

A truncated fragment of p35, the Cdk5 kinase regulatory protein (TFP5), inhibits specifically hyperactive Cdk5/p25 activity and rescues the Alzheimer’s disease and Parkinson’s disease phenotype in model mice. To account for the selective inhibition of Cdk5/p25 activity, the p10 N-terminal domain of p35, absent in p25, spares Cdk5/p35.
Abstract
In a series of studies, we have identified TFP5, a truncated fragment of p35, the Cdk5 kinase regulatory protein, which inhibits Cdk5/p35 and the hyperactive Cdk5/p25 activities in test tube experiments. In cortical neurons, however, and in vivo in Alzheimer’s disease (AD) model mice, the peptide specifically inhibits the Cdk5/p25 complex and not the endogenous Cdk5/p35. To account for the selective inhibition of Cdk5/p25 activity, we propose that the “p10” N-terminal domain of p35, absent in p25, spares Cdk5/p35 because p10 binds to macromolecules (e.g., tubulin and actin) as a membrane-bound multimeric complex that favors p35 binding to Cdk5 and catalysis. To test this hypothesis, we focused on Munc 18, a key synapse-associated neuronal protein, one of many proteins copurifying with Cdk5/p35 in membrane-bound multimeric complexes. Here we show that, in vitro, the addition of p67 protects Cdk5/p35 and has no effect on Cdk5/p25 activity in the presence of TFP5. In cortical neurons transfected with p67siRNA, we also show that TFP5 inhibits Cdk5/p35 activity, whereas in the presence of p67 the activity is protected. It does so without affecting any other kinases of the Cdk family of cyclin kinases. This difference may be of significant therapeutic value because the accumulation of the deregulated, hyperactive Cdk5/p25 complex in human brains has been implicated in pathology of AD and other neurodegenerative disorders.
INTRODUCTION
Cdk5 bound to its activator p35 or p39 is a tightly regulated neuronal protein kinase that targets more than a dozen substrates that regulate neuronal differentiation, migration, synaptogenesis, and synaptic function (Ohshima et al., 1996; Dhavan and Tsai, 2001; Grant et al., 2001; Bibb, 2003; Cheng and Ip, 2003; Gupta and Tsai, 2003; Giese et al., 2005; Angelo et al., 2006; Cheung et al., 2008; Jessberger et al., 2009). Hyperactivated Cdk5 in human brains, however, complexed with p25, a cleaved fragment of p35, is implicated in the hallmark pathology of Alzheimer’s disease (AD). It has been proposed that the p25 subunit arises in human and rodent brains under neuronal stress (Ahlijanian et al., 2000; Kusakawa et al., 2000; Lee et al., 2000; Nath et al., 2000). Stress leads to increased intracellular Ca ion concentration, activation of a protease, calpain, which cleaves p35 into two fragments, the p10 N-terminal myristoylated domain that binds the cell membrane, and p25, a more stable regulator of Cdk5 that forms a hyperactive cytoplasmic complex. It has been proposed that Cdk5/p25 is in part responsible for the AD phenotypes of hyperphosphorylated neurofilament-tau neurofibrillary tangles (NFTs) and Aβ plaques (Cruz et al., 2003; Guo, 2006; Hamdane and Buee, 2007; Han et al., 2009; Sundaram et al., 2013). We succeeded in identifying two truncated fragments of p35, inhibitory peptide (CIP; 126 amino acids [aa]) and P5 (24 aa), each capable of inhibiting Cdk5/p25 activity in vitro and in vivo (Amin et al., 2002; Zheng et al., 2002, 2010; Shukla et al., 2013; Sundaram et al., 2013). In a proof of concept study, a transgenic p25 mouse mutant (p25Tg), overexpressing hyperactive Cdk5/p25, NFTs, Aβ plaques, and other AD phenotypes, including behavioral defects, could be ameliorated by CIP (the larger peptide) overexpression in the brain (Sundaram et al., 2013). A similar rescue of another AD mutant (5XFAD) was obtained by intraperitoneal injections of TFP5, a P5 (24 aa) peptide modified for penetration of the blood–brain barrier by addition of fluorescein isothiocyanate (FITC) at the N-terminal and a protein transduction domain of TAT protein (11 aa) at the C-terminal (Shukla et al., 2013).
What is most surprising about the action of these peptide inhibitors is that in in vitro test tube experiments, both Cdk5 complexes are inhibited; in cultured cells and in brains in vivo, however, endogenous Cdk5/p35 activity is unaffected, whereas activity of Cdk5/p25 is selectively inhibited.
An obvious initial target to account for the specific inhibition of the Cdk5/p25 activity by the TFP5 inhibitory peptide in vivo is the p10 domain, which is absent in p25. In fact, p10 exhibited a protective effect in neurons and nonneurons made toxic by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridinium (MPP+) (Zhang et al., 2012). The addition of a modified p10 to such cells reduces reactive oxygen species (ROS) and cell death, although the mechanism is not understood. Curiously, a toxic effect of p10 itself has also been reported (Chew et al., 2010). The action of the P5 peptide, however, offers a possible mechanism for the role of the p10 domain in specifically sparing Cdk5/p35 inhibition. P35 interacts with a number of proteins primarily via its p10 domain. It bundles tubulin into microtubules and binds strongly to the polymers (Hou et al., 2007). It also binds and bundles F-actin (He et al., 2011) and neurofilament proteins (Qi et al., 1998), which accounts for the presence of membrane-bound multimeric cytoskeletal complexes containing Cdk5/p35 (Veeranna et al., 2000; Lim et al., 2003; Bhaskar et al., 2004). Various nuclear importins (importin b, 5 and 7) also bind to the p10 domain of p35, serving as “shuttles” of Cdk5 between nucleus and cytoplasm. Importins bind p35, displacing it from Cdk5 (Fu et al., 2006) and thereby inhibiting kinase activity. These properties of p35, attributable to the p10 domain, are absent in p25 complexes and suggest that proteins binding to p35 change its confirmation such that its association and activation of Cdk5 are unaffected by P5. We previously showed that polymerized tubulin, and not tubulin, indeed protects the Cdk5/p35 complex from P5 inhibition, whereas Cdk5/p25 is strongly down-regulated (Zheng et al., 2010). To explore this hypothesis further, we focused on Munc 18 (p67), a key synapse-associated neuronal protein, which is also one of many proteins copurifying with Cdk5 in membrane-bound multimeric complexes (Veeranna et al., 2000). Munc 18 is a Cdk5 substrate at the presynaptic junction, which, when phosphorylated, regulates its binding to syntaxin during exocytosis (Pevsner, 1996). Here we show results of experiments in vitro, in cultured HEK cells, and in cortical neurons transfected with p67 small interfering RNA (siRNA) that the presence of Munc 18 is sufficient to protect Cdk5/p35 activity from TFP5 inhibition, whereas the more toxic Cdk5/p25, lacking p10, is specifically inhibited. This property suggests TFP5 as a potential therapeutic candidate for neurodegenerative disorders in which Cdk5/p25 may play an active role.
RESULTS
Munc 18 (p67) is part of a multimeric (supramolecular) complex containing Cdk5/p35
There is evidence that the p10 N-terminal domain of p35, the Cdk5 activator, targets Cdk5 to various substrates (Lim et al., 2003). This may explain why Cdk5 in brain is associated with p35 in a large (>670 kDa), inactive, membrane-bound multimeric complex containing cytoskeletal and synaptic proteins (Lee et al., 1996; Rosales et al., 2000; Bhaskar et al., 2004). Cdk5/p25, instead, is an active, heterodimeric cytoplasmic complex of Cdk5 and p25. In an earlier study in our lab, cytoskeletal proteins and Cdk5, together with synaptic proteins such as syntaxin, copurified with Munc 18 from rat brain lysates (Bhaskar et al., 2004). To confirm the existence of multimeric complexes containing Munc 18, Cdk5/p35, and cytoskeletal proteins, we carried out reciprocal coimmunoprecipitation experiments with rat brain lysates using antibodies to Munc 18 and to p35 (Figure 1, A and B). The reciprocal pull down indicated that a similar repertoire of cytoskeletal proteins and kinases colocalize with Munc 18 and p35, all, presumably, part of macromolecular complexes associated with the myristoylated p10 domain of p35. We should make it clear that the coimmunoprecipitated proteins are not necessarily part of a single large multimeric complex; we suggest that these pull-down experiments cannot distinguish a single macromolecular complex from a mix of several smaller complexes. Except for tubulin, serum albumin controls with each antibody show no evidence of associated proteins. We stress that p25 was not expressed in both cases, unbound to Cdk5 as one might expect. P25 is not associated with multimeric protein complexes; instead, it exists in the cytoplasm as a heterodimer with Cdk5 (Lee et al., 1996; Rosales et al., 2000).
FIGURE 1:
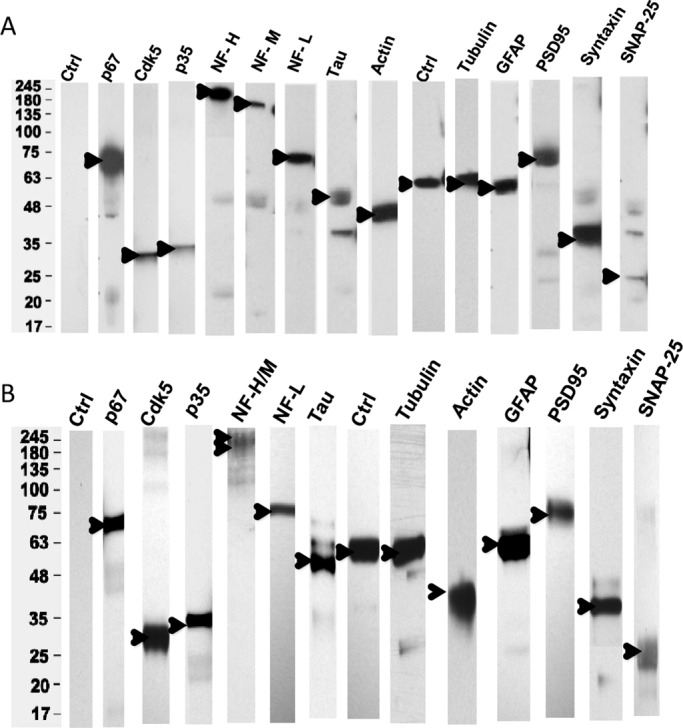
P67 binds to Cdk5/p35, cytoskeletal proteins and synaptic proteins in a rat brain lysate. (A) Rat brain lysate was immunoprecipitated (IP) with p67 antibody and the bound proteins, eluted from washed beads, were immunodetected with Western blot (WB) analyses using respective antibodies as shown. See Materials and Methods for antibody concentrations (n = 3). (B) A reciprocal p35 IP of a similar rat brain lysate shows bound proteins similar to those in a p67 coimmunoprecipitation (n = 3). Rabbit IgG was used as controls for each antibody shown, and, except for tubulin, no proteins were immunoprecipitated. An example of control blots is shown in lane 1. For lane 10 (Ctrl) in A and lane 8 (Ctrl) in B, tubulin was detected with anti-tubulin antibody.
Munc 18 stimulates Cdk5/p35 activity in vitro without affecting the activity of Cdk5/p25
In earlier fractionation studies, we demonstrated that Munc 18 acted as a “regulator” of Cdk5/p35 phosphorylation; it stimulated activity almost sevenfold (Shetty et al., 1995). It was later shown that Munc 18 is a substrate: Cdk5/p35 phosphorylates Munc 18 at the synapse, which dissociates Munc 18 from syntaxin to initiate exocytosis (Shuang et al., 1998). Cdk5/p25 also phosphorylates Munc 18. To examine the interactions of Cdk5/p35 and Cdk5/p25 with Munc 18 in vitro, we studied the activities of the two commercially available complexes in the presence and absence of Munc 18 (1.5 μg) under identical conditions in vitro with histone as substrate (Figure 2). Whereas Munc 18 stimulated Cdk5/p35 activity twofold in this assay, it had no effect on the much greater activity of Cdk5/p25. This suggests that binding of Munc 18 to the N-terminal p10 domain of p35 may induce conformational changes in p35 that favor interaction with the activation site of Cdk5. Although Munc 18 is a substrate, it is more likely that histone was the preferred substrate under these conditions.
FIGURE 2:
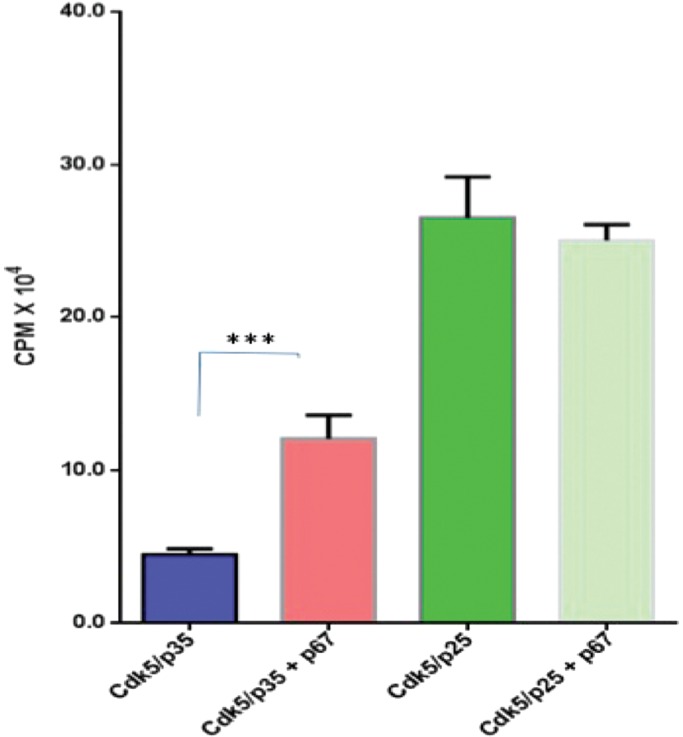
P67 stimulates Cdk5/p35 activity but not Cdk5/p25 activity in an in vitro assay. The assay consists of active Cdk5/p35 or active Cdk5/p25 (5 ng) and histone H1 as a substrate (20 μg), bacterially expressed GST-p67 (1.5 μg), and [γ32P]ATP (0.05 mM). The results represent the values of three experiments and are expressed as ±SEM. ***p < 0.001.
TFP5, a modified truncated peptide from p25, inhibits Cdk5/p35 and Cdk5/p25 in vitro (test tube)
Hyperactivity of Cdk5/p25, a sign of a deregulated kinase, has been invoked as a toxic factor in neurodegenerative disorders (Patrick et al., 1999; Nguyen et al., 2001; Cruz et al., 2003; Cruz and Tsai, 2004; Shukla et al., 2013; Sundaram et al., 2013). We identified a 24-aa truncated fragment of p25, P5, which inhibits AD-like phenotypes in cultured cortical neurons (Zheng et al., 2005, 2010; Kesavapany et al., 2007). To study its effects in AD model mice, we modified P5 to TFP5, which has a FITC tag at the N-terminal end and a TAT sequence at the C-terminal to facilitate passage through the blood–brain barrier (Shukla et al., 2013). By virtue of its inhibition of Cdk5/p25 phosphorylation, its rescue of behavior and AD pathology in 5XFAD model mice without evidence of side effects suggests that the peptide has no effect on endogenous Cdk5/p35 activity. To test the specificity of inhibition, we studied test tube kinase assays of Cdk5/p25 and Cdk5/p35 in the presence of TFP5. The results shown in Figure 3 indicate that both kinase complexes are inhibited equally, ∼75%; inhibition at two concentrations of TFP5 is similar, whereas the scrambled peptide control has no effect. In view of our results in vivo, how do we explain the absence of any side effects due to inhibition of endogenous Cdk5/p35? In what does specificity reside?
FIGURE 3:
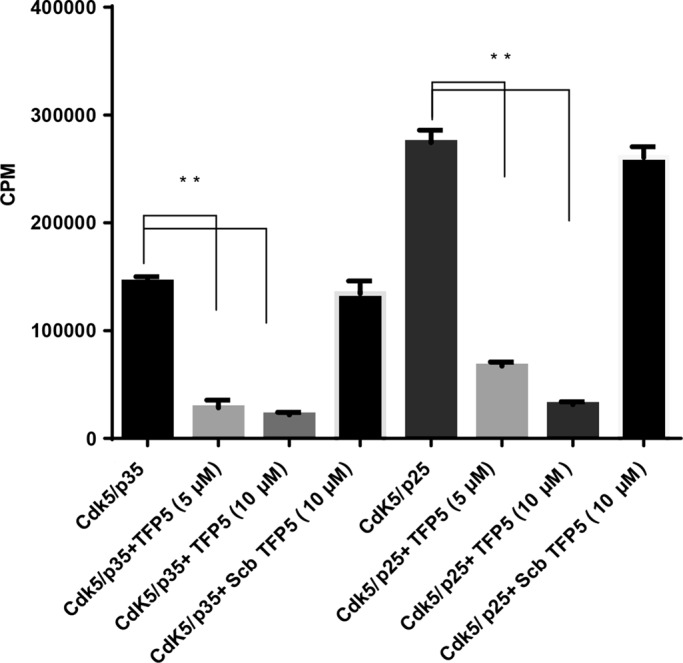
The TFP5 peptide inhibits Cdk5/p35 and Cdk5/p25 in vitro kinase assays. In an in vitro pad assay with histone as substrate, the TFP5 peptide inhibited Cdk5/p35 and Cdk5/p25 activities at 74 and 75%, respectively, at 5 μM concentration. Note that the scrambled peptide (Scb) at 10 μM has no effect on kinase activities. The results represent the value of three experiments and are expressed as ±SEM. **p < 0.01.
In vitro (test tube), in the presence of Munc 18, TFP5 selectively inhibits Cdk5/p25 but not Cdk5/p35 activity
We speculated that Munc18, like microtubules (Zheng et al., 2010), should bind the p10 domain of p35 and spare inhibition of Cdk5/p35 activity by TFP5 in vitro. Activity of Cdk5/p25, however, lacking the p10 domain, should be inhibited. To test these predictions, we set up in vitro assays with histone as substrate as before, in the presence and absence of Munc 18. We studied the effect on activity of three different concentrations of TFP5, 0.5, 1.0, and 2.0 μM (Figure 4). Figure 4, A and C, shows the autoradiographs and Coomassie-stained blots of Cdk5/p35 and Cdk5/p25, respectively. Quantitation of the data from four different experiments is shown in Figure 4, B and D. In the absence of Munc18, TFP5 inhibition is not significantly different between the two complexes (lanes 1–4 in each case, percentage inhibition of Cdk5/p35 varies from 0 to 41% and that of Cdk5/p25 from 11 to 38%). Of note, with the addition of Munc 18, activity of the Cdk5/p25 complex is still inhibited (15–53%) compared with the relatively unaffected Cdk5/p35 activity (7–17%; compare lanes 2–8, respectively). Selective inhibition by TFP5 in the presence of Munc 18 is consistent with the hypothesis; binding of Munc 18 to the p10 domain of p35 restores the regulator’s ability to activate Cdk5 phosphorylation. Without the p10 domain, however, p25 fails to compete with TFP5 binding, and phosphorylation is markedly inhibited.
FIGURE 4:

P67 specifically protects Cdk5/p35 from inhibition by the TFP5 peptide in vitro. The assay consists of active Cdk5/p35 or active Cdk5/p25 (5 ng), histone H1 as substrate (20 μg), bacterially expressed GST-p67 (0.375 μg), TFP5 at three concentrations (0.5, 1.0. and 2.0 μM), and [γ32P]ATP (0.05 mM). (A, C) Top, autoradiographs; bottom, Coomassie-stained gels of the kinase assays. (B, D) Mean densities of histone phosphorylation in the autoradiographs measured using ImageJ software. Results represent values from four experiments and are expressed as ±SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
A similar series of experiments completed with the TFP5 scrambled peptide as control displayed no effects on phosphorylation activity of each Cdk5 complex with or without the addition of Munc 18 (unpublished data). The real test of the hypothesis, however, should be in cortical neurons or in cells cotransfected with the respective active complexes and treated with TFP5.
TFP5 specifically inhibits the binding of p25 to Cdk5 transfected in HEK 293 cells without affecting the binding of transfected p35
The question arises whether TFP5 selectively inhibits Cdk5 kinase activity in cells as it does in vitro in the presence of Munc 18. Initially, we determined whether TFP5 preferentially inhibits p25 binding to Cdk5; we cotransfected HEK 293 cells with Cdk5/p25 or Cdk5/p35. We assumed that proliferating HEK cells should express high levels of cytoskeletal proteins such as actin and microtubules, which are known to be part of the p35 multimeric complex. These should act like Munc 18 and favor formation of an active Cdk5/p35 complex. P25, on the other hand, without the p10 domain, should not compete with TFP5 binding to Cdk5. After transfection, cells were incubated with TFP5 for 24 h. Cells were lysed, and the level of expression was determined in Western blots (Figure 5A). As can be seen, the levels of expression of Cdk5, p35, and p25 were robust in the lysates and unaffected by TFP5 treatment. To assess the competitive binding of TFP5 to Cdk5, we carried out Cdk5 immunoprecipitation using the J3 Cdk5 antibody for all lanes shown and surveyed the blots with the same antibodies as in Figure 5A. The results, shown in Figure 5B, indicate that the level of p25 coimmunoprecipitated with Cdk5 is decreased by TFP5; no significant changes in Cdk5–p35 binding, however, were seen. We suggest that by virtue of the p10 myristoylated domain in p35, binding of p35 to local cytoskeletal proteins such as endogenous microtubules competes more effectively for Cdk5 than TFP5 (Zheng et al., 2010). P25, without the p10 domain, is competitively displaced from Cdk5 by TFP5. As seen in the bar graph in Figure 5C, Cdk5/p25 binding is reduced and significantly inhibited (∼50%) by TFP5 (compare lanes 6 and 8). The scrambled peptide has no effect on binding (lane 7).
FIGURE 5:

TFP5 competes specifically with p25 binding to Cdk5 without affecting p35-Cdk5 binding in transfected HEK 293 cells. HEK 293 cells were transfected with Cdk5/p35 or Cdk5/p25 and incubated in 800 nM TFP5 or scrambled peptide control (SCB) for 24 h. (A, B) Lysates were prepared for Western blots to assess levels of expression (A) and subjected to a Cdk5 immunoprecipitation with J-3 antibody to determine the extent of p25 and p35 binding to Cdk5 by Western blots using the respective antibodies (B). (C) Mean density scans of lanes 3–8 were carried out with the National Institutes of Health ImageJ (n = 3, ±SEM, *p < 0.05)
How, then, do we explain the decreased binding of p25 to Cdk5 in the absence of TFP5, in spite of the robust p25 expression in the lysates (lanes 6 and 7)? It has been shown that most expressed p25 preferentially binds to GSK3 rather than Cdk5 in cotransfected neurons and neuroblastoma cells (Chow et al., 2014). Cells cotransfected with Cdk5, GSK3, p35, and p25 display a strong preferential binding of p25 to GSK3, even in the presence of Cdk5. P35 was shown not to bind to GSK3 under these conditions. In HEK cells, which share many genes with neurons and adrenal cells (Shaw et al., 2002), it is possible that endogenous GSK3 may act like a sponge to soak up most p25, leaving much less available to Cdk5. A Cdk5 immunoprecipitation would show strong p35 binding, while p25 binding to Cdk5 would be markedly reduced. This would account for the reduced p25 binding in lanes 6 and 7 of Figure 5B. Significantly, further reduction in binding is seen in the presence of TFP5 (lane 8), suggesting that the peptide competes successfully against any remaining p25 for the Cdk5 catalytic site. Lane 9, the positive control, is not shown in Figure 5C.
TP5 specifically inhibits Cdk5/p25 activity in transfected HEK 293 cells
If, in fact, as suggested earlier, TP5 inhibited binding of p25 to Cdk5, kinase activity should decrease with increasing concentration of TP5, whereas Cdk5/p35 activity should be unaffected. In a similar series of transfection experiments as described earlier, the Cdk5 immunoprecipitates were subjected to a phosphorylation assay with histone as substrate. The results of three experiments are summarized in Figure 6. In Figure 6A, the phosphorylation activity of Cdk5 IPs, as autoradiograms, is shown after cells were incubated in different concentrations of TP5. Note the reduction in histone phosphorylation by Cdk5/p25 in lanes 7–9. Aliquots of these same assays were used for pad assays, with data quantified in Figure 6B. The activity of the Cdk5/p25 complex is significantly inhibited from 50 to ∼84% with increasing doses of TP5, whereas activity of Cdk5/p35 is relatively unaffected (Figure 6B, lanes 2–5). The data correlate well with the binding data presented in Figure 5. Again, we suggest that conformational changes of p35–p10 binding to endogenous microtubules (Zheng et al., 2010), permit p35 to compete more effectively than TP5 for the catalytic site on Cdk5.
FIGURE 6:

Cdk5/p25 activity is specifically inhibited by TP5 peptide in transfected HEK 293 cells. HEK 293 cells were transfected with Cdk5/p35 or Cdk5/p25. The cells were then incubated in different concentrations of TP5 (0–800 nm) for 24 h, washed, and subjected to a Cdk5 immunoprecipitation procedure. (A) The respective Cdk5 immunoprecipitates were assayed for kinase activity with histone as substrate. Autoradiographs of the respective assays. H1, total histone; pH1, phospho-histone. (B). The identical lysates were used for Cdk5 kinase pad assays. Quantified results of three experiments (± SEM). *p < 0.05.
TFP5 does not inhibit Munc 18 binding to Cdk5/p35 in cotransfected HEK cells
Figure 1B showed that Munc 18 is coimmunoprecipitated with p35 in a rat brain lysate. The question arises as to whether TFP5 affects the binding of Munc 18 to a p35 multimeric complex. To determine whether Munc 18 binding to Cdk5/p35 in cells is unaffected in the presence of the TFP5 peptide, we cotransfected HEK 293 cells with Cdk5/p35-myc-histidine (his) or Cdk5/p25-myc-his plasmid, respectively. The cells were then incubated in different concentrations of TFP5 for 24 h and prepared as lysates (Figure 7A). Western blots to visualize proteins with appropriate antibodies were produced and sowed a robust level of expression of p67, p35 p25, Cdk5, and actin in lysates. Histidine-tagged immunoprecipitations were then carried out, followed by Western blots using the same antibodies as in Figure 7B. The level of p67 bound to Cdk5/p35 cells was unchanged at 250 nM, 500 nM, and 1 μM TFP5 (Figure 7C), whereas p67 bound to Cdk5/p25 was significantly decreased at the same concentrations of TFP5. This suggests that p67 binds to a multimeric complex on p35, whereas p25, without the p10 domain, fails to bind to the same extent. As a result, Cdk5/p25 activity is inhibited by the peptide, whereas Cdk5/p35 activity is spared. These results in transfected cells are consistent with the in vitro results presented in Figure 5.
FIGURE 7:

P67 preferentially binds to CDK5/p35 in the presence of TFP5 in cotransfected HEK 293 cells. HEK 293 cells were cotransfected with Cdk5/p35 (myc-his)/p67 or Cdk5/p25 (myc-his)/p67 and incubated in the presence of different concentrations of TFP5 peptide for 24 h. Lysates were then prepared and subjected to Western blotting to visualize protein expression of transfected plasmids with respective antibodies. (A) Expression of different proteins in the lysates using p67, p35/p25, Cdk5, and β-actin antibodies. (B) Immunoprecipitation using 500 μg of proteins (p35 myc-his and p25 myc-his) with monoclonal his-tag antibody detected with p67, p35, Cdk5, and β-actin antibodies, respectively, after Western blotting. (C) Quantification of immunoblots of p67 and p35 antibodies, respectively, from three experiments (± SEM). **p < 0.01.
Immunoprecipitates of myc-his-p35–transfected cortical neurons significantly bind more proteins than immunoprecipitates of cells transfected with myc-his-p25
Our hypothesis predicts that the p10 domain of p35 is essential in the formation of multimeric complexes. To test this hypothesis further, we overexpressed myc-his-p35 or myc-his-p25 in rat cortical neurons. A comparable pull-down experiment as before using myc antibody should yield multimeric complexes in myc-p35 neurons that are absent in myc-p25–overexpressing neurons (Figure 8). The level of expression in the two populations of transfected neurons is shown in Figure 8A. Expression levels of myc-p35 and myc-p25 are equivalent in both groups. With the same myc antibody, we carried out immunoprecipitations on neuronal lysates and screened for cytoskeletal and synaptic proteins using appropriate specific antibodies. Input shows expression of each protein in the lysate. Except for glial fibrillary acidic protein (GFAP), comparison of the Western blots shows dramatic qualitative and quantitative differences in protein profiles copurified from each, with that from p25-transfected cells expressing lower levels of proteins (Figure 8, B–F). Again, these data are consistent with the hypothesis that a p10 multimeric complex of p35 specifically spares Cdk5/p35 activity from TFP5 inhibition.
FIGURE 8:

The p10 N-terminal domain of p35 preferentially binds multimeric proteins in cortical neurons transfected with p35 myc-his. Rat cortical neurons at 5 days in vitro were transfected with p35 myc-his or p25 myc-his plasmid. After 48 h, lysates were prepared and subjected to SDS–PAGE using 30 μg of lysate for each lane. (A) Protein expression was analyzed using three different antibodies to confirm p35 and p25 expression with p35 (C-19), myc, and his antibodies, respectively. Arrowhead and arrow represent the expression of myc-his p35 and myc-his p25, respectively. (B–F). Both p35 and p25 lysates were used at 500-μg protein concentration for immunoprecipitation with mouse monoclonal myc antibody. Input represents expression level of each protein. After SDS–PAGE, Western blotting was performed using different polyclonal antibodies as follows: p67 (B), PSD95 (C), tau (D), GFAP (E), and syntaxin 1 (F). Data were analyzed with Prism 5.0 software. Tukey’s multiple comparisons test was used. Differences with *p < 0.05 were considered significant. The quantification of bands was done using ImageJ software. The results represent the value of three experiments and are expressed as ±SEM. **p < 0.01 and ***p < 0.001.
Down-regulation of Munc 18 by p67 siRNA in TFP5-treated cortical neurons results in inhibition of Cdk5/p35 binding and phosphorylation activity
If, as we suggest, Munc 18 is essential in sustaining the activity of Cdk5/p35 in the presence of the TFP5, then reducing Munc 18 from cortical neurons should increase sensitivity to TFP5 inhibition, similar to its effect on Cdk5/p25. A converse experiment of the one described in Figure 9 was carried out by reducing or eliminating the expression of Munc 18 in rat cortical neurons by transfection with p67 siRNA, followed by incubation with TFP5 (200 nM) was shown to be effective within neurons (Figure 9). The expression levels for each protein are displayed in Figure 9A. It is clear that the presence of the p67 siRNA results in a reduction in the expression of p67; control siRNA has no effect. Note that the presence or absence of TFP5 or its control scrambled peptide (Scb) has no effect on the expression levels of p35 and Cdk5. A Cdk5 immunoprecipitation with the J3 antibody provides a measure of Cdk5 kinase activity using histone as a substrate (Figure 9B). In the presence of p67 siRNA (lane 5), a greater reduction of histone phosphorylation is seen in the presence of TFP5. This also correlates with reduced p35 binding in lane 5. Scrambled peptide and/or the control siRNA have no effect on binding of either p67 or of p35. The Cdk5 immunoprecipitates in each lane were assayed for activity in a pad assay (Figure 9C,) and a clear inhibition by TFP5 is seen in lane 5, whereas scrambled peptide or p67 control siRNA has no effect on activity (compare lane 5 with lanes 3 and 4). These data are consistent with the hypothesis that Munc 18 binding to the p10 N-terminal domain of p35 spares Cdk5/ activity in vivo while specifically inhibiting Cdk5/p25, which lacks the p10 domain.
FIGURE 9:

TFP5 inhibited p35/Cdk5 activity in cortical neurons transfected with p67 siRNA. P67 siRNA and control siRNA were transfected into rat primary cultured cortical neurons (on day 7). Cells were then incubated with TFP5 (200 nM) or SCB (200 nM) peptides for 72 h. Cells treated with roscovitine (25 μM) for 2 h were used as a positive control. Cells were harvested and subjected to Western blotting and Cdk5 kinase assay. (A) Western blots showing the expression of p67 and p35 in the cells transfected with p67 siRNA and treated with TFP5 or SCB peptides. P67 expression was reduced (lanes 4–7) in the presence of p67 siRNA. P67 siRNA did not affect p35 or Cdk5 expression (lanes 1–7). (B) Radiographs of Cdk5 IP activity in cells transfected with p67 siRNA were inhibited by TFP5 and unaffected by scrambled peptide (compare lanes 4 and 5). Roscovitine showed robust inhibition of Cdk5 activity (lane 7). (C) Activities of the same lysates determined in pad assays. Data are expressed as mean of three separate experiments. *p < 0.01.
FIGURE 10:
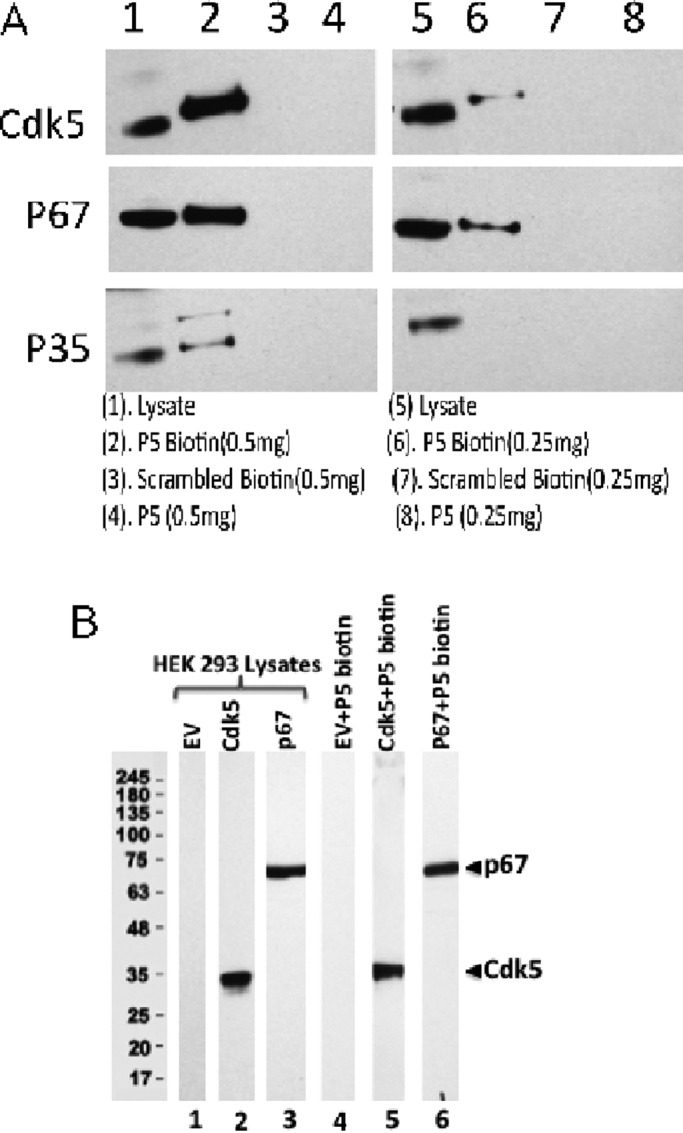
P5-biotin pull-down assays of rat brain lysates and transfected HEK293 cells show P5 binding to Cdk5, p35, and p67. (A) Two different concentrations of P5-biotin were used in affinity binding assays with Scb (scrambled peptide) as negative control. Protein extracts (500 μg in 500 μl) from mouse brain tissues were incubated with different concentrations (0.5 and 0.25 mg/ml) of P5-biotin, scrambled-biotin, or P5 without biotin overnight at 4°C. On the second day, streptavidin magbeads (50 μl) were added to the lysates for 2 h at 4°C. After gentle spinning, streptavidin magbeads were collected and washed three times. The proteins were eluted from streptavidin magbeads by using sample buffer and heating at 96°C for 5 min. The eluted proteins were used for Western blot analysis, where each lane is immunoassayed with the three antibodies. (B) P5-biotin binds directly to both Cdks and p67. Expression of HEK293 cell lysates (25 μg) as transfected with empty vector (EV; lane 1), Cdk5 (lane 2), or p67 (lane 3). A 500-μg amount of each lysate was mixed with 0.5 mg of P5-biotin, followed by end-to-end rotation at 4°C overnight. On the next morning, the complex was pulled down with streptavidin beads. After washing three times with l× Tris-buffered saline, the beads were subjected to SDS–PAGE and Western blot analysis using anti-Cdk5 and anti-p67 antibodies in each lane. The EV negative control indicates that endogenous p67 and Cdk5 in HEK 293 cells are either absent or at levels too low to be detected.
A TFP5-biotin pull down of brain lysates confirms peptide interactions with Cdk5/p35 and p67
Although we showed that a p67 pull down of brain lysates shows interactions with a multimeric protein complex containing Cdk5/p35, synaptic, and cytoskeletal proteins, a reciprocal pull down with P5 (TFP5) might also reveal interactions with proteins of the multimeric complex. Because we have no specific P5 antibody, we used the biotin-streptavidin procedure with a custom-designed P5-biotin preparation (Figure 10 and Supplemental Figure S1). We showed that the binding of biotin to the P5 peptide had no effect on peptide-specific inhibition of Cdk5/p25 or Cdk5/p35 activity in vitro (Supplemental Figure S1). Clearly, addition of the biotin to the P5 at different concentrations showed equivalent inhibition of the Cdk5 active complexes. Scrambled biotin-P5 had no inhibitory effect on kinase activity.
P5-biotin immunoprecipitations of mouse brain lysates colocalize Cdk5/p35 and p67
The pull-down assay with two concentrations of P5-biotin reveal the specific binding of Cdk5, p35, and p67 to P5-biotin; P5 alone and scrambled peptide-biotin have no effect (Figure 10A). In this experiment, it is possible that the P5-biotin bound to p67, which in turn was also bound to Cdk5/p35, or P5-biotin bound directly to a Cdk5/p35/p67 complex. To determine which protein is directly bound by P5-biotin, we carried out the experiment in HEK 293 cells transfected with p67 or Cdk5. In Figure 10B, the P5 peptide binds independently to each of the proteins. P5-biotin binding to Cdk5 is expected because the peptide binds at the activation to inhibit the kinase activity. Its binding to p67 suggests that the peptide, in the presence of proteins of a multimeric complex on the N-terminal domain of p35, may preferentially bind to the complex, reducing its ability to bind Cdk5, favoring p35 binding and activation of the kinase. These results are consistent with the hypothesis that sparing of Cdk5/p35 activity in vivo is due to binding of the inhibitory peptide to proteins of the multimeric complex on the p10 domain of p35.
Glutathione S-transferase p10 pull down of rat brain lysate independently reveals cytoskeletal and synaptic proteins
To demonstrate that the p-10 N-terminal domain of p35 specifically interacts with cytoskeletal proteins in addition to Cdk5, we used a published procedure for glutathione S-transferase (GST) pull down of p35 truncated fragments (Qu et al., 2002).
We prepared a recombinant GST-p10 protein that was incubated with an adult rat brain lysate, prepared as a gel, and visualized in Westerns with several different antibodies as shown in Figure 11. As a control, we used only the GST that pulls down tau, actin, and tubulin, three common proteins (Figure 11A). The GST-p10 pulls down a more robust expression of the cytoskeletal proteins, including NFL, as well as p35 and the synaptic proteins p67 and syntaxin (Figure 11B). These last proteins are similar to those detected in p67 and p35 pull downs, respectively, described in Figure 1.
FIGURE 11:
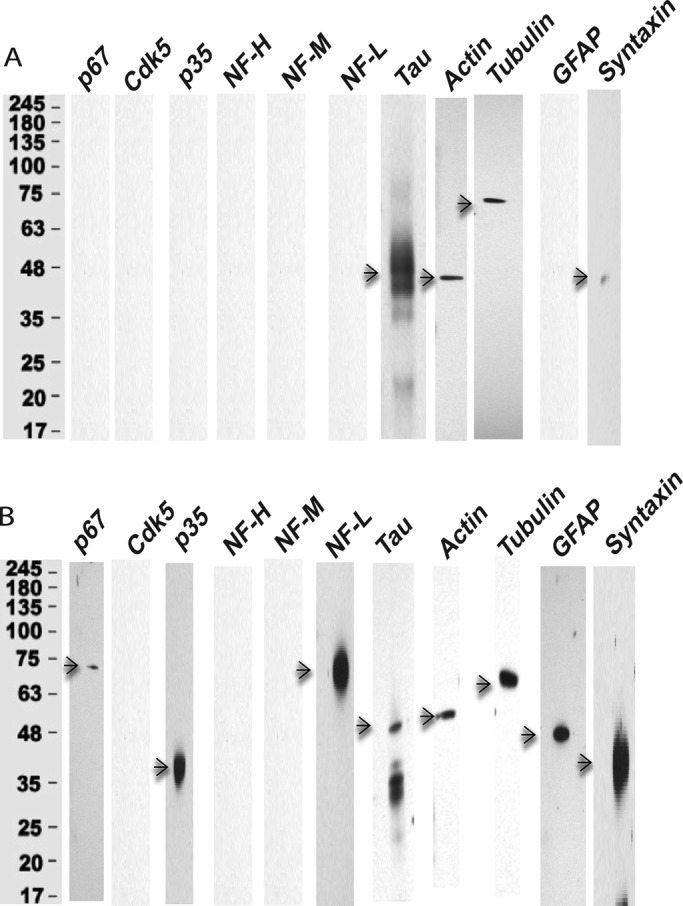
Identification of GST-p10 proteins pulled down from the rat brain lysate. Proteins were isolated from rat brain extracts in pull-down experiments using GST or GST-p10 and separated by SDS–PAGE on 4–20% gels and immunodetected with Western blot (WB) analysis using respective antibodies as shown (n = 3). (A) GST control. Except for the expression of tau, β-actin, and β-tubulin, very few proteins were pulled down from rat brain lysate with GST alone. (B) In contrast, the GST-p10 pull down resulted in the expression of cytoskeletal proteins NFL, tubulin, actin, and tau and included expression of p67, p35, GFAP, and syntaxin.
These results are consistent with our hypothesis that the p10 domain of p35 and its associated multimeric complex of cytoskeletal and synaptic proteins are what alter the p35 conformation such that it successfully activates Cdk5 kinase activity in vivo in the presence of the TFP5 peptide. P25, without the p10 domain, fails to compete with the peptide, which associates with the kinase catalytic site and inhibits activity.
DISCUSSION
Deregulated Cdk5/p25 has been identified as a key player in the production of neurodegenerative pathologies such as paired helical filaments, amyloid plaques, and synaptic dysfunction, leading to neuronal cell death, behavioral abnormalities, and dementia. Consequently the hyperactive complex has been proposed as a principal therapeutic target for AD and other neuronal disorders (Lau et al., 2002; Lau and Ahlijanian, 2003; Tsai et al., 2004). As members of the family of cell cycle cyclin-dependent kinases (Cdks), several potent inhibitors resembling roscovitine have been studied as potential therapeutic candidates to target Cdk5/p25 activity. Although p35 and p39, rather than cyclins, activate Cdk5, kinase inhibition by roscovitine (or the related compounds aloisine and indirubin-3) is nonspecific; it also affects Cdks, as it competes at the ATP-binding site essential for the action of Cdks, as well as of most kinases. A search for specific inhibitors that act at sites other than the ATP-binding site is essential and underway in several laboratories (Lau et al., 2002; Glicksman et al., 2007). For example, a library of 110,000 compounds is being screened using full-length tau as a substrate to distinguish competitive from noncompetitive ATP inhibitors (Glicksman et al., 2007). Our approach, based on p35-truncated peptides, has successfully demonstrated specific inhibition of Cdk5/p25 in cortical neurons and AD mouse models without affecting Cdk5/p35 activity or that of cell cycle Cdks (Amin et al., 2002; Zheng et al., 2010). We believe that results from our experiments with Munc 18 offer a molecular explanation for the specific sensitivity of the deregulated toxic target.
Hyperactive Cdk5/p25 arises in situ from activated, stressed, or aging neurons in which stress factors such as glutamate toxicity, ROS elevation, and mutations in APP and/or presenilins induce calpain activation and cleavage of p35 into p10 and p25. P25 collects in the cytosol and/or nucleus, whereas p10 accumulates in the detergent-resistant fraction of membrane-bound organelles and insoluble membrane proteins with cytoskeletal elements (Kanungo et al., 2009). Our data are consistent in showing that the p10 domain of p35 is essential in 1) binding p35 to the cell membrane, 2) interacting with cytoskeletal proteins (tubulin, actins, neurofilaments, Munc 18, and tau) to regulate their macromolecular assembly, and 3) targeting Cdk5 to specific substrates within multimeric complexes at the membrane, within the cytosol, and in the nucleus. The last result accounts for the presence in bovine brain extracts of three Cdk5 states: Cdk5 as part of a large (670 kDa), inactive multimeric complex, a monomeric Cdk5 that can be complexed to a p35 recombinant and activated, and a highly active Cdk5/p25 heterodimer (Lee et al., 1996). Our data from rat brains confirm the existence of such macromolecular complexes containing tubulin, neurofilaments, and synaptic proteins together with Cdk5, p35, and p67 (Bhaskar et al., 2004; Figure 1). Moreover, a pull-down experiment with P5-biotin showed that the inhibitor peptide did bind to proteins of the multimeric complex, namely Cdk5, p35, and p67. This suggests that the peptide binds to the p10 domain of p35 and, as a result, fails to interfere with p35 binding to the critical catalytic site of Cdk5. In a number of experiments, we demonstrate that this interaction of p35 with macromolecules at its N-terminal p10 domain accounts for the specificity of TFP5 inhibition in cells; p25 without p10 is unable to compete for the Cdk5 active site in the presence of TFP5. Significantly, the compelling result is the demonstration that p10 independently binds proteins of the multimeric complex (Figure 11). Finally, in a critical negative experiment, we demonstrated that TFP5 treatment inhibited endogenous Cdk5/p35 activity when p67 expression was down-regulated in cortical neurons transfected with p67 siRNA (Figure 9). Reduction in the expression of p67, a key protein in the multimeric complex at the p10 domain of p35, is sufficient to inhibit Cdk5/p35 in neurons.
It is noteworthy that Cdk5/p25-induced toxicity in neurons is protected by overexpression of the p10 peptide (Zhang et al., 2012). It was shown that high doses of MPP+-induced toxicity in cortical neurons, a model for Parkinson’s disease, was significantly reduced after p10 overexpression; several defects were inhibited, including Cdk5/p25 activation, ROS accumulation, and DNA strand breakage, which mark neuronal death. The cascade leading to cell death is Cdk5/p25 phosphorylation and inhibition of peroxireductase (Prx2), responsible for ROS accumulation and cell death. p10 inhibition of Cdk5/p25 activates Prx2, restores ROS regulation, and rescues cell survival (Zhang et al., 2012). Zhang et al. (2012) write, “p10 (protein of 10 kDa) is a unique prosurvival domain in p35 essential for normal Cdk5/p35 function in neurons.” Here the effect of p10 seems to require unknown cellular factors to form an inhibitory complex. An alternative study claiming that p10 is toxic rather than protective is difficult to explain, other than attributing it to differences in experimental conditions (Chew et al., 2010).
Our results with Munc 18, however, suggest an alternative mechanism for p10’s effect on Cdk5/p35 activity—the domain is a site of multiple protein interactions that stabilize p35 binding to the Cdk5 catalytic site. The structural configuration of p35, when bound to microtubules and/or Munc 18 in a multimeric complex, which protects its interaction with/activation of Cdk5 in the presence of TFP5, is not understood. Because the Cdk5/p35 crystal structure is not known, an answer is best obtained from studies of the Cdk5/p25 structure in the presence and absence of roscovitine inhibitors (Lim et al., 2001; Tarricone et al., 2001; Mapelli et al., 2005) and/or from a molecular dynamics study of competitive binding to Cdk5 of the large inhibitory peptide CIP derived from p35 (Tan et al., 2009).
The structure of the Cdk5/p25 complex and the details of enzyme–activator molecular interactions, although defined for p25, also apply to its p35 parent. The N-terminal p10 motif is assumed to play no role in the interactions; it may be essential for localization, maintaining stability of the active configuration, and/or docking to other regulators (Tarricone et al., 2001). The active configuration is induced by shared loops and residues on both regulators that appose and bind numerous specific residues of the enzyme to extend the activation loop (T loop), thereby creating a hydrophobic pocket facilitating substrate interaction. We suggest that p25 binding induces a more stable configuration; that is, it may extend the activation loop much more than p35, which may account for its hyperactivity. The Ile-153 residue in the loop renders the specificity of the interaction; cyclins fail to make that contact. This direct and unique association of p35 and p25 with the activation loop is independent of its phosphorylation of the activation loop, as is required for the Cdk2 family of kinases. These fundamental differences between Cdk5 activators and Cdc2-like cyclin activators may account for the strong selectivity of TFP5 inhibition.
A molecular dynamics simulation of the interaction of CIP—the larger inhibitory peptide derived from N- and C-terminal truncations of p25—suggests that significant conformational changes are induced in the hydrophobic pocket, which affects substrate binding and inhibits activity (Tan et al., 2009; Cardone et al., 2010a, b, 2016). Given that P5 is derived from CIP, we speculate that it behaves like CIP and interferes with extension of the activation loop of Cdk5. The P5 sequence includes critical residues essential for direct binding to the activation loop. We suggest that this smaller molecule competes more successfully with p25 but fails to extend the activation loop, leading to an inactive configuration. The p10 N-terminal domain in p35, however, plays no role in binding to Cdk5; myristoylated, it preferentially associates with membranes or other macromolecules (Munc 18, microtubules, neurofilaments), assembling into multimeric complexes, which may stabilize Cdk5 binding. At the same time, such structural displacements may render the p35 into a more competitive configuration, which, even in the presence of P5 (TFP5), succeeds in sustaining T-loop extension and activity. Although our data are consistent with this model, only additional crystallographic studies and molecular dynamics simulations of Cdk5/p35 interactions with substrates, P5 inhibitors, and macromolecules could reveal the nature of the molecular interactions.
Figure 12 summarizes the experimental results and illustrates the hypothesis that the presence of the p10 domain on the p35 regulator successfully protects the activity of the Cdk5/p35 endogenous complex from the TFP5 peptide inhibitor while specifically inhibiting the Cdk5/p25 complex.
FIGURE 12:
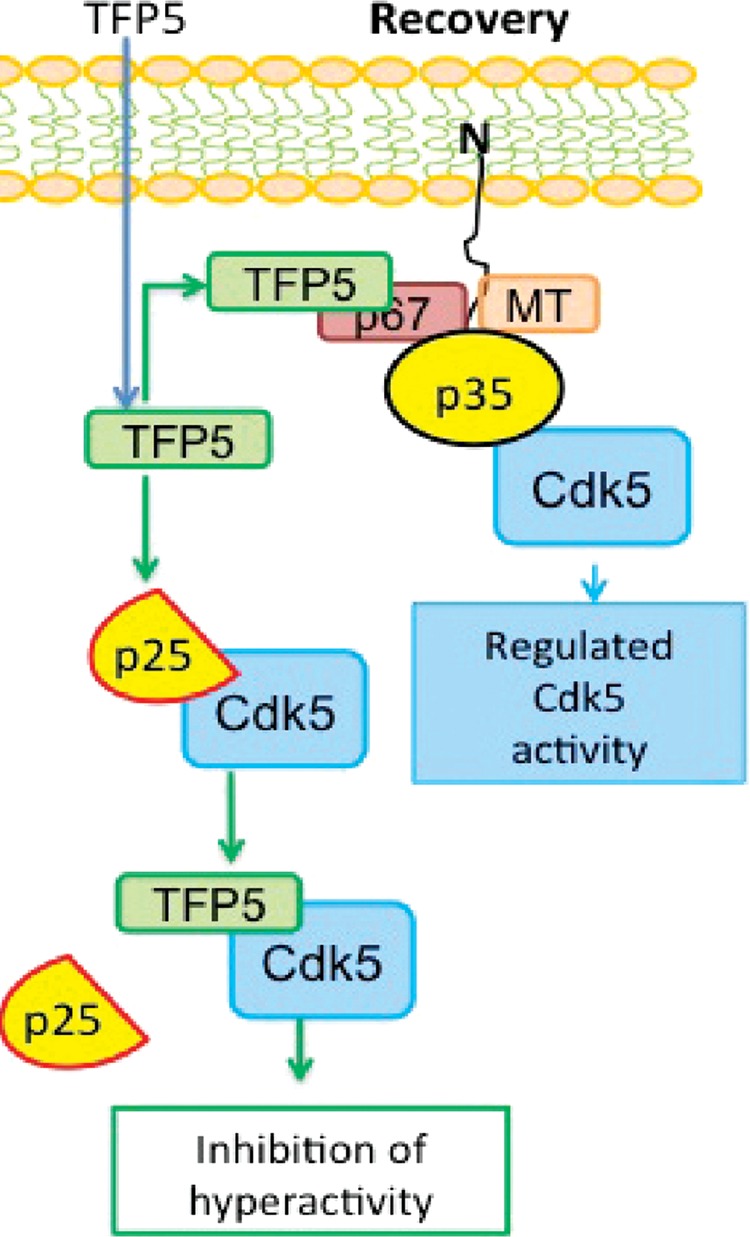
Diagram illustrating the hypothesis to explain the P5 specific inhibition of Cdk5/p25 in vivo. The recovery neuronal state after P5/TFP5 treatment. In recovery, TFP5 entering the neuron preferentially interacts with the endogenous Cdk5/p35 multimeric complex, which prevents peptide association with catalytic sites on Cdk5, whereas in neurons with hyperactive Cdk5/p25, TFP5 readily competes with p25 on Cdk5 catalytic sites and inhibits kinase activity.
MATERIALS AND METHODS
Materials
Cdk5 (J-3, C-8), p35 (C-19), SNAP 25, syntaxin-1, synaptophysin, and synaptotagmin (dilution 1:500), β-actin (1:2000), and p67 (Munc 18) siRNA were obtained from Santa Cruz Biotechnology (Santa Cruz Biotechnology, Santa Cruz, CA). Mouse monoclonal Munc 18 (p67; dilution 1:500) was from BD Biosciences (San Jose, CA). Neurofilament H/M/L, β-tubulin, monoclonal p35 antibodies (dilution 1:500), and active Cdk5/p35/p25 kinases were purchased from Sigma-Aldrich (St. Louis, MO). Rabbit polyclonal tau (1:1000) was from Dako (Carpinteria, CA), and monoclonal tau (1:500) was from Invitrogen (Grand Island, NY). GFAP (1:1000) was from Abcam (Cambridge, MA). Mouse monoclonal Cdk5 (1:500) was from OriGene (Rockville, MD). Secondary horseradish peroxidase–conjugated antibodies (1:2500) were from GE Healthcare (Marlborough, MA). Roscovitine was from BIOMOL Research Laboratories (Plymouth Meeting, PA). TFP5, FITCGGGKEAFWDRCLSVINLMSSKMLQINAYARAARRAARR, and scrambled FITCGGGGGGFWDRCLSGKGKMSSKGGGINAYARAARRAARR peptides were synthesized by GenScript (Piscataway, NJ), and γ-p32 ATP was from PerkinElmer (Waltham, MA). HEK 293 cells were from the American Type Culture Collection (ATCC; Manassas, VA). P5 Scb-biotin peptide was from Government Scientific Source (Reston, VA). Transfection reagents DNA in Neuro and DNA in 293 cells were purchased from MTI-Globalstem (Gaithersburg, MD). Transfection reagent for siRNA was from Signagen Laboratories (Rockville, MD). pcDNA 3.1/myc-his (-) A vector was a kind gift from Sunita Agarwal (National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, MD). T-PER, M-PER, and N-PER lysis buffers and protease inhibitors were purchased from Thermo Fisher Scientific (Grand Island, NY).
The p35 and p25 plasmids were cloned into pcDNA 3.1/myc-his (-) A vector at 5′ NheI and 3′ ApaI sites.
Cell cultures
Primary cultures of rat cortical neurons were prepared from E-18 rat fetuses as described previously (Zheng et al., 2002). Transfection of plasmids was performed at day in vitro 5 with DNA in Neuro following the manufacturer’s instructions unless otherwise stated. At 48 h posttransfection, lysates were prepared for further analysis.
HEK293 cells were obtained from ATCC, cultured in DMEM with 10% calf serum, and supplemented with 100 U/ml penicillin and 100 μg/ml streptomycin. Cells were maintained at 37°C in a humidified atmosphere of 5% CO2. The cells were transiently transfected using DNA in 293 according to the manufacturer’s instructions in the presence of TFP peptide at different concentrations and treated 2 h before transfection unless otherwise stated. At 24 h posttransfection, the cells were lysed with M-per lysis reagent with protease and phosphatase inhibitors. These lysates were subjected to SDS–PAGE and immunoprecipitation for further analysis.
Immunoprecipitation
Immunoprecipitations (IPs) from transfected HEK 293 cells and cortical neurons were performed essentially as described previously (Kesavapany et al., 2007). For rat brain IP studies, 500 μg of protein was used for each IP to ensure maximum coimmunoprecipitation of interactive proteins.
Immunoblotting
Western blot analysis was performed as described previously (Zheng et al., 2002). In brief, cortical neurons and HEK 293 cells were harvested by scraping from dishes, lysed in ice-cold lysis buffer, and incubated for 30 min on ice. After centrifugation for 30 min at 13,000 × g at 4°C, the protein concentrations of the supernatants were determined using bicinchoninic acid protein reagent. An equal amount of total protein (25 μg of protein/lane) was resolved on a 4–20% SDS–polyacrylamide gel and transferred onto a nitrocellulose membrane. This membrane was incubated in blocking buffer containing 20 mM Tris-HCl, pH 7.4, 150 mM NaCl, and 0.1% (vol/vol) Tween 20 (TTBS) plus 5% dry milk (wt/vol) for 1 h at room temperature. This was followed by incubation overnight at 4°C in primary antibodies. The membranes were then washed four times in TTBS (5 min each). This was followed by incubation in secondary antibody (goat anti-mouse or goat anti-rabbit/mouse immunoglobulin G (IgG) with horseradish peroxidase conjugate at a dilution of 1:2500) for 2 h at room temperature. Western blots were analyzed using the GE Healthcare enhanced chemiluminescence kit following the manufacturer’s instructions. Quantitative assay of antigen expression was based on density measurements of protein bands using ImageJ software (http://rsb.info.nih.gov/ij/).
GST-p10 pull down of rat brain lysate
This was based on a procedure published in Qu et al. (2002).
For plasmid construction, GST-p10 was cloned into pGEX 4T-2 at 5′ BamHI and 3′ EcoRI sites using as forward primer GATCGGATCCATGGGCACGGTGCTGTCCCT and as reverse primer GATCGAATTCCTAGAATGTGGACAGGTTGGCGC.
The positive clones were verified by restriction digestion and sequencing.
GST-p10 was expressed and purified essentially as described previously (Amin et al., 2002) and also followed manufacturer’s instructions for purification.
For isolation of GST-p10 interacting proteins, lysate of adult rat brain was prepared by homogenizing in T-Per lysis buffer with protease and phosphatase inhibitors (Thermo Fisher Scientific). GST or GST-p10 was coated on GSH beads, mixed with 1.5 mg of rat brain lysate, and incubated overnight in the cold with end-to-end rotation. The beads were then extensively washed with extraction buffer provided with the kit. The proteins were eluted with reduced glutathione, subjected to SDS–PAGE, and Western blotted with a variety of antibodies for immunodetection.
Kinase assay
Kinase assays were performed as described previously (Veeranna et al., 2000; Zheng et al., 2002).
Statistical analysis
Data were analyzed with Prism 3.0 software (GraphPad Software, San Diego, CA). Bonferroni and Dunnett multiple comparison testing was used. Differences with *p < 0.05 were considered significant.
Supplementary Material
Acknowledgments
We acknowledge Christine Winters (National Institutes of Health, National Institute of Neurological Disorders and Stroke) for providing cortices for the cortical neuron preparations. This research was supported by the Intramural Research Programs of the National Institutes of Health, National Institute of Neurological Disorders and Stroke.
Abbreviations used:
- AD
Alzheimer’s disease
- CIP
inhibitory peptide
- SCB
scrambled peptide control.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E15-12-0857) on September 14, 2016.
REFERENCES
- Ahlijanian MK, Barrezueta NX, Williams RD, Jakowski A, Kowsz KP, McCarthy S, Coskran T, Carlo A, Seymour PA, Burkhardt JE, et al. Hyperphosphorylated tau and neurofilament and cytoskeletal disruptions in mice overexpressing human p25, an activator of cdk5. Proc Natl Acad Sci USA. 2000;97:2910–2915. doi: 10.1073/pnas.040577797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin ND, Albers W, Pant HC. Cyclin-dependent kinase 5 (cdk5) activation requires interaction with three domains of p35. J Neurosci Res. 2002;67:354–362. doi: 10.1002/jnr.10116. [DOI] [PubMed] [Google Scholar]
- Angelo M, Plattner F, Giese KP. Cyclin-dependent kinase 5 in synaptic plasticity, learning and memory. J Neurochem. 2006;99:353–370. doi: 10.1111/j.1471-4159.2006.04040.x. [DOI] [PubMed] [Google Scholar]
- Bhaskar K, Shareef MM, Sharma VM, Shetty AP, Ramamohan Y, Pant HC, Raju TR, Shetty KT. Co-purification and localization of Munc18-1 (p67) and Cdk5 with neuronal cytoskeletal proteins. Neurochem Int. 2004;44:35–44. doi: 10.1016/s0197-0186(03)00099-8. [DOI] [PubMed] [Google Scholar]
- Bibb JA. Role of Cdk5 in neuronal signaling, plasticity, and drug abuse. Neurosignals. 2003;12:191–199. doi: 10.1159/000074620. [DOI] [PubMed] [Google Scholar]
- Cardone A, Albers RW, Sriram RD, Pant HC. Evaluation of the interaction of cyclin-dependent kinase 5 with activator p25 and with p25-derived inhibitor CIP. J Comput Biol. 2010b;17:707–721. doi: 10.1089/cmb.2009.0202. [DOI] [PubMed] [Google Scholar]
- Cardone A, Brady M, Sriram R, Pant HC, Hassan SA. Computational study of the inhibitory mechanism of the kinase CDK5 hyperactivity by peptide P5 and derivation of a pharmacophore. J Comput Aided Mol Des. 2016;30:513–521. doi: 10.1007/s10822-016-9922-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardone A, Hassan SA, Albers RW, Sriram RD, Pant HC. Structural and dynamic determinants of ligand binding and regulation of cyclin-dependent kinase 5 by pathological activator p25 and inhibitory peptide CIP. J Mol Biol. 2010a;401:478–492. doi: 10.1016/j.jmb.2010.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng K, Ip NY. Cdk5: a new player at synapses. Neurosignals. 2003;12:180–190. doi: 10.1159/000074619. [DOI] [PubMed] [Google Scholar]
- Cheung ZH, Gong K, Ip NY. Cyclin-dependent kinase 5 supports neuronal survival through phosphorylation of Bcl-2. J Neurosci. 2008;28:4872–4877. doi: 10.1523/JNEUROSCI.0689-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chew J, Chen MJ, Lee AY, Peng ZF, Chong KW, He L, Bay BH, Ng JM, Qi RZ, Cheung NS. Identification of p10 as a neurotoxic product generated from the proteolytic cleavage of the neuronal Cdk5 activator. J Cell Biochem. 2010;111:1359–1366. doi: 10.1002/jcb.22864. [DOI] [PubMed] [Google Scholar]
- Chow HM, Guo D, Zhou JC, Zhang GY, Li HF, Herrup K, Zhang J. CDK5 activator protein p25 preferentially binds and activates GSK3beta. Proc Natl Acad Sci USA. 2014;111:E4887–E4895. doi: 10.1073/pnas.1402627111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz JC, Tsai LH. Cdk5 deregulation in the pathogenesis of Alzheimer’s disease. Trends Mol Med. 2004;10:452–458. doi: 10.1016/j.molmed.2004.07.001. [DOI] [PubMed] [Google Scholar]
- Cruz JC, Tseng HC, Goldman JA, Shih H, Tsai LH. Aberrant Cdk5 activation by p25 triggers pathological events leading to neurodegeneration and neurofibrillary tangles. Neuron. 2003;40:471–483. doi: 10.1016/s0896-6273(03)00627-5. [DOI] [PubMed] [Google Scholar]
- Dhavan R, Tsai LH. A decade of CDK5. Nat Rev. 2001;2:749–759. doi: 10.1038/35096019. [DOI] [PubMed] [Google Scholar]
- Fu X, Choi YK, Qu D, Yu Y, Cheung NS, Qi RZ. Identification of nuclear import mechanisms for the neuronal Cdk5 activator. J Biol Chem. 2006;281:39014–39021. doi: 10.1074/jbc.M512663200. [DOI] [PubMed] [Google Scholar]
- Giese KP, Ris L, Plattner F. Is there a role of the cyclin-dependent kinase 5 activator p25 in Alzheimer’s disease. Neuroreport. 2005;16:1725–1730. doi: 10.1097/01.wnr.0000185019.67434.d2. [DOI] [PubMed] [Google Scholar]
- Glicksman MA, Cuny GD, Liu M, Dobson B, Auerbach K, Stein RL, Kosik KS. New approaches to the discovery of cdk5 inhibitors. Curr Alzheimer Res. 2007;4:547–549. doi: 10.2174/156720507783018181. [DOI] [PubMed] [Google Scholar]
- Grant P, Sharma P, Pant HC. Cyclin-dependent protein kinase 5 (Cdk5) and the regulation of neurofilament metabolism. Eur J Biochem. 2001;268:1534–1546. [PubMed] [Google Scholar]
- Guo Q. When good Cdk5 turns bad. Sci Aging Knowledge Environ. 2006;2006:pe5. doi: 10.1126/sageke.2006.5.pe5. [DOI] [PubMed] [Google Scholar]
- Gupta A, Tsai LH. Cyclin-dependent kinase 5 and neuronal migration in the neocortex. Neurosignals. 2003;12:173–179. doi: 10.1159/000074618. [DOI] [PubMed] [Google Scholar]
- Hamdane M, Buee L. The complex p25/Cdk5 kinase in neurofibrillary degeneration and neuronal death: the missing link to cell cycle. Biotechnol J. 2007;2:967–977. doi: 10.1002/biot.200700059. [DOI] [PubMed] [Google Scholar]
- Han YQ, Sun AM, Liu QL, Chen LH, Yuan YW. p35 and p25 expressions and Cdk5 kinase activity in primary cultured rat hippocampal neurons with X-ray exposure [in Chinese] Nan Fang Yi Ke Da Xue Xue Bao [J Southern Med Univ] 2009;29:405–407. [PubMed] [Google Scholar]
- He L, Zhang Z, Yu Y, Ahmed S, Cheung NS, Qi RZ. The neuronal p35 activator of Cdk5 is a novel F-actin binding and bundling protein. Cell Mol Life Sci. 2011;68:1633–1643. doi: 10.1007/s00018-010-0562-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Z, Li Q, He L, Lim HY, Fu X, Cheung NS, Qi DX, Qi RZ. Microtubule association of the neuronal p35 activator of Cdk5. J Biol Chem. 2007;282:18666–18670. doi: 10.1074/jbc.C700052200. [DOI] [PubMed] [Google Scholar]
- Jessberger S, Gage FH, Eisch AJ, Lagace DC. Making a neuron: Cdk5 in embryonic and adult neurogenesis. Trends Neurosci 32, 575–582. 2009. [DOI] [PMC free article] [PubMed]
- Kanungo J, Zheng YL, Amin ND, Pant HC. Targeting Cdk5 activity in neuronal degeneration and regeneration. Cell Mol Neurobiol. 2009;29:1073–1080. doi: 10.1007/s10571-009-9410-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kesavapany S, Zheng YL, Amin N, Pant HC. Peptides derived from Cdk5 activator p35, specifically inhibit deregulated activity of Cdk5. Biotechnol J. 2007;2:978–987. doi: 10.1002/biot.200700057. [DOI] [PubMed] [Google Scholar]
- Kusakawa G, Saito T, Onuki R, Ishiguro K, Kishimoto T, Hisanaga S. Calpain-dependent proteolytic cleavage of the p35 cyclin-dependent kinase 5 activator to p25. J Biol Chem. 2000;275:17166–17172. doi: 10.1074/jbc.M907757199. [DOI] [PubMed] [Google Scholar]
- Lau LF, Ahlijanian MK. Role of cdk5 in the pathogenesis of Alzheimer’s disease. Neurosignals. 2003;12:209–214. doi: 10.1159/000074622. [DOI] [PubMed] [Google Scholar]
- Lau LF, Seymour PA, Sanner MA, Schachter JB. Cdk5 as a drug target for the treatment of Alzheimer’s disease. J Mol Neurosci. 2002;19:267–273. doi: 10.1385/JMN:19:3:267. [DOI] [PubMed] [Google Scholar]
- Lee MS, Kwon YT, Li M, Peng J, Friedlander RM, Tsai LH. Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature. 2000;405:360–364. doi: 10.1038/35012636. [DOI] [PubMed] [Google Scholar]
- Lee KY, Rosales JL, Tang D, Wang JH. Interaction of cyclin-dependent kinase 5 (Cdk5) and neuronal Cdk5 activator in bovine brain. J Biol Chem. 1996;271:1538–1543. doi: 10.1074/jbc.271.3.1538. [DOI] [PubMed] [Google Scholar]
- Lim AC, Qu D, Qi RZ. Protein-protein interactions in Cdk5 regulation and function. Neurosignals. 2003;12:230–238. doi: 10.1159/000074625. [DOI] [PubMed] [Google Scholar]
- Lim HY, Seow KT, Li Q, Kesuma D, Wang JH, Qi RZ. Structural insights into Cdk5 activation by a neuronal Cdk5 activator. Biochem Biophys Res Commun. 2001;285:77–83. doi: 10.1006/bbrc.2001.5086. [DOI] [PubMed] [Google Scholar]
- Mapelli M, Massimiliano L, Crovace C, Seeliger MA, Tsai LH, Meijer L, Musacchio A. Mechanism of CDK5/p25 binding by CDK inhibitors. J Med Chem. 2005;48:671–679. doi: 10.1021/jm049323m. [DOI] [PubMed] [Google Scholar]
- Nath R, Davis M, Probert AW, Kupina NC, Ren X, Schielke GP, Wang KK. Processing of cdk5 activator p35 to its truncated form (p25) by calpain in acutely injured neuronal cells. Biochem Biophys Res Commun. 2000;274:16–21. doi: 10.1006/bbrc.2000.3070. [DOI] [PubMed] [Google Scholar]
- Nguyen MD, Lariviere RC, Julien JP. Deregulation of Cdk5 in a mouse model of ALS: toxicity alleviated by perikaryal neurofilament inclusions. Neuron. 2001;30:135–147. doi: 10.1016/s0896-6273(01)00268-9. [DOI] [PubMed] [Google Scholar]
- Ohshima T, Ward JM, Huh CG, Longenecker G, Veeranna, Pant HC, Brady RO, Martin LJ, Kulkarni AB. Targeted disruption of the cyclin-dependent kinase 5 gene results in abnormal corticogenesis, neuronal pathology and perinatal death. Proc Natl Acad Sci USA. 1996;93:11173–11178. doi: 10.1073/pnas.93.20.11173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrick GN, Zukerberg L, Nikolic M, de la Monte S, Dikkes P, Tsai LH. Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature. 1999;402:615–622. doi: 10.1038/45159. [DOI] [PubMed] [Google Scholar]
- Pevsner J. The role of Sec1p-related proteins in vesicle trafficking in the nerve terminal. J Neurosci Res. 1996;45:89–95. doi: 10.1002/(SICI)1097-4547(19960715)45:2<89::AID-JNR1>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Qi Z, Tang D, Zhu X, Fujita DJ, Wang JH. Association of neurofilament proteins with neuronal Cdk5 activator. J Biol Chem. 1998;273:2329–2335. doi: 10.1074/jbc.273.4.2329. [DOI] [PubMed] [Google Scholar]
- Qu D, Qing L, Lim H-Y, Cheung NS, Li R, Wang JH, Qi RZ. The protein SET binds the neuronal Cdk5 activator p35 and modulates Cdk5/p35 activity. J Biol Chem. 2002;277:7324–7332. doi: 10.1074/jbc.M107270200. [DOI] [PubMed] [Google Scholar]
- Rosales JL, Nodwell MJ, Johnston RN, Lee KY. Cdk5/p25(nck5a) interaction with synaptic proteins in bovine brain. J Cell Biochem. 2000;78:151–159. doi: 10.1002/(sici)1097-4644(20000701)78:1<151::aid-jcb14>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- Shaw G, Morse S, Ararat M, Graham FL. Preferential transformation of human neuronal cells by human adenoviruses and the origin of HEK 293 cells. FASEB J. 2002;16:869–871. doi: 10.1096/fj.01-0995fje. [DOI] [PubMed] [Google Scholar]
- Shetty KT, Kaech S, Link WT, Jaffe H, Flores CM, Wray S, Pant HC, Beushausen S. Molecular characterization of a neuronal-specific protein that stimulates the activity of Cdk5. J Neurochem. 1995;64:1988–1995. doi: 10.1046/j.1471-4159.1995.64051988.x. [DOI] [PubMed] [Google Scholar]
- Shuang R, Zhang L, Fletcher A, Groblewski GE, Pevsner J, Stuenkel EL. Regulation of Munc-18/syntaxin 1A interaction by cyclin-dependent kinase 5 in nerve endings. J Biol Chem. 1998;273:4957–4966. doi: 10.1074/jbc.273.9.4957. [DOI] [PubMed] [Google Scholar]
- Shukla V, Zheng YL, Mishra SK, Amin ND, Steiner J, Grant P, Kesavapany S, Pant HC. A truncated peptide from p35, a Cdk5 activator, prevents Alzheimer’s disease phenotypes in model mice. FASEB J. 2013;27:174–186. doi: 10.1096/fj.12-217497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundaram JR, Poore CP, Sulaimee NH, Pareek T, Asad AB, Rajkumar R, Cheong WF, Wenk MR, Dawe GS, Chuang KH, et al. Specific inhibition of p25/Cdk5 activity by the Cdk5 inhibitory peptide reduces neurodegeneration in vivo. J Neurosci. 2013;33:334–343. doi: 10.1523/JNEUROSCI.3593-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan VB, Zhang B, Lim KM, Tay TE. Explaining the inhibition of cyclin-dependent kinase 5 by peptides derived from p25 with molecular dynamics simulations and MM-PBSA. J Mol Model 16, 1–8. 2009 doi: 10.1007/s00894-009-0514-1. [DOI] [PubMed] [Google Scholar]
- Tarricone C, Dhavan R, Peng J, Areces LB, Tsai LH, Musacchio A. Structure and regulation of the CDK5-p25(nck5a) complex. Mol Cell. 2001;8:657–669. doi: 10.1016/s1097-2765(01)00343-4. [DOI] [PubMed] [Google Scholar]
- Tsai LH, Lee MS, Cruz J. Cdk5, a therapeutic target for Alzheimer’s disease. Biochim Biophys Acta. 2004;1697:137–142. doi: 10.1016/j.bbapap.2003.11.019. [DOI] [PubMed] [Google Scholar]
- Veeranna, Shetty KT, Takahashi M, Grant P, Pant HC. Cdk5 and MAPK are associated with complexes of cytoskeletal proteins in rat brain. Brain Res. 2000;76:229–236. doi: 10.1016/s0169-328x(00)00003-6. [DOI] [PubMed] [Google Scholar]
- Zhang L, Liu W, Szumlinski KK, Lew J. p10, the N-terminal domain of p35, protects against CDK5/p25-induced neurotoxicity. Proc Natl Acad Sci USA. 2012;109:20041–20046. doi: 10.1073/pnas.1212914109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng YL, Amin ND, Hu YF, Rudrabhatla P, Shukla V, Kanungo J, Kesavapany S, Grant P, Albers W, Pant HC. A 24-residue peptide (P5), derived from p35, the Cdk5 neuronal activator, specifically inhibits Cdk5-p25 hyperactivity and tau hyperphosphorylation. J Biol Chem. 2010;285:34202–34212. doi: 10.1074/jbc.M110.134643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng YL, Kesavapany S, Gravell M, Hamilton RS, Schubert M, Amin N, Albers W, Grant P, Pant HC. A Cdk5 inhibitory peptide reduces tau hyperphosphorylation and apoptosis in neurons. EMBO J. 2005;24:209–220. doi: 10.1038/sj.emboj.7600441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng YL, Li BS, Amin ND, Albers W, Pant HC. A peptide derived from cyclin-dependent kinase activator (p35) specifically inhibits Cdk5 activity and phosphorylation of tau protein in transfected cells. Eur J Biochem. 2002;269:4427–4434. doi: 10.1046/j.1432-1033.2002.03133.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.