

A Drosophila model system is used to show that the hereditary spastic paraplegia proteins spastin and atlastin help axons but not dendrites regenerate. The endoplasmic reticulum concentrates at tips of regenerating axons but not dendrites, and this depends on spastin and atlastin.
Abstract
Mutations in >50 genes, including spastin and atlastin, lead to hereditary spastic paraplegia (HSP). We previously demonstrated that reduction of spastin leads to a deficit in axon regeneration in a Drosophila model. Axon regeneration was similarly impaired in neurons when HSP proteins atlastin, seipin, and spichthyin were reduced. Impaired regeneration was dependent on genetic background and was observed when partial reduction of HSP proteins was combined with expression of dominant-negative microtubule regulators, suggesting that HSP proteins work with microtubules to promote regeneration. Microtubule rearrangements triggered by axon injury were, however, normal in all genotypes. We examined other markers to identify additional changes associated with regeneration. Whereas mitochondria, endosomes, and ribosomes did not exhibit dramatic repatterning during regeneration, the endoplasmic reticulum (ER) was frequently concentrated near the tip of the growing axon. In atlastin RNAi and spastin mutant animals, ER accumulation near single growing axon tips was impaired. ER tip concentration was observed only during axon regeneration and not during dendrite regeneration. In addition, dendrite regeneration was unaffected by reduction of spastin or atlastin. We propose that the HSP proteins spastin and atlastin promote axon regeneration by coordinating concentration of the ER and microtubules at the growing axon tip.
INTRODUCTION
Hereditary spastic paraplegias (HSPs) are a group of neurodegenerative disorders characterized by axonal degeneration of upper motor neurons in the corticospinal tract. These neurons indirectly control voluntary movement, and hence patients suffering from HSP display spasticity and weakness of lower limbs (Blackstone, 2012; Fink, 2013). Postmortem studies in HSP patients reveal a vulnerability of the longest axons to degeneration, suggesting deficits in long-term axon maintenance (Deluca et al., 2004; Salinas et al., 2008).
More than 50 HSP-causative genes have been identified. Rather than being involved exclusively in neuron-specific pathways such as synaptogenesis or axon path finding, many proteins encoded by these genes are involved in universal cellular processes, including membrane trafficking, microtubule organization, and lipid metabolism (Blackstone, 2012; Fink, 2013; Lo Giudice et al., 2014; Noreau et al., 2014).
About 40% of HSP cases are linked to mutations in the gene that encodes spastin (Lo Giudice et al., 2014). In vitro, spastin severs microtubules (Evans et al., 2005; Roll-Mecak and Vale, 2005; Salinas et al., 2005), and its activity can be regulated by microtubule posttranslational modifications (Valenstein and Roll-Mecak, 2016). Spastin plays important roles in neuronal development, perhaps in association with microtubule regulation. In Drosophila mutants or animals with neuron-specific RNA interference (RNAi), axon terminals at the neuromuscular junction are abnormal, and microtubule stability is altered (Sherwood et al., 2004; Trotta et al., 2004; Orso et al., 2005). Development of dendrite arbors is also abnormal in Drosophila neurons with reduced levels of spastin (Jinushi-Nakao et al., 2007). In fish, reduction of spastin impairs axon outgrowth (Wood et al., 2006), perhaps through regulation of microtubule dynamics (Butler et al., 2010). Spastin function in mature neurons is less studied, although spastin continues to be expressed in adult rats (Solowska et al., 2008), and adult spastin mutant flies have a progressive decline in motor skills over the long term (Sherwood et al., 2004). At the cellular level, reduction in spastin has been associated with failure of mature neurons to regenerate axons in Drosophila (Stone et al., 2012); however, how spastin promotes regeneration is not known.
Whereas most mechanistic studies of spastin function in neurons have focused on microtubules, both mammals and flies have spastin isoforms that are associated with membranes. In mammals, the long M1 spastin isoform has an N-terminal region that contains a hydrophobic domain, and this isoform is an integral membrane protein (Park et al., 2010) believed to localize to the endoplasmic reticulum (ER), endosomes (Sanderson et al., 2006), and lipid droplets (Papadopoulos et al., 2015). In Drosophila, there are also spastin isoforms with an N-terminal hydrophobic region predicted to be a transmembrane domain (Trotta et al., 2004), and this region directs overexpressed spastin to punctate membranes (Roll-Mecak and Vale, 2005).
Mammalian spastin physically interacts with the ER proteins atlastin (Evans et al., 2006; Sanderson et al., 2006) and REEP1 (Park et al., 2010), and Drosophila spastin interacts physically and genetically with atlastin (Lee et al., 2009). Intriguingly, mutations in the genes that encode atlastin and REEP1 also lead to HSP (Lo Giudice et al., 2014). Like spastin, the enzymatic activity of atlastin is well established; it is a GTPase that mediates homotypic fusion of ER membranes (Hu et al., 2009; Orso et al., 2009).
Based on the interactions between this subset of HSP proteins and their established functions in shaping the ER and microtubule cytoskeleton, they may work together to coordinate ER shape and position along microtubules (Park et al., 2010). However, so far there is no evidence to support this role in mature neurons. Atlastin and spastin also regulate lipid droplet formation (Klemm et al., 2013; Papadopoulos et al., 2015) and BMP signaling (Tsang et al., 2009; Fassier et al., 2010; Zhao and Hedera, 2013; Summerville et al., 2016), and so these are other potential common functions that could be relevant to HSP. Beyond this group of interacting proteins, other HSP genes encode proteins broadly involved in ER morphogenesis, endosomal trafficking, microtubule-based transport, and mitochondrial function (Noreau et al., 2014), but most are not as well characterized as spastin and atlastin.
In our previous study, we identified a link between spastin and axon regeneration, but we did not find any differences in injury-induced microtubule rearrangements between control and spastin-reduced neurons (Stone et al., 2012). To try to understand the relationship between spastin and regeneration, we now use several different approaches. We first tested whether other HSP proteins might also be involved in axon regeneration, focusing mostly on atlastin. We also tried to identify intracellular reorganizations that coincided with axon outgrowth during regeneration. We found that the ER was highly concentrated in patches near tips of regenerating axons but not dendrites. Moreover, this redistribution of the ER during regeneration was disrupted in spastin mutants and atlastin RNAi neurons. Involvement of both spastin and atlastin in regeneration and ER reorganization suggests a model in which spastin and atlastin promote regeneration by coupling microtubule and ER concentration at tips of regenerating axons.
RESULTS
Multiple HSP proteins are specifically required for injury-induced axon regeneration
HSP is a postdevelopmental degenerative disease; therefore functions of HSP associated with maintenance of mature neurons are of particular interest. We previously found that axon regeneration was impaired in Drosophila neurons missing a single copy of the spastin gene (Stone et al., 2012). We thus wanted to test whether other proteins associated with HSP might also be involved in axon regeneration.
Drosophila larval dendritic arborization (da) neurons are conducive to injury studies because axons of single cells can be easily severed using a pulsed ultraviolet (UV) laser, and regeneration can be tracked up to 4 d postinjury in whole, live animals (Stone et al., 2010). Axon regeneration in these cells is initiated by the DLK kinase cascade (Stone et al., 2014), as in worms (Hammarlund et al., 2009; Yan et al., 2009) and mice (Shin et al., 2012). Complete removal of the axon or proximal axotomy in the ddaE neuron, which is one of the class I da neurons (Grueber et al., 2002), leads to conversion of a dendrite to an axon, and regrowth can be reliably quantified (Stone et al., 2010). Spastin is required for this type of regeneration, as well as for the more familiar regeneration of an axon from a remaining stump (Stone et al., 2012).
To determine whether other HSP proteins might also play a role in regeneration, we used cell-type specific RNAi (Dietzl et al., 2007) to knock down atlastin, spichthyin, and seipin in class I neurons. As a control, we used RNAi targeting γtubulin37C, a maternal protein not present after initial development (Wilson and Borisy, 1998; Wiese, 2008) that we previously showed has no effect on axon regeneration (Stone et al. 2012). Class I da neurons were labeled by expressing green fluorescent protein (GFP)–tagged EB1, a microtubule end–binding protein, under control of the 221-GAL4 driver. Proximal axotomy was performed on the ddaE neuron, and regrowth was measured 96 h after injury.
In control neurons, one dendrite typically converts from a minus-end-out microtubule polarity to plus-end-out polarity based on the direction of EB1-GFP comet movement and extends beyond its normal territory (Figure 1A). The length of this neurite at 0 and 96 h is measured and normalized to one of the other dendrites to account for overall increase in size as the animal grows. A zero-regeneration measurement means that none of the dendrites converted grew more than the expected based on expansion due to animal size increase. In neurons expressing control RNAi, the average amount of outgrowth beyond that of the nonregenerating neurites over the 96-h period was >300 μm (Figure 1E). Axon regeneration was significantly lower in neurons expressing atlastin, spichthyin, or seipin RNAi hairpins (Figure 1, B–E), suggesting that, like spastin, these HSP proteins play a role in axon regeneration. For atlastin, two independently generated RNAi lines were used. Although they had similar effects, slightly stronger phenotypes were observed with the line obtained from the Bloomington Drosophila Stock Center (Bloomington, IN; atlastin BL), and so we used this line in most other experiments.
FIGURE 1:

Axon regeneration in neurons with reduced levels of HSP proteins. Class I neurons were labeled with UAS-EB1-GFP under the control of 221-GAL4. The axon of the ddaE neuron was severed using a pulsed UV laser (0-h time point), and the same neuron was imaged after 96 h. A white arrowhead in each image indicates the new axon. (A) In neurons expressing control RNAi (γtub37C RNAi), one of the dendrites was converted into an axon as indicated. (B–D) In neurons expressing atlastin, seipin, or spichthyin RNAi, regrowth was significantly reduced. Arrows indicate cut site. Scale bars, 50 μm. (E) Regrowth after axon injury is quantified. Long and short bars indicate averages and SDs, respectively. (F) Quantification of axon regeneration in spastin mutants using mCD8-GFP as a cell-shape marker. (G) Quantification of axon regeneration in neurons expressing HSP RNAis using mCD8-GFP as a cell-shape marker.
We previously showed that axon regeneration was impaired when only one copy of the spastin gene was mutant. Because atlastin mutants have also been characterized (Lee et al., 2009; Orso et al., 2009), we tested whether this gene was haploinsufficient for regeneration. Indeed, in animals with one mutant and one wild-type copy of atlastin, regeneration was reduced (Figure 1E).
The sensitivity of axon regeneration to HSP protein reduction depends on genetic background
While we were testing different RNAi lines to determine whether they had regeneration phenotypes, we also tried several different cell shape markers. In many of our previous studies on axon regeneration, we used EB1-GFP as a combined cell shape marker and reporter of microtubule dynamics and polarity (Stone et al., 2010, 2012). However, cell outlines are clearer when a membrane marker, mCD8-GFP or mCD8-RFP, is used. EB1-GFP is a dominant-negative protein, and at high levels of expression, it disrupts dendrite microtubule polarity (Mattie et al., 2010). At low levels of expression like those used here, it does not result in defects in our assays. For example, regeneration in control neurons is similar in the mCD8-GFP background and EB1-GFP background (Figure 2, E and G). We also performed regeneration assays in neurons labeled with mCD8-RFP and expressing HSP RNAis (Figure 1G). Surprisingly, there was no difference between most control and HSP RNAis in this background; the only exception was atlastin RNAi. In an effort to pinpoint the key difference between the experiments with EB1-GFP and mCD8-RFP, we compared axon regeneration with one mutant copy of spastin using the two markers. This result was very clear: regeneration was reduced in spastin heterozygotes only when EB1-GFP was used as a cell marker (Figure 1F). Note that when the null 5.75 mutant is combined with a hypomorph, regeneration is defective in other backgrounds too (see later discussion of Figure 6E; Stone et al., 2012), so EB1-GFP seems to sensitize the neurons to reduction of spastin. This synthetic effect of partial spastin reduction in the heterozygous background and EB1-GFP suggests that the spastin function in regeneration relates in some way to microtubules.
FIGURE 6:

ER distribution during axon and dendrite regeneration. (A) The axon of a class I ddaE neuron expressing UAS-Rtnl1-GFP was severed and imaged immediately after injury (0 h) and 96 h later. Arrows indicate cut site. An accumulation of Rtnl1-GFP was observed at the regrowing axon tip. (B) Average fold change in Rtnl1-GFP intensity from axon base to tip compared with the fold change in intensity in a dendrite in the same cell. Error bars denote SD. (C) A 100-μm region at the end of the new axon was considered as the “tip,” and the proximal region was considered as the “base.” Rtnl1-GFP intensity was measured in the brightest 10-μm region within the tip and the base of the new axon. For comparison, the tip/base ratio of Rtnl1-GFP fluorescence in a nongrowing dendrite in each cell was also calculated. Error bars indicate the SD. (D) Dendrites of class I ddaE neurons expressing Rtnl1-GFP were removed (arrows show cut sites), and the injured neuron was imaged 48 and 96 h postinjury to track dendrite regeneration. There was no accumulation of Rtnl1-GFP in the tips of regrowing dendrites. (E) Class I ddaE neurons expressing control RNAi and Rtnl1-GFP were proximally axotomized, and ER localization was tracked at 24, 48, and 96; an example is shown. White arrows in the 48- and 96-h images show ER accumulation in the new axon. (F) Class I ddaE neurons expressing Rtnl1-GFP and atlastin BL RNAi were subjected to proximal axotomy (0 h) and imaged after 96 h. White arrowhead indicates regenerating neurite. (G) Quantification of regeneration in control and atlastin knockdown neurons. (H) Quantification of Rtnl1-GFP accumulation in axon tips in atlastin knockdown neurons and control neurons. The error bars are SDs, and an unpaired t test was used to calculate p values.
Dendrite regeneration is not sensitive to reduction of HSP proteins
Like axons, dendrites regenerate after injury (Stone et al., 2014). If the defect in regeneration observed when HSP proteins are knocked down is due to an overall deficit in growth capacity or health of the cell, then we would also expect dendrite regeneration to be impaired. We therefore tested dendrite regeneration in the same genetic backgrounds used for axon regeneration. When dendrites of class I ddaE neurons are removed, new dendrites are generated that regain the same complexity as before injury (Stone et al., 2014). Counting the branch points can therefore be used as a quantitative assay to measure dendrite regeneration in this cell type.
All dendrites of ddaE neurons expressing either EB1-GFP (Figure 2, A–E) or mCD8-GFP (Supplemental Figure S1, A–D) were severed using a pulsed UV laser, and the injured neuron was imaged 96 h after injury. In control neurons expressing γtubulin37C RNAi, dendrites regenerated to similar complexity as uninjured neurons by 96 h. In neurons expressing HSP RNAis and in spastin heterozygotes, injury-induced dendrite regeneration occurred normally (Figure 2F and Supplemental Figure S1E). This suggests that HSP proteins are specifically required for injury-induced axon regrowth but not dendrite regrowth and that the partial reduction of HSP proteins we used does not impair many cellular functions, including large-scale responses to injury.
FIGURE 2:
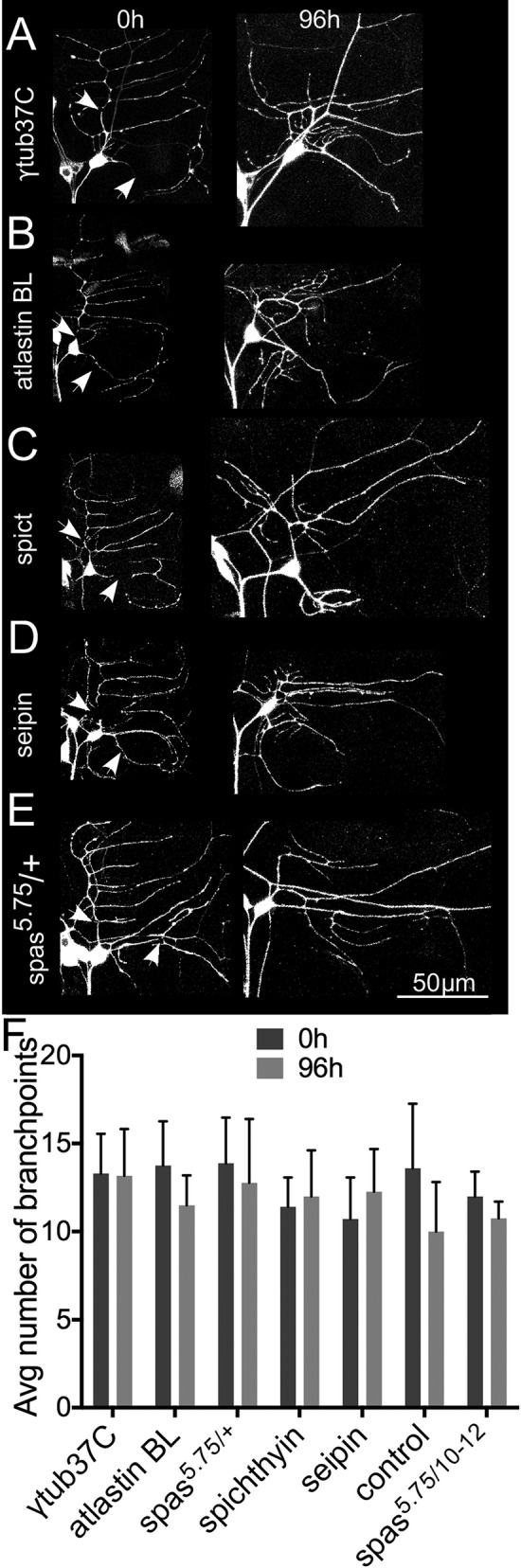
Dendrite regeneration in neurons with reduced levels of HSP proteins. (A–E) Class I ddaE neurons were labeled with EB1-GFP, and all dendrites were removed by laser ablation. Arrows indicate laser cut sites. Injured neurons were imaged after 96 h to track dendrite regrowth. (F) Number of dendrite branch points counted before (0 h) and after injury (96 h). Columns indicate average number of branch points. Error bars denote SD.
Early responses to axon injury are not perturbed by the loss of HSP proteins
Axon injury is followed by a massive up-regulation of microtubule dynamics, meaning here the number of growing microtubule plus ends, in Drosophila and mammalian neurons (Stone et al., 2010; Kleele et al., 2014). Despite the fact that spastin can sever microtubules and could therefore result in such an increase, it is not required for this response (Stone et al., 2012), which relies instead on microtubule nucleation (Chen et al., 2012). To determine whether other HSP proteins were similarly dispensable for increased microtubule dynamics, we used the same genetic backgrounds in which regeneration was impaired (Figure 1E) and assayed the number of microtubule plus ends labeled with EB1-GFP. First, EB1 comets were counted in uninjured neurons expressing either control RNAi (γtubulin37C RNAi) or HSP RNAis (Figure 3, A–C; 0 h). The average number of comets in a 10-μm region of the main dendrite trunk was comparable in control and HSP RNAis, indicating that microtubule dynamics in these backgrounds is normal in the absence of injury (Figure 3D; 0 h).
FIGURE 3:

Early responses to axon injury are not perturbed by the loss of HSP proteins. (A–C) Examples of microtubule dynamics in control, atlastin, and bsk RNAi in uninjured neurons (0 h) and 24 h after injury. Microtubule dynamics was quantified by counting the number of EB1-GFP comets in a 10-μm region of the class I ddaE dendrite in uninjured neurons (0 h) and 24 h after axon injury. White arrows indicate location of cell body, and black arrowheads show individual EB1-GFP comets. Scale bars, 10 μm. (D) Average number of EB1-GFP comets for each time point. Error bars show SD. (E–G) Class I ddaE neurons (white arrows) expressing mCD8-RFP and puc-GFP were subjected to proximal axotomy in control neurons or neurons expressing HSP RNAis. Intensity of puc-GFP in the nucleus was measured at the time of injury (0 h) and 24 h after injury and normalized to that of the uninjured ddaD neuron (arrowheads). Scale bars, 10 μm. (H) Average fold change in puc-GFP intensity from 0 to 24 h. Error bars indicate SD. (I) Class I ddaE neurons expressing EB1-GFP and HSP RNAis were axotomized, and microtubule polarity was analyzed in the dendrites 96 h postinjury. Percentage of neurons in each genotype that were injured. Black regions of the columns denote neurons with at least one plus-end-out neurite, and gray regions denote neurons with all minus-end-out neurites. ***p < 0.001.
Next we performed proximal axotomy in neurons expressing control RNAi, HSP, or JNK (bsk in Drosophila) RNAis and counted EB1-GFP comets 24 h after injury (Figure 3, A–D; 24 h). Only JNK/bsk RNAi reduced the injury-induced increase in microtubule dynamics (Figures 3, C and D), which is expected because this response is known to be JNK dependent (Stone et al., 2010). Therefore, like spastin, other HSP proteins do not seem to be required for the up-regulation of microtubule dynamics after axon injury.
To test more generally whether the HSP proteins were required for the initial response to axon injury, we used a JNK reporter that is activated by axon severing. Downstream of DLK/JNK/fos, the mitogen-activated protein kinase phosphatase puc is transcriptionally up-regulated, and this can be visualized by lacZ (Xiong et al., 2010) or GFP (Stone et al., 2014) insertions in the puc gene. At 24 h after axon injury, puc-GFP in the nucleus of the injured ddaE neuron was more than twofold higher than before injury in all backgrounds except bskDN (dominant-negative JNK; Figure 3, E–H).
A prerequisite for axon regeneration after proximal axotomy is the switching of microtubule polarity in one of the dendrites from minus-end-out to plus-end-out (Mattie et al. 2010; Stone et al. 2010). In spastin mutants, the number of plus-end-out microtubules added to dendrites postaxotomy was comparable to that in control neurons (Stone et al. 2012); however, it was unclear whether polarity switching is complete. To this end, we analyzed microtubule polarity using EB1-GFP as a marker in neurons expressing HSP RNAis or spas5.75/+ neurons 96 h postaxotomy (Figure 3I). In all cases, at least one dendrite converted from minus-end-out to plus-end-out microtubule polarity, confirming that polarity reversal is complete and does not require HSP proteins.
Thus early axon injury responses were not detectably altered in any of these HSP knockdowns, and conversion of a dendrite to axonal microtubule polarity was normal, although the amount of regeneration was reduced. On the basis of these results and our previous analysis of spastin mutants (Stone et al., 2012), we hypothesized that at least some of the HSP proteins may function during the outgrowth stage of axon regeneration rather than during the initial response or polarity conversion, which precedes outgrowth (Stone et al., 2010).
The ER preferentially accumulates in the regenerating axon after injury
To understand cellular events that occur during regenerative axon outgrowth and might involve HSP proteins, we used a panel of markers to visualize intracellular rearrangements. We paired each marker with mCD8-RFP to highlight the plasma membrane. Endosomes were labeled using Rab5–yellow fluorescent protein (YFP), a classic marker of early endosomes (Elkin et al., 2016). In uninjured class I da neurons, Rab5-YFP punctae were localized to the cell body, axon, and dendrites (Supplemental Figure S2). After axon removal, Rab5-YFP distribution did not change dramatically (Figure 4D). Mitochondria were labeled with mito-GFP. As with endosomes, mitochondria were distributed throughout the neuron before and after axon injury (Supplemental Figures S2 and 4A). Ribosomes were tagged with YFP-L10 (Rolls et al., 2007; Hill et al., 2012). Before injury, ribosomes were mainly present in the cell body and at dendrite branch points (Supplemental Figure S2). In axotomized neurons, a few YFP-L10 punctae were observed in the initial part of the regrowing axon that was converted from a dendrite, but none was observed in the region beyond the previously dendritic part (Figure 4B).
FIGURE 4:

The ER preferentially accumulates in the regenerating axon after injury. Axons of class I ddaE neurons were severed (0 h), and regeneration was tracked in the injured neurons 96 h later. mCD8-RFP was used as a cell-shape marker in all cases. Arrows indicate cut site close to the cell body. (A) Mitochondria were labeled using mito-GFP. (B) Ribosomes were labeled using YFP-tagged L10. Asterisks indicate positions of YFP-L10 punctae in the new axon. (C) Smooth ER was labeled with Rtnl1-GFP. (D) Endosomes were labeled using Rab5-YFP. Asterisks indicate positions of Rab5-YFP punctae in the new axon. (E) Superresolution images of class I ddaE neurons labeled with mCD8-RFP and Rtnl1-GFP. Insets show close-up view (3.3× zoom) of Rtnl1-GFP labeling within the cell. Scale bars, 5 μm (inset).
The organelle with the most striking localization pattern during axon regeneration was the ER. Reticulon-like-1 (Rtnl1) is the major Drosophila reticulon protein and is expressed in neurons and localizes to the smooth ER (SER; Wakefield and Tear, 2006; O’Sullivan et al., 2012). To visualize the SER, which, unlike the primarily somatic rough ER, extends throughout the neuron (Krijnse-Locker et al., 1995; Rolls et al., 2002), we generated transgenic Drosophila containing a UAS-Rtnl1-GFP transgene. In the ddaE neuron, continuous Rtnl1-GFP labeling was seen throughout the axon, dendrites, and cell body (Supplemental Figure S3A). To ensure that the structure being labeled was really ER, we used superresolution microscopy in living animals to examine the neurons. The Rtnl1-GFP signal was in very clear tubular structures contained within the bounds of the plasma membrane labeled with mCD8-RFP (Figure 4E, insets). After ddaE neurons were axotomized, an often dramatic accumulation of Rtnl1-GFP signal was observed near the growing axon tip (Figure 4C).
We considered that this concentration of Rtnl1-GFP signal either could be due to a specific distribution of Rtnl1 or represent more generally movement of the SER to the tip of the growing axon. To distinguish between these two possibilities, we generated an additional SER marker, a GFP-tagged IP3 receptor (UAS-GFP-IP3R). IP3R is a large protein of >2500 amino acids. To test whether the GFP-tagged version was functional, it was crossed into a line that contained a lethal point mutation in the gene that encodes IP3R (Itprug3) (Joshi et al., 2004). Flies from this line were then crossed to flies that contained the Act5C-Gal4, which drives expression globally, and Itprsv35, which encodes a null allele of the IP3R (Joshi et al., 2004). Without the GFP-IP3R transgene, no ug3/sv35 animals survived, but when the transgene was expressed, adults with this allelic combination were recovered. Thus the tagged IP3R is functional.
In neurons, the localization pattern of GFP-IP3R was not identical to that of Rtnl1-GFP; it was much dimmer even when expressed by the same Gal4 driver and had a slightly more punctate distribution (Figure 5). Again, we examined its localization with superresolution microscopy and observed it in intracellular membranes (Figure 5D). After axotomy, GFP-IP3R was often seen to accumulate near the regenerating axon tip (Figure 5B, inset). This suggested that the smooth ER in general was redistributing to regenerating axon tips rather than Rtnl1-GFP specifically. Because atlastin (Rismanchi et al., 2008), spastin (Park et al., 2010), and seipin (Windpassinger et al., 2004) all localize to the ER, we hypothesized that at least a subset of HSP proteins may contribute to axon regeneration by helping the ER concentrate at the growing axon tip.
FIGURE 5:

GFP-IP3R labels the ER and accumulates in regenerating axon tips. (A, B) A class I ddaE neuron labeled with mCD8-RFP and imaged 96 h after proximal axotomy. Arrows indicate the tip of the new axon. The boxed area and inset show GFP-IP3R labeling at the axon tip. Scale bar, 25 μm. (C) Quantification of the percentage of neurons with GFP-IP3R signal at the tip of the regenerating axon (n = 19). (D) Peptidergic neurons (white arrow) were labeled with GFP-IP3R under the control of 386-GAL4. Superresolution microscopy was used to visualize the distribution of GFP-IP3R in the cell body.
To test whether a link between ER concentration at regenerating axon tips and HSP proteins was likely, we examined the ER in regenerating dendrites compared to regenerating axons. Because we did not find a role for HSP proteins in dendrite regeneration (Figure 2F), if the ER also concentrated at tips of regenerating dendrites, it would suggest that HSP proteins are not likely to be involved in ER redistribution. To quantitate ER redistribution, we selected a 100-μm region at the end of the new axon that was considered the “tip” and a 100-μm proximal region considered the “base” (Figure 6C). Rtnl1-GFP intensity was measured in the brightest 10-μm region within these tip and base segments. ER accumulation was calculated as fold increase in the Rtnl1-GFP signal at the new axon tip versus the base. On average, the Rtnl1-GFP signal was more than twofold higher at the tip of the regenerating axon than at the base of the same process in control neurons (Figure 6, A and B). However, during dendrite regeneration, Rtnl1-GFP was not concentrated at the tips of the new dendrites but instead was evenly distributed throughout the new arbor (Figure 6D). Thus axon regeneration was distinguished from dendrite regeneration by 1) a requirement for some of the HSP proteins and 2) accumulation of ER at growing tips.
To further characterize ER changes during regeneration, we tracked the timing of ER localization at the tips of newly regenerating axons by imaging injured class I ddaE neurons expressing Rtnl1-GFP and the control γtub37C RNAi at 24, 48, 72, and 96 h (Figure 6E). Of eight injured neurons, a dramatic accumulation of Rtnl1-GFP signal was observed at 24 h in one neuron, at 48 h in six additional neurons, and at 72 h in the remaining neuron. Therefore, in the majority of neurons tested, ER accumulation occurred after increased microtubule dynamics and polarity reversal but before outgrowth.
Reduction of spastin or atlastin is associated with defects in the accumulation of the ER in regenerating axons
To test directly whether HSP proteins might be involved in ER concentration at regenerating axon tips, we analyzed ER repositioning in spastin null/hypomorph and atlastin RNAi animals. These two genotypes were chosen because they were the only ones that resulted in a regeneration phenotype without EB1-GFP in the background.
In atlastin RNAi neurons labeled with Rtnl1-GFP, regeneration was suppressed relative to neurons expressing control γtub37C RNAi (Figure 6, F and G). Moreover, the concentration of ER at the tip of the growing axons was reduced compared with control neurons (Figure 6H). Thus, unlike the early injury responses examined with the puc-GFP reporter and EB1-GFP, which were completely normal in atlastin RNAi neurons (Figure 3), redistribution of the ER was impaired. Although accumulation of the ER at regenerating axon tips was reduced, we did not observe any discontinuities in the ER in these neurons or in uninjured ones (Supplemental Figure S3B).
To generate animals with two mutant copies of spastin in which Rtnl1-GFP was expressed in class I neurons, we had to use a different Gal4 driver than in other experiments. IGI1-Gal4 is on the second chromosome and drives expression in class I neurons (Shimono et al., 2009). The chromosome that contains this Gal4 driver is homozygous lethal due to the IGI1 insertion site or another mutation. In genetic backgrounds that contained this Gal4 driver, regeneration was slightly lower than in other genotypes (see control data in Figure 7E). However, the null/hypomorph combination of spastin mutant alleles (Sherwood et al., 2004) resulted in a significant reduction in regeneration even compared with this lower control (Figure 7E). When we examined the ER distribution in these cells, we noticed that Rtnl1-GFP was often concentrated at multiple tips (Figures 7A), and the number of neurons with this pattern of localization was significantly increased in spastin mutants compared with control (Figure 7F, p < 0.05 using Fisher’s exact test). To understand how this distribution might relate to regeneration, we classified neurons as successful growth (>75 μm over the course of the experiment) or no growth (<75 μm). In both control and spastin mutants, neurons with ER concentration in multiple tips rarely grew (Figure 7G). In both groups, there were also some cells that failed to grow even with ER concentrated at one tip. The group that was most obviously different between control and spastin mutants was the number with multiple ER at the tips that did not grow (4 of 36 in control and 11 of 31 in spastin mutant; p < 0.05 using Fisher’s exact test; Figure 7G). Thus there seemed to be a defect in ER distribution during axon regeneration in spastin mutants.
FIGURE 7:

ER distribution during regeneration in spastin mutant neurons. ER accumulation patterns in class I ddaE neurons during injury-induced axon regeneration. White arrows indicate the ddaE cell body. (A) An injured neuron that failed to regrow and has Rtnl1-GFP accumulation in multiple neurites (red arrows), (B) an injured neuron with <75 μm of regrowth and Rtnl1-GFP accumulation in a single neurite (orange arrow), (C) an injured neuron with >75 μm of regrowth and Rtnl1-GFP accumulation in the regrowing neurite (green arrow; green dashed lines trace the new axon. White dotted lines show a dendrite from a neighboring neuron), and (D) an injured neuron with multiple neurites regrowing, each with Rtnl1-GFP accumulation in the tips (blue arrows). (E) Quantification of regeneration in control (yw) and spas5.75/10-12 neurons. Long and short horizontal bars indicate mean and SD respectively, and an unpaired t test was used to calculate significance. (F) Percentage of neurons with ER accumulation in single or multiple neurite tips in control and spas5.75/10-12 neurons. (G) Percentage of neurons with different categories of growth and ER accumulation patterns. Colors correspond to arrows in A–D.
Microtubules accumulate at regenerating axon tips
Because 1) spastin is a microtubule-severing protein, 2) regeneration phenotypes were exacerbated by the inclusion of EB1-GFP in the background, and 3) the ER slides along microtubules, we also examined the distribution of microtubules in regenerating neurons. To label microtubules, we used tdEOS-αtubulin because this was used to clearly visualize microtubules in cultured Drosophila neurons (Lu et al., 2013). Of interest, during injury-induced axon regeneration in wild-type ddaE neurons, we observed that the tdEOS signal was brighter in the newly specified axon compared with other neurites, and there was considerable accumulation near the growing tip (Figure 8, A and B), reminiscent of the ER accumulation pattern. This prompted us to test whether the microtubule accumulation pattern within the axon would change if spastin levels were reduced. We performed proximal axotomy in spas5.75/10-12 neurons expressing tdEOS-αtubulin. Surprisingly, these neurons did not show a defect in axon regeneration (Figure 8C). Because regeneration was impaired with this allele combination when Rtnl1-GFP (Figure 7) was used to label the cells, these data are most consistent with suppression of the spastin phenotype by overexpression of tdEOS-αtubulin.
FIGURE 8:
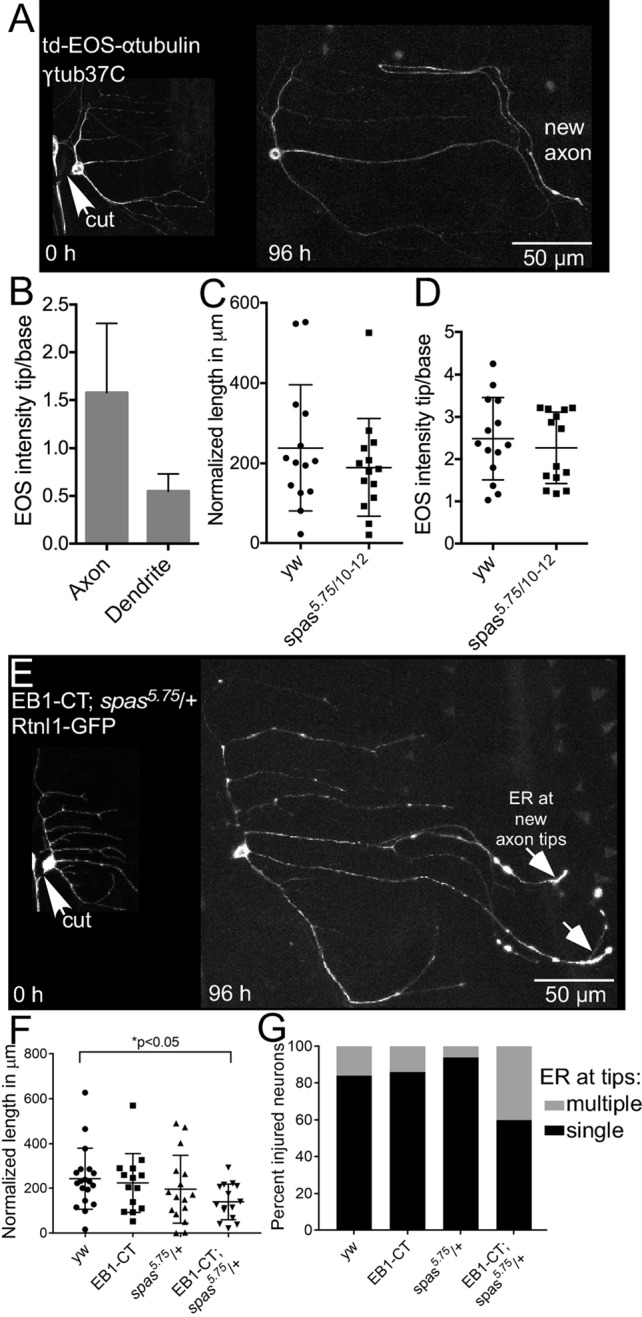
Microtubules accumulate in regenerating axon tips. (A) Class I ddaE neurons expressing tdEOS-αtubulin and control RNAi (γtub37C RNAi) were subjected to proximal axotomy and imaged 96 h after injury. The new axon has an accumulation of tdEOS near the growing tip. (B) Microtubule accumulation in the axon was measured and compared with a dendrite in the same cell. Average fold increase in tdEOS intensity in the axon and dendrite tips. Error bars show SD. (C) Axon regeneration was measured in control (yw) and spastin mutant neurons. (D) tdEOS intensity in the growing axon tips of spastin mutant and control neurons. (E) An example of a class I ddaE neuron expressing Rtnl1-GFP and EB1-CT and harboring the spas5.75/+ allele, subjected to proximal axotomy and imaged 96 h after injury. Rtnl1-GFP accumulation in axon tips is indicated by white arrows. (F) Quantification of regeneration in class I ddaE neurons expressing Rtnl1-GFP combined with either EB1-CT only, spas5.75/+ only, or both EB1-CT and spas5.75/+. Long and short bars indicate averages and SDs, respectively, and the p value was calculated with an unpaired t test. (G) Quantification of percentage of injured neurons in F with ER accumulation at a single axon tip (black) or at multiple axon tips (gray).
Next we further tested the hypothesis that the presence of a dominant-negative microtubule regulator sensitizes the neurons to a reduction in levels of spastin. We used Rtnl1-GFP to label class I ddaE neurons expressing UAS-EB1-CT, an untagged dominant-negative form of EB1 (Mattie et al., 2010). EB1-CT consists of the last 129 amino acids of Drosophila EB1 (Mattie et al., 2010). This region of mammalian EB1 contains a dimerization domain that can act alter catastrophe rates when overexpressed (Komarova et al., 2009). Compared to control neurons (yw), regeneration was not significantly affected by the presence of EB1-CT alone (Figure 8F). In addition, spas5.75/+ neurons expressing Rtnl1-GFP also did not exhibit significant defects in axon regeneration. However, when EB1-CT was expressed in spas5.75/+ neurons, axon regeneration was significantly reduced. This defect was accompanied by Rtnl1-GFP accumulation in multiple neurites (Figure 8G), similar to the phenotypes observed in spas5.75/10-12 neurons (Figure 7F). Taken together, these data suggest a role for microtubules and the ER in regulation of regeneration by spastin.
DISCUSSION
We previously showed that axon regeneration was impaired when one copy of spastin was mutant (Stone et al., 2012). We now show that atlastin is also haploinsufficient for axon regeneration and that reduction of several other HSP proteins using RNAi impairs regeneration. Thus axon regeneration seems to be a postdevelopmental process that involves at minimum a subset of HSP proteins.
The sensitivity of axon regeneration to partial reduction of HSP proteins, however, depends on the genetic background. In our previous study, EB1-GFP was used as a dual-purpose marker of cell shape and microtubule dynamics (Stone et al., 2012). This fusion protein is not, however, completely neutral. GFP fused to the C-terminus of EB1 can interfere with binding of partner proteins to EB1 (Skube et al., 2010). Because EB1 acts as a dynamic platform at growing microtubule ends that recruits other proteins (Akhmanova and Steinmetz, 2008), the presence of large amounts of EB1-GFP could reduce recruitment of other plus end–binding proteins. Indeed, in Drosophila, neurons EB1 binds Apc, which in turn brings kinesin-2 to growing dendritic microtubules to help maintain minus-end-out polarity (Mattie et al., 2010; Weiner et al., 2016), and high levels of EB1-GFP result in mixed polarity (Mattie et al., 2010). Because of this, we express EB1-GFP at low levels, but it is still possible that there is a subtle defect in microtubule growth or organization. Under normal circumstances, this does not result in any defects in regeneration, which is indistinguishable in control neurons expressing EB1-GFP (Figure 1E), mCD8-RFP (Figure 1F), or Rtnl1-GFP (Figure 6G). Only when combined with partial reduction of HSP proteins did we see a difference among neurons expressing different markers. This difference was most clearly demonstrated in spastin heterozygotes, which had a very strong reduction in regenerative growth in neurons labeled with EB1-GFP but not with mCD8-RFP (Figure 1F). The synthetic interaction between EB1-GFP and spastin suggests that even though the early microtubule changes triggered by axon injury were normal, with reduced levels of HSP proteins (Figure 3; Stone et al., 2012), microtubules were somehow involved in the phenotype. This conclusion was supported by a similar effect of EB1-CT, a dominant-negative form of EB1, and the fact that introduction of tdEOS-αtubulin suppressed the spastin phenotype (Figure 8C).
To probe in more depth how HSP protein function related to regenerative axon growth, we took several other approaches. First, we examined dendrite regeneration. Complete regeneration of dendrites after removal of the entire arbor involves very rapid outgrowth (Stone et al., 2014), and so we reasoned that if HSP proteins were generally involved in facilitating growth of neuronal processes, they should be required for dendrite regeneration. No defects in dendrite regeneration were observed, and this suggested that the cells were healthy and that a process specific to axon regeneration was sensitive to HSP protein reduction. Second, we examined a catalogue of intracellular markers to look for rearrangements associated with regenerative axon growth. The ER was most dramatically different in regenerating axons and accumulated near the growing tip. Moreover, this was specific to axon regeneration and not observed in dendrite regeneration.
We assembled all of these observations into a model for HSP protein function during regeneration. We propose that a subset of HSP proteins is involved in concentrating the ER, together with underlying microtubules, at tips of axons undergoing regenerative growth. This model makes particular sense for spastin and atlastin. Because spastin is a microtubule regulator that in flies and mammals also has a transmembrane domain and binds the ER regulatory protein atlastin, their combined action could facilitate concentration of the ER at growing axon tips. In support of this idea, reduction of either protein disrupted ER accumulation at single growing axon tips (Figures 6 and 7).
Although we believe that atlastin and spastin help to concentrate ER at growing axon tips by linking the ER to microtubules, which also accumulate at growing tips, we do not know what mediates the microtubule redistribution. We do suspect, however, that microtubule polarity is involved in setting up tip accumulation of tubulin and ER. Tip accumulation is seen only during regenerative axon growth, which requires microtubules in the growing process to be plus-end-out (Mattie et al., 2010), and not during regenerative dendrite growth, when microtubules are largely minus-end-out (Stone et al., 2014). In initiation of regenerative axon outgrowth in cultured Drosophila neurons, kinesin-mediated microtubule sliding is important (Lu et al., 2015), and so one possibility is that motors slide short pieces of microtubules out to the new tip.
It is intriguing that ER accumulates at regenerating axon but not dendrite tips. This suggests that increased amounts of local ER are specifically important for promoting maximal axon growth. Local SER could promote axon growth by increasing local lipid production or increasing availability of intracellular calcium. A recent study in Caenorhabditis elegans suggests that it is the calcium storage function of SER that is important in this context. In this system, release of ER calcium through ryanodine receptors is required for maximal axon outgrowth, and high levels of calcium were seen at axon tips up to 5 h (the latest time point examined) after axon injury (Sun et al., 2014). Thus perhaps atlastin and spastin help concentrate ER at growing axon tips to provide a local source of intracellular calcium stores, which in turn facilitate regenerative growth.
It is difficult to know how a function for atlastin and spastin, and potentially other HSP proteins, in ER localization during regenerative axon growth relates to the axon degeneration that occurs in the disease. All HSP proteins seem to have important basic cellular functions that are quite universal. For example, atlastin is biochemically an ER fusion protein, but only a subset of disease-causing atlastin mutations affect ER fusion (Ulengin et al., 2015). Perhaps the function of atlastin, spastin, and other HSP proteins important for disease is not the core function but a subtler role these proteins play in very long neurons. ER relocalization during regenerative growth of axons is worth considering as a disease-relevant function for several reasons: 1) it is important for mature neurons, 2) at least two different HSP proteins contribute to it, 3) repeated small failures of regeneration could lead to accumulated axon loss over a long time period, and 4) at least in some genetic backgrounds, the capacity for regenerative growth is reduced when only one allele of the gene is mutant. An interesting further speculation is how this function might relate to cell-type susceptibility to regeneration. The hallmark of HSP is degeneration of upper motor neurons. As in many neurodegenerative diseases, it is unclear why these cells might be more sensitive than others. One possibility raised by our data is that the suite of microtubule regulators expressed in different neurons could influence sensitivity of regeneration to reduction of HSP proteins. For example, slightly higher levels of a microtubule-stabilizing protein might eliminate the need for full HSP protein function during regeneration in the same way that tdEOS-αtubulin bypassed the requirement for spastin and atlastin for regeneration. Similarly, a different set or ration of microtubule plus end–binding proteins could make a particular neuron type more sensitive to partial reduction of HSP proteins in the same way that EB1-GFP and EB1-CT did here.
Although the idea that the function we identified for HSP proteins during axon regeneration is appealing in many ways, it is not a perfect fit. It is not known whether axon regeneration is triggered during normal wear and tear of axons in the spinal cord, and this is a critical missing piece of information necessary to evaluate whether reduction in regeneration might lead to long-term degeneration.
Independently of potential relevance to disease, the differential effect of reduction of HSP proteins on axon and dendrite regeneration is intriguing. One might expect that proteins with core cellular functions like ER and microtubule regulation would be equally required for both types of outgrowth. Similarly, if the ER is concentrated at growing axon tips to provide an extra calcium reservoir, why is this not important for dendrite regenerative growth? It will be extremely interesting to learn what promotes dendrite regeneration in future studies.
MATERIALS AND METHODS
Drosophila stocks
Seipin (104326), spichthyin (110180), atlastin (6719), and γtubulin37C (25271) RNAi lines were from the Vienna Drosophila Research Center (Vienna, Austria). Atlastin (36736) was obtained from the Bloomington Drosophila Stock Center. Seungbok Lee (Seoul National University, Seoul, Korea) provided the atlastin mutant, atl2. Spastin mutant lines (Spas5.75/TM6 and Spas10-12/TM6) were generated by Nina Sherwood (Duke University, Durham, NC).
In axon regeneration assays, the tester lines UAS-dicer2; 221-GAL4, UAS-EB1-GFP and UAS-dicer2; 221-GAL4, UAS-mCD8-RFP were crossed to RNAi lines. Analysis of atlastin or spastin heterozygous mutants was done by crossing to 221-GAL4, UAS-EB1-GFP or UAS-mCD8-RFP; 221-GAL4. Microtubule dynamics was analyzed by crossing UAS-dicer2; 221-GAL4, UAS-EB1-GFP to RNAi lines. To visualize puckered-GFP expression, UAS-dicer2, UAS-mCD8-RFP; 221-GAL4, puc-GFP was crossed to RNAi lines. ER localization assays were done by crossing UAS-dicer2; 221-GAL4, UAS-Rtnl1-GFP to γtubulin37C or atlastin RNAi. Visualization of the ER in spastin mutants was achieved by crossing spas10-12/TM6 into IGI1-GAL4. The resulting fly line IGI1-GAL4/CyO; spas10-12/TM6 was then crossed to UAS-Rtnl1-GFP, spas5.75/TM6. To visualize microtubules with td-EOS-αtubulin, IGI1-GAL4, UAS- td-EOS-αtubulin/CyO was crossed to yw (control) or IGI1-GAL4, UAS- td-EOS-αtubulin/CyO; spas5.75/TM6 was crossed to spas10-12/TM6. Non-tubby larvae (no TM6) were selected for analysis.
Generation of UAS-GFP-IP3R and UAS-Rtnl1-GFP transgenic Drosophila
To generate a tagged version of Rtnl1 that would label the ER well, we started by visualizing existing GFP protein trap lines in the Rtnl1 gene. A trap line generated by the Cooley lab, G00199 (Quinones-Coello et al., 2007), was selected based on the clear reticular pattern of GFP. The UAS version of Rtnl1-GFP was derived from this line. For cloning, total RNA from larval brain from G00199 flies was extracted using TRIzol (Invitrogen, Waltham, MA), cDNA was synthesized with reverse transcriptase, and reverse transcriptase–PCR was performed using primers based on RTNL1 isoform B. Sequencing showed that the flytrap insertion was after amino acid 37. The resultant sequence was first cloned into the PC5-Kan shuttle vector and finally into the pUAS-C5 plasmid for generating transgenic Drosophila (BestGene, Chino Hills, CA).
To generate GFP-tagged IP3R as an alternate way to visualize the ER, we started with the embryonic Drosophila IP3R cDNA in the pFastBac1 vector (Srikanth et al., 2004), which was kindly provided by Ilya Bezprozvanny (University of Texas Southwestern Medical Center, Dallas, TX). An Emerald GFP (EMD) PCR product and the IP3R were cloned into the PC5-Kan shuttle vector to generate the N-terminal-tagged EMD-IP3R. The construct was then subcloned into pUAS-C5 AttB, which was kindly provided by Richard Daniels and Barry Ganetzky (University of Wisconsin, Madison, WI), for generating transgenic Drosophila (Bestgene).
Live imaging of Drosophila larvae
Adult Drosophila crosses were maintained at 25°C, and embryos were raised at 20°C. Two-day-old larvae were used for axotomy assays. Larvae were mounted between a slide and a coverslip for the duration of the imaging process and recovered in food caps in between imaging sessions.
Axon regeneration assay
The axons of class I ddaE neurons were severed close to the cell body (proximal axotomy) using a pulsed UV laser (Photonics Instruments, St. Charles, IL). Time-lapse movies were acquired immediately after injury (0 h) and 96 h after injury using a Zeiss LSM510 confocal microscope (Carl Zeiss, Thornwood, NY). A neurite was defined as plus-end-out if at least three out of four comets moved away from the cell body. Z-projections were generated using ImageJ software (National Institutes of Health, Bethesda, MD), and length of regeneration was calculated as described previously (Stone et al., 2010) and normalized to growth that occurs due to expansion of the larval body wall.
Dendrite regeneration assay
Dendrites of class I ddaE neurons were severed, and time-lapse movies were acquired immediately after injury (0 h) and 96 h after injury using a Zeiss LSM510 confocal microscope. Z-projections were generated using ImageJ software, and the number of dendritic branch points was manually counted from the 0- and 96-h images. Average number of branch points at each time point was plotted in the graph, and statistical significance was calculated via unpaired t test using GraphPad Prism software.
Microtubule dynamics assay
Microtubule plus ends were visualized by tracking GFP-labeled EB1 comets in uninjured neurons (0 h) and 24 h after axon injury. Time-lapse movies were acquired on the Zeiss LSM510 confocal microscope at a frame rate of 2/s. Microtubule dynamics was quantified as the number of comets passing through a 10-μm length of neurite for the entire duration of the movie. Only comets that were in focus for at least three consecutive frames were counted. The average number of EB1 comets was plotted in a graph, and statistical significance was calculated via unpaired t test using GraphPad Prism software.
puc-GFP reporter assay
Injury-induced activation of the JNK signaling pathway was visualized by crossing the disease RNAi lines with a puckered protein trap line under the control of the 221-GAL4 promoter. Proximal axotomies of class I ddaE neurons were performed, and images were acquired immediately after injury (0 h) and 24 h after injury. Nuclear GFP intensity of the injured class I ddaE neuron was measured using ImageJ and normalized to nuclear GFP intensity of the neighboring uninjured class I ddaD neurons. Increase in nuclear GFP intensity at the 24-h time point serves as an indicator of JNK pathway activation.
Superresolution microscopy
Visualization of the ER markers was performed using the Zeiss LSM 880 with Airyscan (Carl Zeiss). Three-day-old old larvae expressing either UAS-Rtnl1-GFP or UAS-GFP-IP3R were mounted as described and imaged with a 63 × oil objective (3 × or 3.3× zoom). Z-projections were assembled in ImageJ using the Bio-Formats plug-in.
Measuring ER and microtubule accumulation
Proximal axotomies were performed in class I ddaE neurons as described, and regrowth was tracked. A 100-μm region at the end of the regenerating axon was considered as the tip, and a 100-μm region at the proximal edge was considered as the base. Using the Plot Profile function in ImageJ, intensities along the brightest 10-μm regions of the axon tip and base were measured in the images from the 96-h time point. Average intensity at the tip was divided by the average intensity at the base to obtain fold increase in intensity. Fold increase in Rtnl1-GFP in the axon was compared with that in a dendrite in the same neuron.
Supplementary Material
Acknowledgments
We are very grateful to Seungbok Lee and Nina Sherwood for fly stocks. We also acknowledge the tremendous resource provided to the Drosophila community by the Bloomington Drosophila Stock Center and the Vienna Drosophila RNAi Center. We also thank Dinara Bulgari for initial tests of the two transgenes used to label the ER and members of the Rolls lab for their thoughts on the project as it developed. This work was funded by a Spastic Paraplegia Foundation grant to M.M.R. and National Institutes of Health Grants NS032385 to E.L. and GM085115 to M.M.R.
Abbreviations used:
- ER
endoplasmic reticulum
- HSP
hereditary spastic paraplegia
- RNAi
RNA interference
- Rtnl1
reticulon-like-1
- SER
smooth ER.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E16-05-0287) on September 7, 2016.
REFERENCES
- Akhmanova A, Steinmetz MO. Tracking the ends: a dynamic protein network controls the fate of microtubule tips. Nat Rev Mol Cell Biol. 2008;9:309–322. doi: 10.1038/nrm2369. [DOI] [PubMed] [Google Scholar]
- Blackstone C. Cellular pathways of hereditary spastic paraplegia. Annu Rev Neurosci. 2012;35:25–47. doi: 10.1146/annurev-neuro-062111-150400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler R, Wood JD, Landers JA, Cunliffe VT. Genetic and chemical modulation of spastin-dependent axon outgrowth in zebrafish embryos indicates a role for impaired microtubule dynamics in hereditary spastic paraplegia. Dis Model Mech. 2010;3:743–751. doi: 10.1242/dmm.004002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Stone MC, Tao J, Rolls MM. Axon injury and stress trigger a microtubule-based neuroprotective pathway. Proc Natl Acad Sci USA. 2012;109:11842–11847. doi: 10.1073/pnas.1121180109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deluca GC, Ebers GC, Esiri MM. The extent of axonal loss in the long tracts in hereditary spastic paraplegia. Neuropathol Appl Neurobiol. 2004;30:576–584. doi: 10.1111/j.1365-2990.2004.00587.x. [DOI] [PubMed] [Google Scholar]
- Dietzl G, Chen D, Schnorrer F, Su KC, Barinova Y, Fellner M, Gasser B, Kinsey K, Oppel S, Scheiblauer S, et al. A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature. 2007;448:151–156. doi: 10.1038/nature05954. [DOI] [PubMed] [Google Scholar]
- Elkin SR, Lakoduk AM, Schmid SL. Endocytic pathways and endosomal trafficking: a primer. Wien Med Wochenschr. 2016;166:196–204. doi: 10.1007/s10354-016-0432-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans KJ, Gomes ER, Reisenweber SM, Gundersen GG, Lauring BP. Linking axonal degeneration to microtubule remodeling by Spastin-mediated microtubule severing. J Cell Biol. 2005;168:599–606. doi: 10.1083/jcb.200409058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans K, Keller C, Pavur K, Glasgow K, Conn B, Lauring B. Interaction of two hereditary spastic paraplegia gene products, spastin and atlastin, suggests a common pathway for axonal maintenance. Proc Natl Acad Sci USA. 2006;103:10666–10671. doi: 10.1073/pnas.0510863103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fassier C, Hutt JA, Scholpp S, Lumsden A, Giros B, Nothias F, Schneider-Maunoury S, Houart C, Hazan J. Zebrafish atlastin controls motility and spinal motor axon architecture via inhibition of the BMP pathway. Nat Neurosci. 2010;13:1380–1387. doi: 10.1038/nn.2662. [DOI] [PubMed] [Google Scholar]
- Fink J. Hereditary spastic paraplegia: clinico-pathologic features and emerging molecular mechanisms. Acta Neuropathol. 2013;126:307–328. doi: 10.1007/s00401-013-1115-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grueber WB, Jan LY, Jan YN. Tiling of the Drosophila epidermis by multidendritic sensory neurons. Development. 2002;129:2867–2878. doi: 10.1242/dev.129.12.2867. [DOI] [PubMed] [Google Scholar]
- Hammarlund M, Nix P, Hauth L, Jorgensen EM, Bastiani M. Axon regeneration requires a conserved MAP kinase pathway. Science. 2009;323:802–806. doi: 10.1126/science.1165527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill SE, Parmar M, Gheres KW, Guignet MA, Huang Y, Jackson FR, Rolls MM. Development of dendrite polarity in Drosophila neurons. Neural Dev. 2012;7:34. doi: 10.1186/1749-8104-7-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Shibata Y, Zhu PP, Voss C, Rismanchi N, Prinz WA, Rapoport TA, Blackstone C. A class of dynamin-like GTPases involved in the generation of the tubular ER network. Cell. 2009;138:549–561. doi: 10.1016/j.cell.2009.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinushi-Nakao S, Arvind R, Amikura R, Kinameri E, Liu AW, Moore AW. Knot/Collier and cut control different aspects of dendrite cytoskeleton and synergize to define final arbor shape. Neuron. 2007;56:963–978. doi: 10.1016/j.neuron.2007.10.031. [DOI] [PubMed] [Google Scholar]
- Joshi R, Venkatesh K, Srinivas R, Nair S, Hasan G. Genetic dissection of itpr gene function reveals a vital requirement in aminergic cells of Drosophila larvae. Genetics. 2004;166:225–236. doi: 10.1534/genetics.166.1.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleele T, Marinkovic P, Williams PR, Stern S, Weigand EE, Engerer P, Naumann R, Hartmann J, Karl RM, Bradke F, et al. An assay to image neuronal microtubule dynamics in mice. Nat Commun. 2014;5:4827. doi: 10.1038/ncomms5827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemm RW, Norton JP, Cole RA, Li CS, Park SH, Crane MM, Li L, Jin D, Boye-Doe A, Liu TY, et al. A conserved role for atlastin GTPases in regulating lipid droplet size. Cell Rep. 2013;3:1465–1475. doi: 10.1016/j.celrep.2013.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komarova Y, De Groot CO, Grigoriev I, Gouveia SM, Munteanu EL, Schober JM, Honnappa S, Buey RM, Hoogenraad CC, Dogterom M, et al. Mammalian end binding proteins control persistent microtubule growth. J Cell Biol. 2009;184:691–706. doi: 10.1083/jcb.200807179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krijnse-Locker J, Parton RG, Fuller SD, Griffiths G, Dotti CG. The organization of the endoplasmic reticulum and the intermediate compartment in cultured rat hippocampal neurons. Mol Biol Cell. 1995;6:1315–1332. doi: 10.1091/mbc.6.10.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M, Paik SK, Lee MJ, Kim YJ, Kim S, Nahm M, Oh SJ, Kim HM, Yim J, Lee CJ, et al. Drosophila Atlastin regulates the stability of muscle microtubules and is required for synapse development. Dev Biol. 2009;330:250–262. doi: 10.1016/j.ydbio.2009.03.019. [DOI] [PubMed] [Google Scholar]
- Lo Giudice T, Lombardi F, Santorelli FM, Kawarai T, Orlacchio A. Hereditary spastic paraplegia: clinical-genetic characteristics and evolving molecular mechanisms. Exp Neurol. 2014;261:518–539. doi: 10.1016/j.expneurol.2014.06.011. [DOI] [PubMed] [Google Scholar]
- Lu W, Fox P, Lakonishok M, Davidson MW, Gelfand VI. Initial neurite outgrowth in Drosophila neurons is driven by Kinesin-powered microtubule sliding. Curr Biol. 2013;23:1018–1023. doi: 10.1016/j.cub.2013.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Lakonishok M, Gelfand VI. Kinesin-1-powered microtubule sliding initiates axonal regeneration in Drosophila cultured neurons. Mol Biol Cell. 2015;26:1296–1307. doi: 10.1091/mbc.E14-10-1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattie FJ, Stackpole MM, Stone MC, Clippard JR, Rudnick DA, Qiu Y, Tao J, Allender DL, Parmar M, Rolls MM. Directed microtubule growth, +TIPs, and kinesin-2 are required for uniform microtubule polarity in dendrites. Curr Biol. 2010;20:2169–2177. doi: 10.1016/j.cub.2010.11.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noreau A, Dion PA, Rouleau GA. Molecular aspects of hereditary spastic paraplegia. Exp Cell Res. 2014;325:18–26. doi: 10.1016/j.yexcr.2014.02.021. [DOI] [PubMed] [Google Scholar]
- Orso G, Martinuzzi A, Rossetto MG, Sartori E, Feany M, Daga A. Disease-related phenotypes in a Drosophila model of hereditary spastic paraplegia are ameliorated by treatment with vinblastine. J Clin Invest. 2005;115:3026–3034. doi: 10.1172/JCI24694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orso G, Pendin D, Liu S, Tosetto J, Moss TJ, Faust JE, Micaroni M, Egorova A, Martinuzzi A, McNew JA, Daga A. Homotypic fusion of ER membranes requires the dynamin-like GTPase atlastin. Nature. 2009;460:978–983. doi: 10.1038/nature08280. [DOI] [PubMed] [Google Scholar]
- O’Sullivan NC, Jahn TR, Reid E, O’Kane CJ. Reticulon-like-1, the Drosophila orthologue of the hereditary spastic paraplegia gene reticulon 2, is required for organization of endoplasmic reticulum and of distal motor axons. Hum Mol Genet. 2012;21:3356–3365. doi: 10.1093/hmg/dds167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos C, Orso G, Mancuso G, Herholz M, Gumeni S, Tadepalle N, Jungst C, Tzschichholz A, Schauss A, Honing S, et al. Spastin binds to lipid droplets and affects lipid metabolism. PLoS Genet. 2015;11:e1005149. doi: 10.1371/journal.pgen.1005149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SH, Zhu PP, Parker RL, Blackstone C. Hereditary spastic paraplegia proteins REEP1, spastin, and atlastin-1 coordinate microtubule interactions with the tubular ER network. J Clin Invest. 2010;120:1097–110. doi: 10.1172/JCI40979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinones-Coello AT, Petrella LN, Ayers K, Melillo A, Mazzalupo S, Hudson AM, Wang S, Castiblanco C, Buszczak M, Hoskins RA, Cooley L. Exploring strategies for protein trapping in Drosophila. Genetics. 2007;175:1089–1104. doi: 10.1534/genetics.106.065995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rismanchi N, Soderblom C, Stadler J, Zhu PP, Blackstone C. Atlastin GTPases are required for Golgi apparatus and ER morphogenesis. Hum Mol Genet. 2008;17:1591–1604. doi: 10.1093/hmg/ddn046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roll-Mecak A, Vale RD. The Drosophila homologue of the hereditary spastic paraplegia protein, spastin, severs and disassembles microtubules. Curr Biol. 2005;15:650–655. doi: 10.1016/j.cub.2005.02.029. [DOI] [PubMed] [Google Scholar]
- Rolls MM, Hall DH, Victor M, Stelzer EH, Rapoport TA. Targeting of rough endoplasmic reticulum membrane proteins and ribosomes in invertebrate neurons. Mol Biol Cell. 2002;13:1778–1791. doi: 10.1091/mbc.01-10-0514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolls MM, Satoh D, Clyne PJ, Henner AL, Uemura T, Doe CQ. Polarity and compartmentalization of Drosophila neurons. Neural Dev. 2007;2:7. doi: 10.1186/1749-8104-2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salinas S, Carazo-Salas RE, Proukakis C, Cooper JM, Weston AE, Schiavo G, Warner TT. Human spastin has multiple microtubule-related functions. J Neurochem. 2005;95:1411–1420. doi: 10.1111/j.1471-4159.2005.03472.x. [DOI] [PubMed] [Google Scholar]
- Salinas S, Proukakis C, Crosby A, Warner TT. Hereditary spastic paraplegia: clinical features and pathogenetic mechanisms. Lancet Neurol. 2008;7:1127–1138. doi: 10.1016/S1474-4422(08)70258-8. [DOI] [PubMed] [Google Scholar]
- Sanderson CM, Connell JW, Edwards TL, Bright NA, Duley S, Thompson A, Luzio JP, Reid E. Spastin and atlastin, two proteins mutated in autosomal-dominant hereditary spastic paraplegia, are binding partners. Hum Mol Genet. 2006;15:307–318. doi: 10.1093/hmg/ddi447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherwood NT, Sun Q, Xue M, Zhang B, Zinn K. Drosophila spastin regulates synaptic microtubule networks and is required for normal motor function. PLoS Biol. 2004;2:e429. doi: 10.1371/journal.pbio.0020429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimono K, Fujimoto A, Tsuyama T, Yamamoto-Kochi M, Sato M, Hattori Y, Sugimura K, Usui T, Kimura K, Uemura T. Multidendritic sensory neurons in the adult Drosophila abdomen: origins, dendritic morphology, and segment- and age-dependent programmed cell death. Neural Dev. 2009;4:37. doi: 10.1186/1749-8104-4-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JE, Cho Y, Beirowski B, Milbrandt J, Cavalli V, Diantonio A. Dual leucine zipper kinase is required for retrograde injury signaling and axonal regeneration. Neuron. 2012;74:1015–1022. doi: 10.1016/j.neuron.2012.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skube SB, Chaverri JM, Goodson HV. Effect of GFP tags on the localization of EB1 and EB1 fragments in vivo. Cytoskeleton (Hoboken) 2010;67:1–12. doi: 10.1002/cm.20409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solowska JM, Morfini G, Falnikar A, Himes BT, Brady ST, Huang D, Baas PW. Quantitative and functional analyses of spastin in the nervous system: implications for hereditary spastic paraplegia. J Neurosci. 2008;28:2147–2157. doi: 10.1523/JNEUROSCI.3159-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srikanth S, Wang Z, Tu H, Nair S, Mathew MK, Hasan G, Bezprozvanny I. Functional properties of the Drosophila melanogaster inositol 1,4,5-trisphosphate receptor mutants. Biophys J. 2004;86:3634–3646. doi: 10.1529/biophysj.104.040121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone MC, Albertson RM, Chen L, Rolls MM. Dendrite injury triggers DLK-independent regeneration. Cell Rep. 2014;6:247–253. doi: 10.1016/j.celrep.2013.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone MC, Nguyen MM, Tao J, Allender DL, Rolls MM. Global up-regulation of microtubule dynamics and polarity reversal during regeneration of an axon from a dendrite. Mol Biol Cell. 2010;21:767–777. doi: 10.1091/mbc.E09-11-0967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone MC, Rao K, Gheres KW, Kim S, Tao J, La Rochelle C, Folker CT, Sherwood NT, Rolls MM. Normal spastin gene dosage is specifically required for axon regeneration. Cell Rep. 2012;2:1340–1350. doi: 10.1016/j.celrep.2012.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summerville JB, Faust JF, Fan E, Pendin D, Daga A, Formella J, Stern M, McNew JA. The effects of ER morphology on synaptic structure and function in Drosophila melanogaster. J Cell Sci. 2016;129:1635–1648. doi: 10.1242/jcs.184929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Shay J, McLoed M, Roodhouse K, Chung SH, Clark CM, Pirri JK, Alkema MJ, Gabel CV. Neuronal regeneration in C. elegans requires subcellular calcium release by ryanodine receptor channels and can be enhanced by optogenetic stimulation. J Neurosci. 2014;34:15947–15956. doi: 10.1523/JNEUROSCI.4238-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trotta N, Orso G, Rossetto MG, Daga A, Broadie K. The hereditary spastic paraplegia gene, spastin, regulates microtubule stability to modulate synaptic structure and function. Curr Biol. 2004;14:1135–1147. doi: 10.1016/j.cub.2004.06.058. [DOI] [PubMed] [Google Scholar]
- Tsang HT, Edwards TL, Wang X, Connell JW, Davies RJ, Durrington HJ, O’Kane CJ, Luzio JP, Reid E. The hereditary spastic paraplegia proteins NIPA1, spastin and spartin are inhibitors of mammalian BMP signalling. Hum Mol Genet. 2009;18:3805–3821. doi: 10.1093/hmg/ddp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulengin I, Park JJ, Lee TH. ER network formation and membrane fusion by atlastin1/SPG3A disease variants. Mol Biol Cell. 2015;26:1616–1628. doi: 10.1091/mbc.E14-10-1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenstein ML, Roll-Mecak A. Graded control of microtubule severing by tubulin glutamylation. Cell. 2016;164:911–921. doi: 10.1016/j.cell.2016.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakefield S, Tear G. The Drosophila reticulon, Rtnl-1, has multiple differentially expressed isoforms that are associated with a sub-compartment of the endoplasmic reticulum. Cell Mol Life Sci. 2006;63:2027–2038. doi: 10.1007/s00018-006-6142-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner AT, Lanz MC, Goetschius DJ, Hancock WO, Rolls MM. Kinesin-2 and Apc function at dendrite branch points to resolve microtubule collisions. Cytoskeleton (Hoboken) 2016;73:35–44. doi: 10.1002/cm.21270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiese C. Distinct Dgrip84 isoforms correlate with distinct gamma-tubulins in Drosophila. Mol Biol Cell. 2008;19:368–377. doi: 10.1091/mbc.E07-08-0801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson PG, Borisy GG. Maternally expressed gamma Tub37CD in Drosophila is differentially required for female meiosis and embryonic mitosis. Dev Biol. 1998;199:273–290. doi: 10.1006/dbio.1998.8900. [DOI] [PubMed] [Google Scholar]
- Windpassinger C, Auer-Grumbach M, Irobi J, Patel H, Petek E, Horl G, Malli R, Reed JA, Dierick I, Verpoorten N, et al. Heterozygous missense mutations in BSCL2 are associated with distal hereditary motor neuropathy and Silver syndrome. Nat Genet. 2004;36:271–276. doi: 10.1038/ng1313. [DOI] [PubMed] [Google Scholar]
- Wood JD, Landers JA, Bingley M, McDermott CJ, Thomas-McArthur V, Gleadall LJ, Shaw PJ, Cunliffe VT. The microtubule-severing protein Spastin is essential for axon outgrowth in the zebrafish embryo. Hum Mol Genet. 2006;15:2763–2771. doi: 10.1093/hmg/ddl212. [DOI] [PubMed] [Google Scholar]
- Xiong X, Wang X, Ewanek R, Bhat P, Diantonio A, Collins CA. Protein turnover of the Wallenda/DLK kinase regulates a retrograde response to axonal injury. J Cell Biol. 2010;191:211–223. doi: 10.1083/jcb.201006039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan D, Wu Z, Chisholm AD, Jin Y. The DLK-1 kinase promotes mRNA stability and local translation in C. elegans synapses and axon regeneration. Cell. 2009;138:1005–1018. doi: 10.1016/j.cell.2009.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Hedera P. Hereditary spastic paraplegia-causing mutations in atlastin-1 interfere with BMPRII trafficking. Mol Cell Neurosci. 2013;52:87–96. doi: 10.1016/j.mcn.2012.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.