 3, structural features of the nucleus visible by differential interference contrast (DIC) imaging allowed us to detect rotation of the nucleus. We observed general rotation of the whole cell that was accompanied by rotation of the nucleus. The Golgi, on the other hand, moved at a greater speed around the nucleus toward the wound edge. By measuring the rotation speed of the Golgi and the nucleus, we noted that the Golgi exhibited a significantly higher rotation speed compared with the nucleus (Figure 6C). Thus we demonstrate that the nucleus, too, moves differently from the Golgi in response to wounding.
Finally, we examined lysosome movement in response to wounding. Unexpectedly, as shown in Supplemental Movie 4, the Golgi and lysosomes localized close to each other and reoriented in a concerted manner upon wounding. By 5 h, both the Golgi and lysosomes were pointed toward the wound edge and remained colocalized (Figure 6D). Quantification showed that lysosome reorientation was significantly enhanced by overexpression of wild-type GOLPH3, but not GOLPH3-R90L, similar to the effect on reorientation of the Golgi (Figure 6E). Moreover, GOLPH3’s ability to enhance both Golgi and lysosome reorientation is through the PtdIns(4)P/GOLPH3/MYO18A/F-actin pathway, because, in addition to GOLPH3-R90L being ineffective, either siRNA depletion of MYO18A or depolymerization of F-actin by latrunculin B significantly impaired reorientation and abolished the ability of GOLPH3 to promote reorientation of both lysosomes and the Golgi (Figure 7, A–D).
FIGURE 7:

GOLPH3 drives Golgi and lysosome reorientation via the GOLPH3/MYO18A/F-actin pathway. (A) Representative IF images of MDA-MB-231 cells overexpressing GFP (control), GOLPH3, or GOLPH3-R90L at wound edge with Golgi labeled in green, lysosome in red, and the nucleus in blue. Cells were transfected with control siRNA or MYO18A siRNA for 48 h before monolayer wounding and fixed 5 h after wounding. (B) Quantification of Golgi and lysosome orientation angles from A. Depletion of MYO18A significantly impairs Golgi and lysosome reorientation, rendering them insensitive to overexpression of GOLPH3. (C) Representative IF images of MDA-MB-231 cells at wound edge treated with DMSO (vehicle control) or 250 nM latrunculin B at time of monolayer wounding and fixed 5 h after wounding. (D) Quantification of Golgi and lysosome orientation angles from C. Depolymerizing F-actin significantly impairs Golgi and lysosome reorientation, rendering them insensitive to overexpression of GOLPH3. (E) Representative IF images of NRK cells at wound edge, transfected with control siRNA or siRNA targeting GOLPH3 or MYO18A 48 h before wounding and fixed 5 h after wounding. (F) Quantification of Golgi and lysosome orientation angles from E. Golgi and lysosome reorientation toward the leading edge upon wounding is significantly impaired by interference with GOLPH3 or MYO18A. (B, D, F) Graphed are mean ± SEM pooled from two independent experiments, with numbers of cells measured (n) and p values (t test with Holm-Bonferroni correction) indicated.
We also tested whether GOLPH3 and MYO18A are required for lysosome reorientation in response to a monolayer scratch. In NRK cells, knockdown of either GOLPH3 or MYO18A not only impaired Golgi reorientation, as observed previously, but also significantly impaired lysosome reorientation (Figure 7, E and F). In HeLa cells, we observed a similar requirement for both GOLPH3 and MYO18A for reorientation of both the Golgi and lysosomes following cell culture wounding (Supplemental Figure 4, A and B). Taking all of our results together, we come to the surprising conclusion that lysosomes move with the Golgi to polarize toward the leading edge in response to wounding, which is dependent on, and also driven by, the PtdIns(4)P/GOLPH3/MYO18A/F-actin pathway.
Lysosome reorientation is indirect, through microtubule tethering to the Golgi
The question follows how GOLPH3, a protein found at the Golgi, regulates lysosome reorientation in response to wounding. As published previously (Wu et al., 2000; Bell et al., 2001; Dippold et al., 2009; Ng et al., 2013; Farber-Katz et al., 2014), we confirmed that GOLPH3 localizes to the Golgi, not lysosomes (Figure 8A). Thus we hypothesized that GOLPH3’s effect on lysosomes may be indirect, via the Golgi. To test this possibility, we first determined whether lysosome reorientation depends on the Golgi. We treated MDA-MB-231 cells with brefeldin A (BFA) to disrupt the Golgi or methanol (vehicle control) at the time of monolayer wounding and measured lysosome reorientation after 5 h (Figure 8, B and C). In control cells, we recapitulated the enhanced reorientation of both the Golgi and lysosomes in response to overexpression of GOLPH3. In BFA-treated cells, the Golgi was effectively disrupted within 15 min of drug treatment (Figure 8B; unpublished data). Consequently, lysosome reorientation was significantly impaired. Moreover, the ability of overexpression of GOLPH3 to augment lysosome reorientation was abolished by BFA disruption of the Golgi. We conclude that GOLPH3 acts on lysosomes indirectly through the Golgi.
FIGURE 8:

GOLPH3 acts indirectly on lysosome reorientation via the Golgi. (A) Representative IF image of cell stained for GOLPH3 (green), with Golgi marked by GM130 (magenta), lysosomes labeled with LysoTracker (red), and the nucleus marked by DAPI (blue). The staining pattern of GOLPH3 closely mimics GM130, but is distinct from LysoTracker. (B) Representative IF images of cells at wound edge treated with MeOH (vehicle) or BFA at time of wounding, with Golgi labeled in green, lysosome in red, and nucleus in blue. (C) Quantification of lysosome orientation angles from B. Lysosome reorientation toward the wound is significantly impaired by disruption of the Golgi. Likewise, the ability of overexpressed GOLPH3 to accelerate lysosome reorientation is abolished by BFA treatment. Graphed are mean ± SEM pooled from two independent experiments. Numbers of cells measured (n) and p values (t test with Holm-Bonferroni correction) are indicated. (D) Measurement of overlap of the Golgi labeled by ManII-GFP and lysosome labeled by LysoTracker from time-lapse imaging upon treatment with latrunculin B to depolymerize F-actin (see Supplemental Movie 5) or nocodazole to depolymerize microtubules (see Supplemental Movie 6). The overlap between the Golgi and lysosome compartments remains intact with latrunculin B treatment, but significantly decreases with nocodazole treatment. Graphed are mean ± SEM, and p values (two-way ANOVA with Holm-Bonferroni correction) are indicated.
Given the close association between the Golgi and lysosomes (Supplemental Movie 4), we reasoned that a possible explanation for the GOLPH3-driven, Golgi-dependent reorientation of lysosomes could be due to structural tethering of lysosomes to the Golgi. Top candidates that could mediate this tethering would be actin or microtubule cytoskeletal elements. Thus we first investigated whether F-actin is responsible for the link between the Golgi and lysosomes. We depolymerized actin with latrunculin B, and examined the effect on the relationship between the Golgi and lysosomes (Supplemental Movie 5). Treatment with latrunculin B led to condensation of the Golgi, consistent with previous observations (Dippold et al., 2009). Lysosomes condensed simultaneously with the Golgi, and the two organelles remained similarly colocalized after drug treatment (Figure 8D), suggesting that F-actin is not required to link lysosomes to the Golgi.
Next we investigated the role of microtubules in tethering lysosomes to the Golgi. Published data indicate that lysosomes are associated with and depend on microtubules to maintain normal morphology (Collot et al., 1984; Matteoni and Kreis, 1987; Scheel et al., 1990; Harada et al., 1998). The Golgi also requires microtubules to maintain normal ribbon morphology (Rogalski and Singer, 1984; Sandoval et al., 1984; Thyberg and Moskalewski, 1999) and is known to be able to initiate de novo microtubule polymerization (Chabin-Brion et al., 2001; Efimov et al., 2007; Rivero et al., 2009; Petry and Vale, 2015). However, little is known about the structural relationship between the two organelles. To examine a possible physical association between the Golgi and lysosomes mediated by microtubules, we observed the consequences of depolymerization of microtubules. Treatment with nocodazole to depolymerize microtubules caused fragmentation and dispersal of both the Golgi and lysosomes, as expected (Supplemental Movie 6). More importantly, we noted that, upon nocodazole treatment, the Golgi and lysosome puncta moved away from each other, becoming spatially distinct. Measurement of the overlap between the Golgi and lysosome compartments showed significant dissociation upon treatment with nocodazole (Figure 8D), in contrast to the effect of latrunculin B treatment. These data suggest that microtubules serve to link the Golgi and lysosomes. Overall we conclude that GOLPH3’s effect on lysosome reorientation is secondary to its effect on the Golgi, mediated by microtubule tethering of lysosomes to the Golgi.
GOLPH3 overexpression promotes directional trafficking toward the leading edge
Having discovered that GOLPH3 drives both the Golgi and lysosomes to reorient toward the leading edge upon wounding, we examined the functional requirement for each organelle for cell migration. We used inhibitors of Golgi or lysosome function to ask whether they interfere with monolayer scratch healing by NRK and MDA-MB-231 cells. Consistent with previous findings (Tseng et al., 2014), inhibition of Golgi function with BFA significantly reduced monolayer wound healing in both NRK and MDA-MB-231 cells (Figure 9, A and B). We note that the reduction in wound healing caused by treatment with BFA is comparable to that due to siRNA depletion of either GOLPH3 or MYO18A (Figure 2, A and B). By contrast, inhibition of lysosomal function by NH4Cl or bafilomycin A1 had a minimal, nonsignificant effect on scratch wound healing (Figure 9, A and B). The effectiveness of lysosome inhibition was verified by loss of LysoTracker staining upon treatment with NH4Cl or bafilomycin A1 (see Supplemental Movie 7). Therefore we conclude that, while the lysosome is dispensable, the Golgi plays an important role in cell migration that is enhanced by overexpression of GOLPH3.
FIGURE 9:

GOLPH3 promotes directional trafficking to the leading edge in wound healing. (A, B) Quantification of scratch wound healing in NRK and MDA-MB-231 cells treated with inhibitors of Golgi or lysosome function at time of wounding. Graphs indicate cell migration into the wound relative to vehicle control. Treatment with Golgi inhibitor BFA results in a significant reduction in cell migration, while treatment with lysosome inhibitors NH4Cl or bafilomycin A1 has minimal effect. (C) Representative IF images of migrating cells expressing ts045-VSVG-GFP at wound. Surface VSVG (red) was detected by using an exofacial VSVG antibody on unpermeablized cells. The Golgi is shown in green and the nucleus in blue. (D) Quantification of surface VSVG at the leading and trailing edges of cells from C. GOLPH3 overexpression results in a significant increase in surface VSVG in both leading and trailing edges compared with GFP (control) or GOLPH3-R90L. (E) Quantification of directional VSVG trafficking expressed as the ratio of leading over trailing surface VSVG intensity. (A, B, D, E) Graphed are mean ± SEM pooled from two independent experiments. Numbers of fields (A and B) or numbers of cells (D and E) measured (n) and p values (t test with Holm-Bonferroni correction) are indicated.
Finally, we sought to elucidate the functional significance of GOLPH3-driven Golgi polarization in directional cell migration. Given that directed trafficking toward the leading edge occurs in migrating cells (Bergmann et al., 1983; Yadav et al., 2009) and that the GOLPH3 pathway is critical for Golgi–to–plasma membrane trafficking (Dippold et al., 2009; Bishé et al., 2012; Ng et al., 2013; Farber-Katz et al., 2014), we examined the effect of overexpression of GOLPH3 on trafficking from the Golgi to the leading edge in wound healing. We transfected MDA-MB-231 cells overexpressing GOLPH3 or control cells with the trafficking reporter ts045-VSVG-GFP (temperature-sensitive mutant of the vesicular stomatitis virus G envelope glycoprotein fused to green fluorescent protein, subsequently referred to as VSVG), which is released from the endoplasmic reticulum and traffics through the Golgi to the plasma membrane upon a shift from the restrictive temperature (40°C) to the permissive temperature (32°C) (Presley et al., 1997). We created a monolayer scratch while cells were at the restrictive temperature and then allowed the Golgi to reorient for 4 h. We then shifted to the permissive temperature for 1 h to allow trafficking of VSVG. By using an exofacial antibody to VSVG on unpermeablized cells, we measured surface VSVG at the front and back of cells at the edge of the monolayer wound (Figure 9C). We defined the front of the cell by a vector drawn from the centroid of the nucleus to the centroid of the Golgi. In GOLPH3-overexpressing cells, we observed significantly higher surface VSVG compared with control cells, especially at the front of the cell, with a smaller increase at the back of the cell (Figure 9, C and D). We calculated the ratio of surface VSVG at the front versus at the back of the cell as a measure of directional trafficking and found a significant increase in directional trafficking in response to overexpression of GOLPH3 (Figure 9E). The ability of GOLPH3 to drive increased trafficking of VSVG to the plasma membrane and increased directional trafficking to the front of the cell is dependent on GOLPH3’s function at the Golgi, since the R90L mutant is inactive. Taking all of our results together, we conclude that overexpression of GOLPH3 promotes directional trafficking from the Golgi to the leading edge in wound healing.
DISCUSSION
GOLPH3-dependent Golgi polarization
A signaling pathway involving Cdc42 regulation of centrosomes and the microtubule cytoskeleton is well established to play an important role in Golgi reorientation and directional cell migration (Etienne-Manneville and Hall, 2001; Yadav et al., 2009; Millarte and Farhan, 2012). In light of the known association of the Golgi with microtubules (Chabin-Brion et al., 2001; Petry and Vale, 2015; Sanders and Kaverina, 2015), it is perhaps not surprising that microtubules can impart a force that leads to repositioning of the Golgi.
Our data indicate an independent role for the actin cytoskeleton to drive Golgi reorientation and directional cell migration. The PtdIns(4)P/GOLPH3/MYO18A/F-actin pathway serves to link the Golgi to the actin cytoskeleton (Dippold et al., 2009; Ng et al., 2013; Farber-Katz et al., 2014). Here we observe that this linkage imparts a force to reposition the Golgi in a manner that is independent of Cdc42 and centrosomes. Thus our data indicate that microtubule and microfilament cytoskeletal elements can each act independently to drive Golgi reorientation, but that efficient Golgi reorientation depends on the concerted efforts of both systems.
A question remains as to how the PtdIns(4)P/GOLPH3/MYO18A/F-actin linkage to the Golgi imparts a force that preferentially moves the Golgi toward the leading edge, despite the fact that PtdIns(4)P and GOLPH3 appear uniformly distributed on the trans-Golgi. One possibility would involve hypothetical signaling to preferentially activate MYO18A near the leading edge, resulting in an imbalance of forces to move the Golgi toward the leading edge. Our data cannot rule out this possibility.
However, we favor a simpler, and therefore more appealing, model. In this model, PtdIns(4)P/GOLPH3/MYO18A is equally present and active along the Golgi. It is well-known that, in polarized cell migration, a gradient of F-actin is established, with the highest concentration toward the leading edge (Nobes and Hall, 1999; Cau and Hall, 2005; Li and Gundersen, 2008; Lam et al., 2014). Increased concentration of F-actin density toward the leading edge provides increased opportunity for interaction with MYO18A. Thus we suggest the increased F-actin density toward the leading edge generates an imbalance of forces applied to the Golgi, via the PtdIns(4)P/GOLPH3/MYO18A/F-actin pathway, leading to movement of the Golgi. Notably, only when the Golgi faces the leading edge would we expect both halves of the Golgi to be in the vicinity of similar concentrations of F-actin, and thus only when the Golgi faces the leading edge would we expect the forces to reach stable equilibrium, resulting in the Golgi stably facing the leading edge. The simplicity of this model and consistency with all available data make this model particularly attractive (Figure 10).
FIGURE 10:
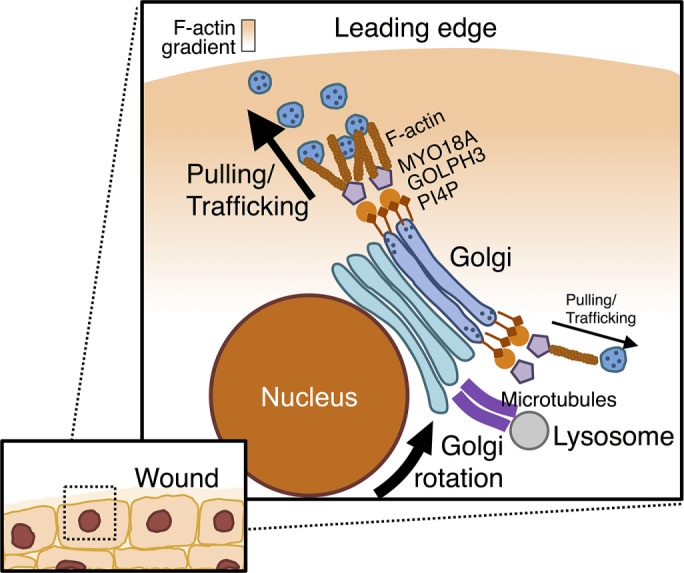
Model for GOLPH3-dependent Golgi reorientation and directional trafficking in wound healing. After cell monolayer wounding, an imbalance across the Golgi in the GOLPH3/MYO18A/F-actin machinery pulls the PtdIns(4)P-rich Golgi to reorient toward the leading edge. Lysosomes move in concert with the Golgi due to linkage to the Golgi mediated by microtubules. As a functional consequence of Golgi polarization, directional trafficking occurs toward the leading edge, which is required for cell migration. Overexpression of GOLPH3 drives enhanced reorientation of the Golgi toward the leading edge, increased overall Golgi–to–plasma membrane trafficking, and increased trafficking preferentially toward the front of the cell. All three contribute to increased directional trafficking to the wound edge, driving enhanced cell migration.
GOLPH3 drives Golgi–to–plasma membrane trafficking toward the wound edge
Our data demonstrate three consequences of overexpression of GOLPH3 that drive increased trafficking from the Golgi to the plasma membrane at the wound edge. First, overexpression of GOLPH3 increases overall trafficking to the plasma membrane. While the PtdIns(4)P/GOLPH3/MYO18A/F-actin pathway has previously been shown to be required for the majority of Golgi–to–plasma membrane trafficking (Dippold et al., 2009; Bishé et al., 2012; Ng et al., 2013), this is the first demonstration that overexpression of GOLPH3 is sufficient to drive increased Golgi–to–plasma membrane trafficking. Second, we also observe that overexpression of GOLPH3 promotes trafficking preferentially toward the front of the cell, as defined by the position of the Golgi relative to the nucleus. Third, overexpression of GOLPH3 promotes reorientation of the Golgi toward the wound edge, thus causing the front of the cell to point toward the wound. All three of these effects contribute toward driving trafficking to the wound edge.
Golgi cargoes are important for directed cell migration
Our VSVG reporter experiments demonstrate that GOLPH3 drives increased trafficking from the Golgi to the plasma membrane at the monolayer wound edge. VSVG serves as a general reporter for trafficking, suggesting that many cargoes undergo enhanced delivery to the plasma membrane at the wound edge. A few cargoes have been reported to be important for cell migration. General delivery of membrane lipids to build the plasma membrane at the leading edge has been suggested to be important to allow the generation of plasma membrane protrusions (lamellipodia and filopodia) that contribute to cell migration (Bergmann et al., 1983). In addition, the delivery of integrins to the leading edge is also known to be important for directional cell migration (Etienne-Manneville and Hall, 2001; Danen et al., 2005). We suspect that there is not a single cargo but rather an ensemble of cargoes important for cell migration that are delivered to the leading edge via the Golgi GOLPH3 pathway. Future work is required to identify critical cargoes.
GOLPH3-dependent cell migration
Several models have been proposed for the role of GOLPH3 in cell migration (Zhou et al., 2013; Isaji et al., 2014; Zhang et al., 2015). Here we provide evidence that it is the GOLPH3 complex at the Golgi, involving the PtdIns(4)P/GOLPH3/MYO18A/F-actin linkage responsible for applying the tensile force needed for Golgi–to–plasma membrane trafficking, that is required for cell migration and is capable of promoting cell migration when GOLPH3 is overexpressed. This is consistent with the observations of Tokuda et al. (2014), who also show that PtdIns(4)P-binding is required for GOLPH3 to promote cell migration.
A previous study has reported a correlation between defective cell migration and compaction of the normally extended ribbon morphology of the Golgi (Millarte et al., 2015). This observation is consistent with our data demonstrating the role for GOLPH3 in cell migration and previous work demonstrating that the Golgi becomes compact upon disruption of any component of the PtdIns(4)P/GOLPH3/MYO18A pathway (Dippold et al., 2009). We note that Millarte et al. (2015) observed reduced cell migration and Golgi PtdIns(4)P upon PLCG1 knockdown, fitting with our model (and with that of Tokuda et al., 2014) that PtdIns(4)P binding is critical for GOLPH3’s ability to drive enhanced cell migration. We propose that the same GOLPH3 linkage of the Golgi to actin is also the linkage important for Golgi repositioning, directional trafficking, and cell migration. Overexpression of GOLPH3 enhances all of these. It will be interesting to test whether the relative contributions of GOLPH3 in Golgi reorientation, directional trafficking, and cell migration can be teased apart from one another.
Role of the GOLPH3 pathway in cancer
A growing body of evidence indicates that GOLPH3 is an oncogene, frequently amplified and overexpressed in human cancers (Scott et al., 2009; Buschman et al., 2015a). Furthermore, high levels of GOLPH3 predict poor prognosis in a wide variety of cancers, independent of other prognostic indicators. Cancer morbidity and mortality are largely a consequence of invasion and metastasis, processes driven by cell migration. Thus the ability of GOLPH3 to drive cell migration may provide mechanistic insight into GOLPH3’s contribution to the pathophysiology of cancer. GOLPH3 is also known to play a central role in the Golgi response to DNA damage that promotes cell survival following DNA damage (Farber-Katz et al., 2014). This provides another mechanism by which GOLPH3 can contribute to cancer progression. GOLPH3, like many clinically important oncogenes (Bieging et al., 2014; Gabay et al., 2014; Stephen et al., 2014), appears to have multiple mechanisms by which it can contribute to the pathophysiology of cancer. In addition to providing insights into the mechanism of important physiology and pathophysiology, the GOLPH3 pathway may provide new targets for novel cancer therapeutic agents.
MATERIALS AND METHODS
Cell culture and DNA construction
MDA-MB-231, NRK, and HeLa S3 cell lines were grown according to American Type Culture Collection (Manassas, VA) guidelines. The stable MDA-MB-231 cell lines that overexpress GOLPH3-IRES-GFP, GOLPH3-R90L-IRES-GFP, or IRES-GFP were constructed with the pBABE-Puro retroviral system (Morgenstern and Land, 1990). Empty retroviral expression vector pBABE-Puro and packaging vectors pUMVC and pVSV-G were kind gifts from Jing Yang (University of California, San Diego). The expression vector of wild-type GOLPH3 was generated by serial cloning to combine GOLPH3 (Dippold et al., 2009) and IRES-hrGFP (Stratagene/Agilent, Santa Clara, CA) into the BamHI and EcoRI sites of pBABE-Puro vector. Expression and packaging vectors were transfected into HEK293T packaging cells using TransIT-LT1 (Mirus, Madison, WI). Viral supernatants harvested at 48 and 72 h after transfection were filtered through a 0.45 μm filter (Thermo Fisher Scientific, Waltham, MA) and incubated with target MDA-MB-231 cells in the presence of 6 μg/ml protamine (Sigma, St. Louis, MO). 24 h after the second viral infection, 0.5 μg/ml puromycin (InvivoGen, San Diego, CA) was used to select and maintain stable cell pools. The cell lines overexpressing IRES-GFP and R90L-IRES-GFP were generated in similar manner.
Cell proliferation assay
On day 0, cells were seeded in replicate wells at 20,000 cells/well in six-well plates, and grown in normal medium. On days 1, 3, and 5, triplicate wells for each cell line were trypsinized and counted using a hemocytometer. Growth rate was measured by taking the binary logarithm of cell counts, normalized to day 1.
Scratch wound healing assay
Cells were seeded in 35 mm gridded glass-bottom culture dishes (MatTek, Ashland, MA) and maintained in normal growth medium until reaching a confluent monolayer. A 200 μl pipette tip was used to scratch the cell monolayer; this was followed by two rinses with phosphate-buffered saline. For endpoint analysis, random fields at the scratch were selected and captured by 10× phase-contrast imaging with an Olympus (Tokyo, Japan) IX81-ZDC microscope. Cells were then incubated in normal growth medium for 15 h before fixation, followed by permeabilization and 4′,6-diamidino-2-phenylindole (DAPI) staining. DAPI images were taken at the same selected fields. For each field, the cells that migrated into the predefined scratch area were counted, and the mean cell density in the scratch area for n fields was normalized to the mean confluent density, and values were scaled relative to the control to calculate relative scratch healing. For time-lapse imaging, cells were incubated in normal growth medium containing calcein AM (Life Technologies/Thermo Fisher Scientific, Waltham, MA) after wounding. Time-lapse images were taken at 15 min intervals for 15 h.
For scratch assays with siRNA knockdowns, siRNA oligos were transfected into cells 48 h before monolayer wounding. For scratch assays with Golgi or lysosome inhibitors, after the scratch was made, cells were incubated in normal growth media containing 0.1% DMSO (Sigma), 0.1% MeOH, 5 ng/ml BFA (Sigma), 10 mM NH4Cl (Sigma), or 100 nM bafilomycin A1 (Sigma) for 8 h before fixation. DMSO was used as vehicle control for bafilomycin A1. MeOH was used as vehicle control for BFA. Because DMSO and MeOH gave identical results, their data are pooled in Figure 9, A and B. For scratch assays with centrinone (synthesized by Sundia MediTech, Shanghai, China; Wong et al., 2015), cells were grown for more than 2 wk in the presence of 100 nM centrinone or 0.1% DMSO and replaced with fresh media containing 100 nM centrinone or 0.1% DMSO at the time of the scratch. For scratch assays with ML141 (Tocris, Bristol, United Kingdom), media was replaced 1 h before scratch with normal growth media containing 10 μM ML141 or 0.1% DMSO and replaced with fresh media containing 10 μM ML141 or 0.1% DMSO at the time of the scratch.
Fluorescence microscopy
Fluorescence microscopy was performed on an Olympus IX81-ZDC spinning-disk confocal microscope, and images were analyzed with Slidebook and ImageJ software. All images shown represent maximum projections in z of confocal stacks with spacing of 0.5 μM. Movies show a single confocal plane.
Measurement of cell migration speed
Time-lapse images of scratch wound healing assay were analyzed in ImageJ of actively migrating cells, defining the center of the nucleus between consecutive frames, and measuring the traveling distance of the nucleus divided by the time interval.
Measurement of Golgi/lysosome/mitochondria orientation angle
IF images of cells at the wound edge stained with a Golgi marker and DAPI were analyzed in ImageJ, by drawing a first line from the centroid of the nucleus to the wound edge and a second line from the centroid of the nucleus to the centroid of the Golgi. The angle formed in between the two lines was measured. The orientation angles of mitochondria and lysosomes were measured in similar manner. For cells before wounding, a fixed direction was chosen arbitrarily, and the angle between said direction and the Golgi was measured for each cell.
Time-lapse imaging of cellular organelles after wounding
Cells were transfected with the expression vector for α-mannosidase II-GFP (ManII-GFP) to mark the Golgi and grown to confluence. Before the scratch wound was made, cells were incubated with MitoTracker Deep Red or LysoTracker Red (Life Technologies), following the manufacturer’s instructions to label mitochondria and lysosomes, respectively. A wound was made in the same way as described for the scratch wound healing assay. After the scratch wound was made, time-lapse images were taken at 3 min intervals for 5 h.
Measurement of Golgi/nucleus rotation speed
Time-lapse fluorescence and DIC images of cells at the wound edge were analyzed in ImageJ by identifying in each frame the direction of the nucleus with the aid of structural features visible by DIC imaging, and identifying the direction of the Golgi by drawing a line through the center of the nucleus and the center of the Golgi (marked by ManII-GFP). Rotation speed was determined by measuring the rotation angle between consecutive frames divided by the time interval.
Measurement of surface ts045-VSVG-GFP in wound healing
Cells were transiently transfected with an expression vector for temperature-sensitive, GFP-tagged ts045-VSVG-GFP and grown to a confluent monolayer. At 36 h after transfection, cells were maintained at the restrictive temperature (40°C) for 12 h before a razor blade was used to create a wound. Cells were returned to 40°C for 4 h to allow Golgi reorientation and then shifted to the permissive temperature (32°C) for 1 h to allow VSVG trafficking before fixation. An exofacial antibody specific to VSVG was used on unpermeabilized cells to detect surface VSVG, followed by permeabilization and staining with GOLPH3 antibody to label the Golgi. IF images were taken of transfected cells at the edge of the wound. Surface VSVG was analyzed in ImageJ by drawing a line with a width of 10 pixels through the nucleus and the Golgi and measuring the average intensity at the plasma membrane at the front and back of the cell (seen as two peaks in the histogram), with background subtracted.
siRNA knockdown
siRNA oligonucleotides containing Stealth modifications were synthesized by LifeTechnologies and transfected using RNAiMAX according to the manufacturer’s instructions. Negative control, GOLPH3, and MYO18A sequences were the same as those described previously (Dippold et al., 2009). If not otherwise specified in the figures, GOLPH3 siRNA #3, and MYO18A siRNA #1 were used. Knockdowns were verified by Western blots performed in parallel for each experiment.
Antibodies
The GOLPH3 rabbit polyclonal antibody was described previously (Dippold et al., 2009). The MYO18A antibody was a generous gift from Zissis Chroneos (University of Texas, Tyler, TX). The GM130 antibody was from BD Biosciences (San Jose, CA). The YAP antibody H-125 was from Santa Cruz Biotechnologies (Dallas, TX). The ATP synthase subunit IF1 antibody 5E2D7 used to mark mitochondria was from Invitrogen/Thermo Fisher Scientific. The LAMP1 and glyceraldehyde-3-phophate dehydrogenase (GAPDH) antibodies were from Cell Signaling Technology (Danvers, MA). The VSVG exofacial monoclonal antibody 8G5F11 was from KeraFAST (Boston, MA). The γ-tubulin antibody GTU-88 was from Sigma. The Cep-192 antibody was described previously (Wong et al., 2015). The TGN46 antibody was from Bio-Rad (Hercules, CA). The horseradish peroxidase–conjugated mouse anti-rabbit secondary antibody for Western blot was from Jackson ImmunoReasearch (West Grove, PA). Alexa Fluor (488, 594, and 647)-conjugated secondary antibodies for IF were from Invitrogen.
Statistics
A two-tailed Student’s unpaired t test was calculated using Microsoft (Redmond, WA) Excel. Multiple comparisons were corrected for family-wise type 1 error using the Holm-Bonferroni correction (Holm, 1979). Analysis of variance (ANOVA) and least-squares linear regression were performed using PastProject statistical software (Hammer et al., 2001).
Supplementary Material
Acknowledgments
We thank Karen Messer, chief of the Division of Biostatistics and Bioinformatics at the University of California, San Diego, for assistance with statistical analyses. We thank Ana Zubiaga, Alexandra Newton, Joan Heller Brown, Jing Yang, and Kun-Liang Guan for insightful comments and discussions. We thank Spencer Wei, Jing Yang, Corina Antal, and Mike Whitney for helpful reagents. Special thanks to Nuo Wang for help drafting the model diagram. This work was supported by Department of Defense Breast Cancer Research Program Era of Hope Award W81XWH-10-1-0822 and a Burroughs Wellcome Fund Career Award in the Biomedical Sciences to S.J.F., a National Institutes of Health grant to K.O. (GM074207), and funds from the Hilton Ludwig Cancer Prevention Initiative to A.K.S. and R.L.D. K.O. and A.K.S. receive salaries and other support from the Ludwig Institute for Cancer Research.
Abbreviations used:
- ANOVA
analysis of variance
- BFA
brefeldin A
- DAPI
4′,6-diamidino-2-phenylindole
- DIC
differential interference contrast
- DMSO
dimethyl sulfoxide
- GAPDH
glyceraldehyde-3-phophate dehydrogenase
- GFP
green fluorescent protein
- IF
immunofluorescence
- ManII-GFP
α-mannosidase II-GFP
- MYO18A
myosin 18A
- NRK
normal rat kidney
- PtdIns(4)P
phosphatidylinositol-4-phosphate
- siRNA
small interfering RNA
- VSVG
temperature-sensitive mutant (ts045) of the vesicular stomatitis virus G envelope glycoprotein fused to green fluorescent protein.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E16-01-0005) on October 5, 2016.
REFERENCES
- Bell AW, Ward MA, Blackstock WP, Freeman HN, Choudhary JS, Lewis AP, Chotai D, Fazel A, Gushue JN, Paiement J, et al. Proteomics characterization of abundant Golgi membrane proteins. J Biol Chem. 2001;276:5152–5165. doi: 10.1074/jbc.M006143200. [DOI] [PubMed] [Google Scholar]
- Bergmann JE, Kupfer A, Singer SJ. Membrane insertion at the leading edge of motile fibroblasts. Proc Natl Acad Sci USA. 1983;80:1367–1371. doi: 10.1073/pnas.80.5.1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieging KT, Mello SS, Attardi LD. Unravelling mechanisms of p53-mediated tumour suppression. Nat Rev Cancer. 2014;14:359–370. doi: 10.1038/nrc3711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishé B, Syed GH, Field SJ, Siddiqui A. Role of phosphatidylinositol 4-phosphate (PI4P) and its binding protein GOLPH3 in hepatitis C virus secretion. J Biol Chem. 2012;287:27637–27647. doi: 10.1074/jbc.M112.346569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buschman MD, Rahajeng J, Field SJ. GOLPH3 links the Golgi, DNA damage, and cancer. Cancer Res. 2015a;75:624–627. doi: 10.1158/0008-5472.CAN-14-3081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buschman MD, Xing M, Field SJ. The GOLPH3 pathway regulates Golgi shape and function and is activated by DNA damage. Front Neurosci. 2015b;9:362. doi: 10.3389/fnins.2015.00362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cau J, Hall A. Cdc42 controls the polarity of the actin and microtubule cytoskeletons through two distinct signal transduction pathways. J Cell Sci. 2005;118:2579–2587. doi: 10.1242/jcs.02385. [DOI] [PubMed] [Google Scholar]
- Chabin-Brion K, Marceiller J, Perez F, Settegrana C, Drechou A, Durand G, Poüs C. The Golgi complex is a microtubule-organizing organelle. Mol Biol Cell. 2001;12:2047–2060. doi: 10.1091/mbc.12.7.2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collot M, Louvard D, Singer SJ. Lysosomes are associated with microtubules and not with intermediate filaments in cultured fibroblasts. Proc Natl Acad Sci USA. 1984;81:788–792. doi: 10.1073/pnas.81.3.788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czuchra A, Wu X, Meyer H, van Hengel J, Schroeder T, Geffers R, Rottner K, Brakebusch C. Cdc42 is not essential for filopodium formation, directed migration, cell polarization, and mitosis in fibroblastoid cells. Mol Biol Cell. 2005;16:4473–4484. doi: 10.1091/mbc.E05-01-0061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danen EHJ, van Rheenen J, Franken W, Huveneers S, Sonneveld P, Jalink K, Sonnenberg A. Integrins control motile strategy through a Rho–cofilin pathway. J Cell Biol. 2005;169:515–526. doi: 10.1083/jcb.200412081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dippold HC, Ng MM, Farber-Katz SE, Lee S-K, Kerr ML, Peterman MC, Sim R, Wiharto PA, Galbraith KA, Madhavarapu S, et al. GOLPH3 bridges phosphatidylinositol-4-phosphate and actomyosin to stretch and shape the Golgi to promote budding. Cell. 2009;139:337–351. doi: 10.1016/j.cell.2009.07.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efimov A, Kharitonov A, Efimova N, Loncarek J, Miller PM, Andreyeva N, Gleeson P, Galjart N, Maia ARR, McLeod IX, et al. Asymmetric CLASP-dependent nucleation of noncentrosomal microtubules at the trans-Golgi network. Dev Cell. 2007;12:917–930. doi: 10.1016/j.devcel.2007.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etienne-Manneville S, Hall A. Integrin-mediated activation of Cdc42 controls cell polarity in migrating astrocytes through PKCζ. Cell. 2001;106:489–498. doi: 10.1016/s0092-8674(01)00471-8. [DOI] [PubMed] [Google Scholar]
- Farber-Katz SE, Dippold HC, Buschman MD, Peterman MC, Xing M, Noakes CJ, Tat J, Ng MM, Rahajeng J, Cowan DM, et al. DNA damage triggers Golgi dispersal via DNA-PK and GOLPH3. Cell. 2014;156:413–427. doi: 10.1016/j.cell.2013.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedl P, Gilmour D. Collective cell migration in morphogenesis, regeneration and cancer. Nat Rev Mol Cell Biol. 2009;10:445–457. doi: 10.1038/nrm2720. [DOI] [PubMed] [Google Scholar]
- Friedl P, Wolf K. Tumour-cell invasion and migration: diversity and escape mechanisms. Nat Rev Cancer. 2003;3:362–374. doi: 10.1038/nrc1075. [DOI] [PubMed] [Google Scholar]
- Gabay M, Li Y, Felsher DW. MYC activation is a hallmark of cancer initiation and maintenance. Cold Spring Harb Perspect Med. 2014;4:a014241. doi: 10.1101/cshperspect.a014241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammer Ø, Harper DAT, Ryan PD. PAST: paleontological statistics software package for education and data analysis. Palaeontol Electron. 2001;4:9. [Google Scholar]
- Harada A, Takei Y, Kanai Y, Tanaka Y, Nonaka S, Hirokawa N. Golgi vesiculation and lysosome dispersion in cells lacking cytoplasmic dynein. J Cell Biol. 1998;141:51–59. doi: 10.1083/jcb.141.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm S. A simple sequentially rejective multiple test procedure. Scand J Stat. 1979;6:65–70. [Google Scholar]
- Hong L, Kenney SR, Phillips GK, Simpson D, Schroeder CE, Nöth J, Romero E, Swanson S, Waller A, Strouse JJ, et al. Characterization of a Cdc42 protein inhibitor and its use as a molecular probe. J Biol Chem. 2013;288:8531–8543. doi: 10.1074/jbc.M112.435941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Insall RH, Machesky LM. Actin dynamics at the leading edge: from simple machinery to complex networks. Dev Cell. 2009;17:310–322. doi: 10.1016/j.devcel.2009.08.012. [DOI] [PubMed] [Google Scholar]
- Isaji T, Im S, Gu W, Wang Y, Hang Q, Lu J, Fukuda T, Hashii N, Takakura D, Kawasaki N, et al. An oncogenic protein Golgi phosphoprotein 3 up-regulates cell migration via sialylation. J Biol Chem. 2014;289:20694–20705. doi: 10.1074/jbc.M113.542688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiwanuka E, Lee CC, Hackl F, Caterson EJ, Junker JP, Gerdin B, Eriksson E. Cdc42 and p190RhoGAP activation by CCN2 regulates cell spreading and polarity and induces actin disassembly in migrating keratinocytes. Int Wound J. 2016;13:372–381. doi: 10.1111/iwj.12315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause M, Gautreau A. Steering cell migration: lamellipodium dynamics and the regulation of directional persistence. Nat Rev Mol Cell Biol. 2014;15:577–590. doi: 10.1038/nrm3861. [DOI] [PubMed] [Google Scholar]
- Kupfer A, Louvard D, Singer SJ. Polarization of the Golgi apparatus and the microtubule-organizing center in cultured fibroblasts at the edge of an experimental wound. Proc Natl Acad Sci USA. 1982;79:2603–2607. doi: 10.1073/pnas.79.8.2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam P-Y, Fischer RS, Shin WD, Waterman CM, Huttenlocher A. Spinning disk confocal imaging of neutrophil migration in zebrafish. Methods Mol Biol. 2014;1124:219–233. doi: 10.1007/978-1-62703-845-4_14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Gundersen GG. Beyond polymer polarity: how the cytoskeleton builds a polarized cell. Nat Rev Mol Cell Biol. 2008;9:860–873. doi: 10.1038/nrm2522. [DOI] [PubMed] [Google Scholar]
- Locascio A, Nieto MA. Cell movements during vertebrate development: integrated tissue behaviour versus individual cell migration. Curr Opin Genet Dev. 2001;11:464–469. doi: 10.1016/s0959-437x(00)00218-5. [DOI] [PubMed] [Google Scholar]
- Matteoni R, Kreis TE. Translocation and clustering of endosomes and lysosomes depends on microtubules. J Cell Biol. 1987;105:1253–1265. doi: 10.1083/jcb.105.3.1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millarte V, Boncompain G, Tillmann K, Perez F, Sztul E, Farhan H. Phospholipase C γ1 regulates early secretory trafficking and cell migration via interaction with p115. Mol Biol Cell. 2015;26:2263–2278. doi: 10.1091/mbc.E15-03-0178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millarte V, Farhan H. The Golgi in cell migration: regulation by signal transduction and its implications for cancer cell metastasis. ScientificWorldJournal. 2012;2012:498278. doi: 10.1100/2012/498278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgenstern JP, Land H. Advanced mammalian gene transfer: high titre retroviral vectors with multiple drug selection markers and a complementary helper-free packaging cell line. Nucleic Acids Res. 1990;18:3587–3596. doi: 10.1093/nar/18.12.3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng MM, Dippold HC, Buschman MD, Noakes CJ, Field SJ. GOLPH3L antagonizes GOLPH3 to determine Golgi morphology. Mol Biol Cell. 2013;24:796–808. doi: 10.1091/mbc.E12-07-0525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobes CD, Hall A. Rho GTPases control polarity, protrusion, and adhesion during cell movement. J Cell Biol. 1999;144:1235–1244. doi: 10.1083/jcb.144.6.1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petry S, Vale RD. Microtubule nucleation at the centrosome and beyond. Nat Cell Biol. 2015;17:1089–1093. doi: 10.1038/ncb3220. [DOI] [PubMed] [Google Scholar]
- Presley JF, Cole NB, Schroer TA, Hirschberg K, Zaal KJM, Lippincott-Schwartz J. ER-to-Golgi transport visualized in living cells. Nature. 1997;389:81–85. doi: 10.1038/38001. [DOI] [PubMed] [Google Scholar]
- Ridley AJ. Life at the leading edge. Cell. 2011;145:1012–1022. doi: 10.1016/j.cell.2011.06.010. [DOI] [PubMed] [Google Scholar]
- Rivero S, Cardenas J, Bornens M, Rios RM. Microtubule nucleation at the cis-side of the Golgi apparatus requires AKAP450 and GM130. EMBO J. 2009;28:1016–1028. doi: 10.1038/emboj.2009.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzo R, Parashuraman S, D’Angelo G, Luini GOLPH3 and oncogenesis: what is the molecular link. Tissue Cell. doi: 10.1016/j.tice.2016.06.008. [DOI] [PubMed] [Google Scholar]
- Rogalski AA, Singer SJ. Associations of elements of the Golgi apparatus with microtubules. J Cell Biol. 1984;99:1092–1100. doi: 10.1083/jcb.99.3.1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders AAWM, Kaverina I. Nucleation and dynamics of Golgi-derived microtubules. Front Neurosci 9. 2015:431. doi: 10.3389/fnins.2015.00431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandoval IV, Bonifacino JS, Klausner RD, Henkart M, Wehland J. Role of microtubules in the organization and localization of the Golgi apparatus. J Cell Biol. 1984;99:113s–118s. doi: 10.1083/jcb.99.1.113s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheel J, Matteoni R, Ludwig T, Hoflack B, Kreis TE. Microtubule depolymerization inhibits transport of cathepsin D from the Golgi apparatus to lysosomes. J Cell Sci. 1990;96:711–720. doi: 10.1242/jcs.96.4.711. [DOI] [PubMed] [Google Scholar]
- Scott KL, Kabbarah O, Liang M-C, Ivanova E, Anagnostou V, Wu J, Dhakal S, Wu M, Chen S, Feinberg T, et al. GOLPH3 modulates mTOR signalling and rapamycin sensitivity in cancer. Nature. 2009;459:1085–1090. doi: 10.1038/nature08109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephen AG, Esposito D, Bagni RK, McCormick F. Dragging ras back in the ring. Cancer Cell. 2014;25:272–281. doi: 10.1016/j.ccr.2014.02.017. [DOI] [PubMed] [Google Scholar]
- Taft MH, Behrmann E, Munske-Weidemann L-C, Thiel C, Raunser S, Manstein DJ. Functional characterization of human myosin-18A and its interaction with F-actin and GOLPH3. J Biol Chem. 2013;288:30029–30041. doi: 10.1074/jbc.M113.497180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thyberg J, Moskalewski S. Role of microtubules in the organization of the Golgi complex. Exp Cell Res. 1999;246:263–279. doi: 10.1006/excr.1998.4326. [DOI] [PubMed] [Google Scholar]
- Tokuda E, Itoh T, Hasegawa J, Ijuin T, Takeuchi Y, Irino Y, Fukumoto M, Takenawa T. Phosphatidylinositol 4-phosphate in the Golgi apparatus regulates cell–cell adhesion and invasive cell migration in human breast cancer. Cancer Res. 2014;74:3054–3066. doi: 10.1158/0008-5472.CAN-13-2441. [DOI] [PubMed] [Google Scholar]
- Tseng C-N, Hong Y-R, Chang H-W, Yu T-J, Hung T-W, Hou M-F, Yuan S-SF, Cho C-L, Liu C-T, Chiu C-C, et al. Brefeldin A reduces anchorage-independent survival, cancer stem cell potential and migration of MDA-MB-231 human breast cancer cells. Molecules. 2014;19:17464–17477. doi: 10.3390/molecules191117464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong YL, Anzola JV, Davis RL, Yoon M, Motamedi A, Kroll A, Seo CP, Hsia JE, Kim SK, Mitchell JW, et al. Reversible centriole depletion with an inhibitor of Polo-like kinase 4. Science. 2015;348:1155–1160. doi: 10.1126/science.aaa5111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CC, Taylor RS, Lane DR, Ladinsky MS, Weisz JA, Howell KE. GMx33: a novel family of trans-Golgi proteins identified by proteomics. Traffic. 2000;1:963–975. [PubMed] [Google Scholar]
- Yadav S, Puri S, Linstedt AD. A primary role for Golgi positioning in directed secretion, cell polarity, and wound healing. Mol Biol Cell. 2009;20:1728–1736. doi: 10.1091/mbc.E08-10-1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Ding Z, Mo J, Sang B, Shi Q, Hu J, Xie S, Zhan W, Lu D, Yang M, et al. GOLPH3 promotes glioblastoma cell migration and invasion via the mTOR-YB1 pathway in vitro. Mol Carcinog. 2015;54:1252–1263. doi: 10.1002/mc.22197. [DOI] [PubMed] [Google Scholar]
- Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J, Xie J, Ikenoue T, Yu J, Li L, et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007;21:2747–2761. doi: 10.1101/gad.1602907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Zhan W, Bian W, Hua L, Shi Q, Xie S, Yang D, Li Y, Zhang X, Liu G, et al. GOLPH3 regulates the migration and invasion of glioma cells through RhoA. Biochem Biophys Res Commun. 2013;433:338–344. doi: 10.1016/j.bbrc.2013.03.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.