

SNAP-25 regulates Ca2+ channels in an unknown manner. Endogenous and exogenous SNAP-25 inhibit Ca2+ currents indirectly by recruiting syntaxin-1 from clusters on the plasma membrane, thereby making it available for Ca2+ current inhibition. Thus the cell can regulate Ca2+ influx by expanding or contracting syntaxin-1 clusters.
Abstract
SNAP-25 regulates Ca2+ channels, with potentially important consequences for diseases involving an aberrant SNAP-25 expression level. How this regulation is executed mechanistically remains unknown. We investigated this question in mouse adrenal chromaffin cells and found that SNAP-25 inhibits Ca2+ currents, with the B-isoform being more potent than the A-isoform, but not when syntaxin-1 is cleaved by botulinum neurotoxin C. In contrast, syntaxin-1 inhibits Ca2+ currents independently of SNAP-25. Further experiments using immunostaining showed that endogenous or exogenous SNAP-25 expression recruits syntaxin-1 from clusters on the plasma membrane, thereby increasing the immunoavailability of syntaxin-1 and leading indirectly to Ca2+ current inhibition. Expression of Munc18-1, which recruits syntaxin-1 within the exocytotic pathway, does not modulate Ca2+ channels, whereas overexpression of the syntaxin-binding protein Doc2B or ubMunc13-2 increases syntaxin-1 immunoavailability and concomitantly down-regulates Ca2+ currents. Similar findings were obtained upon chemical cholesterol depletion, leading directly to syntaxin-1 cluster dispersal and Ca2+ current inhibition. We conclude that clustering of syntaxin-1 allows the cell to maintain a high syntaxin-1 expression level without compromising Ca2+ influx, and recruitment of syntaxin-1 from clusters by SNAP-25 expression makes it available for regulating Ca2+ channels. This mechanism potentially allows the cell to regulate Ca2+ influx by expanding or contracting syntaxin-1 clusters.
INTRODUCTION
Neurosecretion—the release of neurotransmitters from neurosecretory cells or neurons—depends on the rapid exocytosis of transmitter-filled vesicles upon the arrival of a signal. In most excitatory cells, an action potential elicits Ca2+ influx through voltage-gated calcium channels (VGCCs) to trigger release. The machinery driving vesicle fusion with the plasma membrane consists of the ternary SNARE complex (comprised of syntaxin-1, synaptosomal-associated protein of 25 kDa [SNAP-25], and VAMP2/synaptobrevin-2), which forms between the vesicle and the plasma membrane (Jahn and Fasshauer, 2012), and proteins of the Munc18 and Munc13/CAPS families, which play essential roles in SNARE-complex assembly (Rizo and Sudhof, 2012). Vesicular synaptotagmin-1 is the main Ca2+ sensor in many systems, triggering release upon binding to Ca2+ (Sudhof, 2013). Owing to the brief duration of the action potential, the coupling between Ca2+ influx and vesicle fusion takes place within small, diffusion-limited coupling compartments (“microdomains” or “nanodomains”) around a single or a few clustered VGCCs (Neher, 2015). Therefore a mechanism must exist that ensures proper colocalization between VGCCs and secretory vesicles. This is especially pertinent in synapses, where spatial and temporal coupling is very tight (Kochubey et al., 2011); in addition, a certain degree of vesicle:channel coupling must be assumed in neurosecretory cells (Klingauf and Neher, 1997).
VGCCs (especially the P/Q-type [Cav2.1] and N-type [Cav2.2] channels) interact directly with both syntaxin-1 and SNAP-25 in biochemical experiments (Rettig et al., 1996; Kim and Catterall, 1997; Yokoyama et al., 1997, 2005). The main interaction occurs via the synaptic protein interaction (synprint) site in the intracellular loop between domains II and III (Catterall, 1999). The functional consequences of the interaction with syntaxin-1 have been most thoroughly investigated, whereas less is known about the role of SNAP-25. Loading of a peptide including the synprint site into cells (Mochida et al., 1996; Rettig et al., 1997; Keith et al., 2007) or deleting the synprint site from P/Q- or N-type channels (Mochida et al., 2003; Harkins et al., 2004) disrupts neurotransmitter release, presumably due to mislocalization of vesicles away from VGCCs. However, the synprint peptide has been shown to inhibit synaptic transmission even in invertebrates, whose VGCC do not express a synprint site (Spafford et al., 2003).
Coexpression of syntaxin-1 with VGCCs (N, P/Q, or L-type [Cav1.1-1.4]) reduces Ca2+ currents, which in some cases has been attributed to a shift in the inactivation curve of the channels toward negative values (Bezprozvanny and Tsien, 1995; Bezprozvanny et al., 2000; Wiser et al., 1996, 1999; Degtiar et al., 2000; Hurley et al., 2004; Davies et al., 2011). For N-type channels, the mechanism involves potentiation of the inhibition by the Gβγ-subunit (Stanley and Mirotznik, 1997), leading to both a leftward shift in the inactivation curve and tonic inhibition (Jarvis et al., 2000). Conversely, cleaving syntaxin-1 with botulinum neurotoxin serotype C (BoNT/C) in cortical synaptosomes potentiates Ca2+ influx (Bergsman and Tsien, 2000) and in calyx-type chicken ciliary ganglion synapse, shifted the inactivation curve of N-type channels to the right (Stanley, 2003). Although they do not express a classical synprint site, the T-type VGCCs (Cav3.1-3.3) are also negatively regulated by syntaxin-1 (Weiss et al., 2012; Carbone et al., 2014).
The role of SNAP-25 in regulating Ca2+ influx has been investigated in neurons (Verderio et al., 2004; Pozzi et al., 2008; Condliffe et al., 2010; Condliffe and Matteoli, 2011). Whereas SNAP-25 is necessary for synaptic release in both GABAergic and glutamatergic neurons (Tafoya et al., 2006; Bronk et al., 2007; Delgado-Martinez et al., 2007), the native expression level of SNAP-25 is higher in the latter (Verderio et al., 2004; Frassoni et al., 2005), which was correlated with a lower Ca2+ influx into these neurons (Verderio et al., 2004). Furthermore, Ca2+ influx was augmented in neurons from SNAP-25 knockouts or heterozygotes or after knockdown (Verderio et al., 2004; Pozzi et al., 2008; Condliffe et al., 2010; Condliffe and Matteoli, 2011). This was associated with a lower paired-pulse ratio in glutamatergic neurons from SNAP-25 heterozygote animals (Antonucci et al., 2013). The situation is complicated by changing SNAP-25 expression levels during neuronal development, which renders effects of SNAP-25 on VGCCs and paired-pulse ratio transient (Frassoni et al., 2005; Antonucci et al., 2013), and the reported effects were not seen in some studies (Tafoya et al., 2006, 2008; Scullin et al., 2012).
It remains an open question by what mechanism SNAP-25 regulates VGCCs in a native system, since mechanistic studies used heterologous expression of both SNAREs and calcium channels in nonsecretory cell types (Wiser et al., 1996; Zhong et al., 1999; Jarvis and Zamponi, 2001). One study found that SNAP-25 shifts the inactivation curve of P/Q channels toward more hyperpolarized membrane potentials, whereas coexpression of syntaxin-1 and synaptotagmin-1 relieved this inhibition (Zhong et al., 1999). It was thus suggested that the SNAP-25–induced inhibition of P/Q-type channels might form part of the exocytotic cascade by limiting Ca2+ influx locally until a proper SNARE complex has formed (Zhong et al., 1999). In a coexpression experiment using N-type channels, SNAP-25 did not in itself induce G-protein inhibition but was able to partly relieve inhibition caused by syntaxin-1A expression (Jarvis and Zamponi, 2001).
Studies in neurosecretory cells identified native SNAP-25 and syntaxin-1 in distinct membrane clusters (Lang et al., 2001; Sieber et al., 2007; Lopez-Font et al., 2010) from which they have to be recruited in order for productive SNARE-complex assembly to take place (Barg et al., 2010; Knowles et al., 2010). Thus the bulk of SNAP-25 and syntaxin-1 in the membrane does not form a SNARE complex until the arrival of a vesicle, and fusion itself requires only a few SNARE complexes (Mohrmann et al., 2010; Sinha et al., 2011), constituting an infinitesimal fraction of the total SNARE content of the cell. Thus it is unclear how the proposed mechanism of VGCC inhibition and disinhibition would lead to noticeable changes in the macroscopic Ca2+ current. Alternatively, SNAREs might act to regulate VGCCs in parallel to their function in exocytosis. Such an alternative function of SNAREs might be potentially important because a number of diseases have been associated with an aberrant expression level of the SNARE SNAP-25, including attention-deficit hyperactivity disorder and epilepsy (Corradini et al., 2009).
Here we investigated how SNAP-25 regulates VGCCs in mouse adrenal chromaffin cells, which constitute a versatile assay system to study calcium-triggered exocytosis. We were especially interested in understanding whether SNAP-25 regulates VGCC directly or indirectly and whether this regulation takes place as part of the exocytic pathway or in parallel to it.
RESULTS
Inhibition of Ca2+ currents by SNAP-25 requires syntaxin-1
To understand how SNAP-25 regulates the current through voltage-gated Ca2+ channels, we performed patch-clamp experiments and isolated the Ca2+ currents from embryonic mouse chromaffin cells. We applied 5-ms depolarization steps (within a P/-4 protocol to subtract the leak current) using a bath solution containing 10 mM Ca2+ and tetrodotoxin combined with a Cs+-based pipette solution to block currents through Na+ and K+ channels. To prevent intracellular increases in [Ca2+]i, which might activate intracellular signaling, we included 10 mM ethylene glycol tetraacetic acid (EGTA) in the pipette solution. Representative Ca2+ currents from a SNAP-25 knockout (KO) cell are shown in Figure 1A, with arrows indicating current amplitude (a) and tail current (b). We found that deletion of SNAP-25 (i.e., using SNAP-25 KO chromaffin cells) led to significantly larger Ca2+ currents than in wild-type (WT) littermates (Figure 1B). SNAP-25a or SNAP-25b was overexpressed in SNAP-25 KO cells for 6–8 h using a Semliki Forest virus (SFV) harboring a bicistronic message with enhanced green fluorescent protein (EGFP) as expression marker. This led to a depression of the Ca2+ currents. Of note, expression of SNAP-25a reversed the calcium currents to their control (WT) amplitude, whereas expression of SNAP-25b depressed them even further (Figure 1B). This finding is consistent with the predominant expression of the SNAP-25a isoform in the adrenal gland (Bark et al., 1995; Grant et al., 1999). The stronger effect of SNAP-25b in regulating Ca2+ channels has also been observed in neurons (Pozzi et al., 2008). Of importance, cell sizes were unchanged by SNAP-25 expression (Figure 1C). To ensure that these changes were caused by SNAP-25 expression and not by the expression virus or the expression marker, we transfected a group of cells with SFV expressing EGFP only and found no effect of this treatment (Figure 1B). We conclude that endogenous as well as exogenously expressed SNAP-25 down-regulates VGCCs in adrenal chromaffin cells.
FIGURE 1:

Calcium current amplitudes are reduced by both endogenous and exogenous SNAP-25 expression. (A) Calcium currents from mouse chromaffin cells were measured by activation via depolarization steps to 0 mV (for 5 ms). Representative calcium currents from a SNAP-25 KO cell. Peak and tail amplitude are indicated by arrows. (B) Deletion of SNAP-25 (SNAP-25 KO) led to higher calcium current amplitudes, whereas viral expression of SNAP-25a in SNAP-25 KO cells reversed calcium current amplitudes to WT levels. Expression of SNAP-25b had an even stronger depressing effect. Cleavage of syntaxin-1 by BoNT/C1 increased the amplitudes in SNAP-25 WT cells, whereas BoNT/C was without effect in SNAP-25 KO cells, indicating that the effect of SNAP-25 on calcium currents might be mediated via syntaxin-1. The expression of EGFP in SNAP-25 KO cells was without effect. Means ± SEM. *p < 0.05, ***p < 0.001 (one-way ANOVA with Tukey’s post hoc test). N = 3 preparations for each condition; n = 23–29 cells/condition. (C) Cell sizes were unchanged by SNAP-25 expression. (D) Immunoblotting against Cavα2δ4 in SNAP-25 KO, WT, and heterozygote (HT) adrenal glands did not reveal differences in expression level. (E) The I/V curves were depressed but not shifted upon SNAP-25 expression. Activation (F) and inactivation (G) curves were only slightly shifted, if at all, by SNAP-25 expression or deletion.
To understand whether SNAP-25 acts directly on the VGCCs, or indirectly via syntaxin-1, we expressed BoNT/C light chain. BoNT/C cleaves syntaxin-1 near its transmembrane anchor (Blasi et al., 1993), and in addition it cleaves SNAP-25 between Arg-198 and Arg-199, resulting in a C-terminal deletion of eight amino acids (Foran et al., 1996; Vaidyanathan et al., 1999). It was previously shown that a SNAP-25 truncated by nine amino acids from the C-terminal end displays normal regulation of calcium influx (Verderio et al., 2004). Effectiveness of BoNT/C treatment was confirmed using electrophysiology and immunostaining against syntaxin-1 (see later discussion of Figure 4, I and J). After expression of BoNT/C in SNAP-25 WT cells, Ca2+ current amplitudes increased, whereas no significant effect was seen in SNAP-25 KO cells (Figure 1B). As a result, Ca2+ current amplitudes were indistinguishable in SNAP-25 KO and WT cells expressing BoNT/C.
FIGURE 4:

Overexpressing SNAP-25 increases syntaxin-1 immunoavailability but not its expression level. (A) Confocal slices of WT and SNAP-25 KO adrenal chromaffin cells with or without SNAP-25a or SNAP-25b overexpression stained for syntaxin-1. Scale bar, 5 μm. (B) Quantification of syntaxin-1 immunostaining (Fluor. int., fluorescence intensity). Data are normalized to the syntaxin-1 signal from SNAP-25 WT cells. ***p < 0.001 (one-way ANOVA, Tukey’s post hoc test). (C) Representative profile plots of syntaxin-1 immunolabeling. The peaks in the plots (arrows) indicate the plasma membrane (PM). Inset, confocal midsection of a SNAP-25 KO cell, indicating how the profile was measured. Scale bar, 5 μm. (D) Quantification of syntaxin-1 immunolabeling at the PM from WT and SNAP-25 KO cells. *p < 0.05, **p < 0.01, ***p < 0.001 (one-way ANOVA, Tukey’s post hoc test). (E) Western blots showing the levels of syntaxin-1 and SNAP-25 in cultured adrenal chromaffin cells before and after overexpression of SNAP-25b. The syntaxin-1 level decreased, although the immunoavailability increased. (F) 3D-SIM showed that viral overexpression of SNAP-25b in SNAP-25 KO cells leads to recruitment of syntaxin-1 from PM clusters. Top, blow-ups of part of the membranes from a SNAP-25 KO cell and a SNAP-25 KO cell overexpressing SNAP-25b. Scale bar, 2 μm. (G) Schematic illustration of the limited immunoavailability of syntaxin-1 in clusters (top) and an increased immunoavailability after overexpression of SNAP-25 (bottom), which results in recruitment of syntaxin from PM clusters. (H) Confocal maximum projections of WT cells with and without BoNT/C1 stained for SNAP-25. Scale bar, 5 μm. (I) The effect of BoNT/C was tested on secretion in SNAP-25 WT cells in a calcium uncaging experiment (flash at arrow), verifying that secretion was strongly depressed. (J) Quantification of the SNAP-25 and syntaxin-1–specific signal after cleavage of syntaxin-1 by BoNT/C. **p < 0.01 (Student’s t test, two-tailed). N = 2, n = 15–19.
Because SNAP-25 expression is without consequence for Ca2+ current amplitudes after cleaving syntaxin-1, either SNAP-25 regulates VGCCs indirectly, via syntaxin-1, or syntaxin-1 and SNAP-25 are both required—perhaps within dimers—to down-regulate channels. To distinguish between these possibilities, we overexpressed syntaxin-1A in SNAP-25 KO cells (Figure 2). This led to strong depression of Ca2+ current amplitudes (Figure 2A). Syntaxin-1 is normally found in the so-called “closed” configuration, in which the Habc domain is folded back on the SNARE (also called H3) domain (Dulubova et al., 1999). Constitutively open syntaxin-1 stimulates secretion (Gerber et al., 2008; Liu et al., 2010). However, expression of the syntaxin-1A LE-mutant (L165A/E166A), which favors the open configuration (Dulubova et al., 1999), also led to strong inhibition of calcium currents (Figure 2A), indicating that blocking of VGCCs is independent of the syntaxin-1 conformation—if anything, inhibition was slightly stronger in the LE mutant.
FIGURE 2:
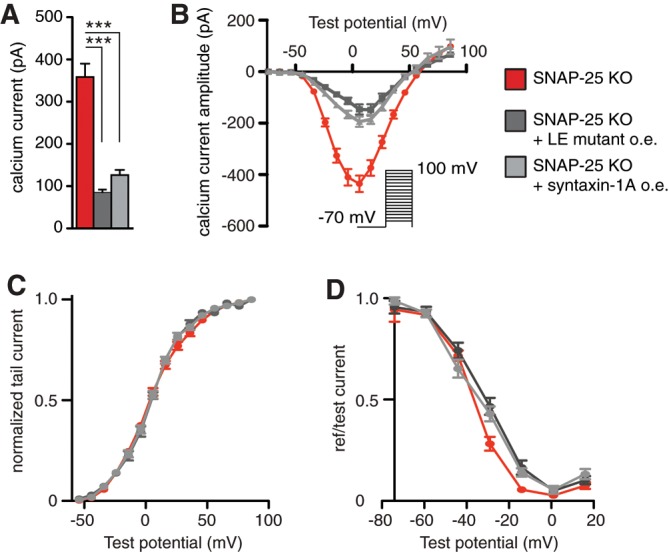
Syntaxin-1 expression strongly reduces calcium currents in SNAP-25 KO cells. (A) Viral overexpression of syntaxin-1A or the LE mutant of syntaxin-1A led to a strong decrease in calcium current amplitudes. Means ± SEM. ***p < 0.001 (one-way ANOVA, Tukey’s post hoc test). N = 3, n = 17–19. (B) The I/V relationship was depressed but not shifted upon syntaxin overexpression. (C) The activation window was not affected by syntaxin overexpression. (D) The inactivation curve was slightly shifted toward more positive potentials upon syntaxin-1A overexpression.
Overall we conclude that syntaxin-1 is a regulator of VGCC acting on its own, or at least independently of SNAP-25, whereas the effects of SNAP-25 are eliminated after expression of BoNT/C, consistent with an indirect effect of SNAP-25 via syntaxin-1.
Activation and inactivation curves are mildly affected by SNAP-25 and syntaxin-1
To understand the effect, if any, of SNAP-25 and syntaxin-1 on the gating properties of VGCCs, we plotted I/V curves and activation and steady-state inactivation curves (Figure 1E-G). The I/V curve was depressed but not noticeably shifted by SNAP-25 expression (Figure 1E) or syntaxin-1 expression (Figure 2C). The activation window shows that cells expressing endogenous or exogenous SNAP-25 were slightly shifted to hyperpolarized values compared with the SNAP-25 KO (Figure 1F), although the shift was small. Nevertheless, the direction of the shift is consistent with results upon knockdown of SNAP-25 in glutamatergic neurons (Condliffe et al., 2010). Syntaxin-1 expression did not change the activation curve (Figure 2C).
Steady-state inactivation curves were also very slightly left-shifted in cells expressing SNAP-25 compared with SNAP-25 KO (Figure 1F), which is qualitatively consistent with silencing data (Condliffe et al., 2010), although the effect seen here was smaller. A strong leftward shift of the steady-state inactivation curve was previously found after coexpression of exogenous SNAP-25 with P/Q channels in HEK cells (Zhong et al., 1999). Conversely, syntaxin-1 expression caused a rightward shift of part of the inactivation curve (Figure 2D).
From these experiments, we conclude that SNAP-25 clearly reduces overall Ca2+ current amplitudes, but the effects on activation and steady-state inactivation properties of VGCC are very mild and might be obscured by the different amplitudes of the measured currents or for other reasons (see later discussion).
Expression of Ca2+ channel types in the absence and presence of SNAP-25
The mild shifts in activation and inactivation curves upon SNAP-25 expression might be due to a selective effect on one or two VGCC types combined with the broad expression of Ca2+ channel types in embryonic or neonatal mouse adrenal chromaffin cells (Albillos et al., 2000; Aldea et al., 2002; Padin et al., 2015). Previous work was carried out in neurons, where N- and P/Q-type channels are much more prevalent (Condliffe et al., 2010), or after heterologous coexpression with P/Q channels alone (Zhong et al., 1999). To understand whether SNAP-25 expression changes the proportion of Ca2+ current through different VGCC types, we applied channel blockers sequentially to SNAP-25 KO and WT cells (Figure 3). Nifedipine was used to block L-type channels, ω-conotoxin to block N-type channels, ω-agatoxin to block P/Q channels, SNX482 to block R-type (i.e., Cav2.3) channels, and Ni2+ to block T-type channels. The results showed that L-type channels were the most prevalent VGCCs in both SNAP-25 KO and WT embryonic chromaffin cells, but N, P/Q, R, and T types also were detected (Figure 3B). It is important to note that the current fractions depend critically on the recording conditions and the exact protocol (Padin et al., 2015). The results are therefore hard to compare among studies. However, we can conclude that if SNAP-25 only affects the activation and steady-state inactivation curves of N- and P/Q-type channels, effects in adrenal chromaffin cells will likely be small, and they might not be detectable. Testing the fractional currents (in percentage of total current, two-tailed Student’s t test) between SNAP-25 KO and WT cells did not reveal significant changes for any of the channel types. Thus SNAP-25 expression does not cause large changes in the relative contributions of different channels types to the Ca2+ current.
FIGURE 3:
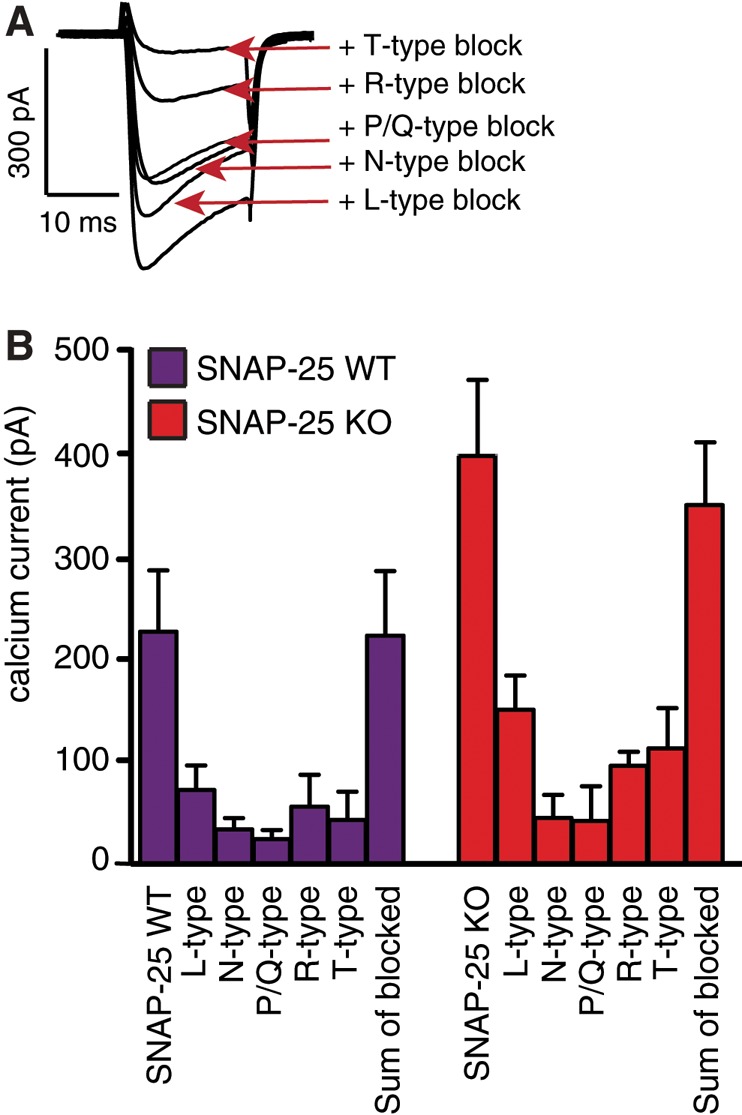
SNAP-25 WT and KO cells show similar relative expression of calcium channel subtypes. (A) Representative traces from a single-KO adrenal chromaffin cell during sequential additive application of calcium channel blockers. Bottom to top: nifedipine (L type, 3 μM), ω-conotoxin (N type, 1 μM), agatoxin (P/Q type, 1 μM), SNX482 (R type, 300 nM), Ni2+ (T type, 40 μM). (B) Sequential application of blockers did not reveal large differences in the fraction of calcium channel subtypes between SNAP-25 WT and KO cells. N = 3, n = 10–13.
Expression of SNAP-25 recruits syntaxin-1 from dense clusters and increases its immunoavailability
The notion that SNAP-25 regulation of VGCCs depends on the expression of syntaxin-1 whereas syntaxin-1 regulates VGCCs independently of SNAP-25 prompted us to examine how SNAP-25 expression affects syntaxin-1 distribution. We expressed SNAP-25a or SNAP-25b in SNAP-25 KO or WT cells and stained against syntaxin-1. Strikingly, overexpression of either SNAP-25 isoform led to a fourfold increase in the syntaxin-1 staining in confocal images (Figure 4, A and B). This was not caused by bleedthrough due to the overexpression of SNAP-25, because these cells were stained only against syntaxin-1, and the (relatively weak) fluorescence of the expressed EGFP was used to identify expressing cells. This increase in syntaxin-1 staining upon SNAP-25 expression was previously found (Hugo et al., 2013). Our staining was specific for syntaxin-1 because after expression of BoNT/C, syntaxin-1 staining was strongly reduced (Figure 4J). In plotting a line profile through a confocal section around the equatorial plane of the cell (avoiding the nucleus), we saw that overexpression of both SNAP-25a and SNAP-25b caused increased syntaxin-1 staining at or near the plasma membrane (Figure 4, C and D).
The increased syntaxin-1 staining in SNAP-25–expressing cells might be caused by up-regulation of the syntaxin-1 expression level per se or a change in syntaxin-1 immunoavailability by recruiting syntaxin-1 from the extremely dense clusters present on the plasma membrane (Sieber et al., 2007); these clusters reduce immunoavailability by epitope masking (Lang et al., 2001; Figure 4G). To distinguish between those possibilities, we performed Western blotting of SNAP-25–expressing cultured mouse adrenal chromaffin cells and compared them with untransfected cells. We found that whereas SNAP-25 was overexpressed (5.8 ± 0.7)-fold (mean ± SEM, n = 3) by the SFV system, the syntaxin-1 level was actually somewhat depressed (Figure 4E). On average, the syntaxin-1 band was reduced to 51 ± 12% (mean ± SEM, n = 3) by overexpression of SNAP-25. The reason for the reduced syntaxin-1 abundance after SNAP-25 overexpression is unknown; possibly, recruiting syntaxin-1 from clusters on the plasma membrane (see later discussion) could target syntaxin-1 for degradation (see Discussion for another possibility). In any case, increased syntaxin-1 expression cannot explain the increase in syntaxin-1 labeling upon SNAP-25 overexpression. Instead, SNAP-25 expression increases the immunoavailability of syntaxin-1.
To investigate directly whether SNAP-25 recruits syntaxin-1 from the clusters on the membrane, we subjected stained cells to three-dimensional structured illumination microscopy (3D-SIM). On examining single optical sections around the equatorial plane of the cell, it became readily apparent that syntaxin-1 is strongly clustered in SNAP-25 KO cells (Figure 4F, left). However, on overexpression of SNAP-25b, those clusters disappeared, and instead, almost continuous and relatively homogeneous staining was present at the plasma membrane (Figure 4F, right). Note that the reconstruction algorithm used here involved scaling, such that intensity differences cannot be appreciated in these images. These data are consistent with the idea that increased staining intensities (when measured in the confocal microscopy) are caused by recruitment of syntaxin-1 from clusters (Figure 4G), increasing immunoavailability.
We investigated whether syntaxin-1 also affects the immunoavailability of SNAP-25. However, after expression of BoNT/C, staining for SNAP-25 was unchanged (Figure 4, H and J), whereas staining for syntaxin-1 was reduced (Figure 4J), and secretion triggered by calcium uncaging was strongly inhibited (Figure 4I), confirming the activity of expressed BoNT/C to cleave syntaxin-1. Furthermore, overexpression of syntaxin-1A did not change the intensity of SNAP-25 staining (Supplemental Figure S1). Therefore SNAP-25 expression regulates syntaxin-1 clustering, but the opposite is not the case.
The differences in syntaxin-1 immunolabeling and clustering after SNAP-25 overexpression were immediately apparent (Figure 4; see also Hugo et al., 2013), probably due to the high SNAP-25 overexpression level induced by the SFV system (Figure 4E). If a similar mechanism exists at endogenous expression levels, then changes in syntaxin-1 labeling and clustering should be detectable when comparing SNAP-25 KO and WT cells, although these changes might be much less conspicuous. We directly compared syntaxin-1 staining in SNAP-25 WT and KO chromaffin cells (Figure 5). Indeed, we found that in confocal images, the total staining intensity for syntaxin-1 was ∼40% lower in SNAP-25 KO than in WT cells (Figure 5, A and B). However, in Western blots of adrenal glands, syntaxin-1 expression was indistinguishable between SNAP-25 KO and WT animals (Figure 5C). Using 3D-SIM, syntaxin-1 clusters could be distinguished in both WT and SNAP-25 KO cells (Figure 5, D and E). Automatized cluster detection (see Materials and Methods) was used to derive the mean cluster size per cell, and the population means of this parameter from WT and SNAP-25 KO cells were compared. This resulted in the conclusion that the SNAP-25 KO has significantly smaller syntaxin-1 clusters (Figure 5F). Thus syntaxin-1 clusters are expanded by endogenous SNAP-25 expression, leading to higher overall immunoavailability.
FIGURE 5:

SNAP-25 KO cells have lower immunoavailability of syntaxin-1 and smaller clusters of syntaxin-1. (A) Confocal micrographs of equatorial planes of a SNAP-25 WT and SNAP-25 KO adrenal chromaffin cells, respectively, stained for syntaxin-1. Scale bar, 2 μm. (B) Quantification of syntaxin-1 fluorescence intensity. ***p < 0.001 (Student’s t test, two-tailed). (C) Immunoblotting shows that the expression of syntaxin-1 in SNAP-25 WT and KO adrenal glands is indistinguishable. Each lane represents data from one animal. (D) 3D-SIM of SNAP-25 WT and SNAP-25 KO cells display clusters of syntaxin-1 at the plasma membrane. Scale bar, 2 μm. Inset, blow-up of plasma membranes from SNAP-25 WT and KO cells with lines for quantification. (E) Line profiles along the lines in inset to D. (F) Quantification of syntaxin-1 cluster size shows a decreased size in SNAP-25 KO cells. **p < 0.01 (Student’s t test, two-tailed).
We conclude that both endogenous and exogenous SNAP-25 expression cause an increase in syntaxin-1 immunoavailability in chromaffin cells through the expansion of syntaxin-1 plasma membrane clusters. This coincides with down-regulation of Ca2+ currents.
Overexpression of Doc2b or ubMunc13-2, but not Munc18-1, increases syntaxin-1 immunoavailability and inhibits Ca2+ channels
We next wanted to understand whether the expansion of syntaxin-1 clusters and the increased immunoavailability of syntaxin-1 are obligatorily linked to Ca2+ current inhibition. We therefore looked for other ways to expand syntaxin-1 clusters and argued that strong overexpression of syntaxin-1–binding proteins might achieve this task, even if those proteins—unlike SNAP-25—are not expressed at sufficient endogenous levels to cause syntaxin-1 cluster expansion under control conditions.
One syntaxin-1 interacting protein in adrenal chromaffin cells is Doc2B (Friedrich et al., 2008). Doc2A and Doc2B interact with syntaxin-1:SNAP-25 dimers and ternary SNARE complexes (Friedrich et al., 2008; Groffen et al., 2010; Sato et al., 2010). We previously showed that overexpression of Doc2b increases the size of the readily releasable vesicle pool (RRP) while depressing sustained release (Pinheiro et al., 2013). During the course of that investigation, we found that overexpression of exogenous Doc2B strongly reduces Ca2+ influx when cells were stimulated by a depolarization protocol (Supplemental Figure S2). In fact, reduction of Ca2+ currents in bovine chromaffin cells after Doc2B expression was noted before (Duncan et al., 1999), although it was unclear whether it was statistically significant. Here we measured the Ca2+ current in newborn Doc2B KO mouse chromaffin before and after overexpression of Doc2B. We indeed found that calcium currents were depressed (Figure 6C). To understand whether a similar mechanism was involved as in the SNAP-25–induced depression of Ca2+ currents, we stained Doc2B WT, Doc2B KO, and Doc2B-overexpressing cells against syntaxin-1. The Doc2B-overexpressing cells displayed a very strong (more than an order of magnitude) increase in syntaxin-1 staining, which appeared to be strongest at or near the plasma membrane (Figure 6, A and B). Western blot analysis showed that Doc2B overexpression did not up-regulate syntaxin abundance (Figure 6D), pointing to increased immunoavailability as the mechanism resulting in down-regulation of Ca2+ currents.
FIGURE 6:

Increasing syntaxin-1 immunoavailability reduces calcium currents in mouse chromaffin cell. (A) Confocal midsections of WT and Doc2B KO adrenal chromaffin cells with and without Doc2B overexpression (o.e.) stained against syntaxin-1. Scale bar, 5 μm. (B) Quantification of the syntaxin-1 immunolabeling indicates a very strong increase upon Doc2B overexpression (note broken axis). ***p < 0.001 (one-way ANOVA with Tukey’s post hoc test). N = 2, n = 21–26. (C) Amplitudes of calcium currents in Doc2B KO cells with and without Doc2B overexpression. (D) Western blotting of cultured chromaffin cells shows that the endogenous expression level of syntaxin-1 is not changed upon overexpression of Doc2B. (E) Confocal midsections of SNAP-25 WT cells with and without ubMunc13-2 overexpression stained for syntaxin-1. Scale bar, 5 μm. (F) Quantification of the immunoavailability of syntaxin-1 reveals an increase upon ubMunc13-2 overexpression. *p < 0.05 (Student’s t test, two-tailed). N = 2, n = 20. (G) Calcium currents in SNAP-25 WT cells and SNAP-25 WT overexpressing ubMunc13-2. ***p < 0.001 (Student’s t test, two-tailed). N = 2, n = 20. (H) Confocal midsections of Munc18-1 KO cells with and without Munc18-1 overexpression, stained for syntaxin-1. Scale bar, 5 μm. (I) Quantification of total syntaxin-1 fluorescence intensity. (J) Calcium currents in Munc18-1 KO cells with and without Munc18-1 overexpression. Means ± SEM. No significance is found (one-way ANOVA, Tukey’s post hoc test). N = 3, n = 21–23. (K) Western blotting showed that the endogenous expression level of syntaxin-1 is not changed upon overexpression of Munc18-1. (L) Confocal midsections of SNAP-25 WT cells with and without MBCD stained against syntaxin-1. Scale bar, 5 μm. (M) Quantification of syntaxin-1 immunolabeling indicates a strong increase upon treatment with MBCD. ***p < 0.001 (Student’s t test, two-tailed). N = 2, n = 26–28. (N) Amplitudes of calcium currents in SNAP-25 WT cells treated with MBCD were decreased compared with untreated WT cells. ***p < 0.001 (Student’s t test, two-tailed). N = 2, n = 26–28.
We also expressed ubMunc13-2, which was recently identified as the endogenous Munc13 isoform in adrenal chromaffin cells (Man et al., 2015). Munc13 proteins interact with syntaxin-1 via their MUN domain and cause the transition from a “closed” to an “open” conformation, which is required for vesicle priming (Basu et al., 2005; Stevens et al., 2005). Expression of ubMunc13-2 in embryonic chromaffin cells significantly increased syntaxin-1 staining (Figure 6, E and F) and resulted in inhibition of Ca2+ currents (Figure 6G). Separate experiments showed that expression of ubMunc13-2 in chromaffin cells induces a massive increase in secretion triggered by Ca2+ (Supplemental Figure S2), confirming expression of ubMunc13-2.
The first protein that binds to syntaxin-1 in the exocytotic cascade is most likely Munc18-1 (Ma et al., 2013). In chromaffin cells, Munc18-1 is necessary for docking vesicles to the plasma membrane, and secretion in Munc18-1–knockout cells is abrogated (Voets et al., 2001). However, Munc18-1 also has an essential postdocking role in exocytosis, probably by stimulating SNARE-complex assembly (Gulyas-Kovacs et al., 2007; Rizo and Sudhof, 2012).
We investigated Ca2+ currents in Munc18-1 KO cells and Munc18-1 KO cells overexpressing Munc18-1. Ca2+ current amplitudes were indistinguishable between the two groups (Figure 6J). Immunostaining and Western blotting against syntaxin-1 did not identify significant changes between the groups (Figure 6, H, I, and K). Note that Munc18 expression has been described to cause increased syntaxin-1 targeting to the plasma membrane (Gulyas-Kovacs et al., 2007); however, in the present context, we always quantified the total syntaxin-1 immunolabeling of the cell, which is not significantly changed during short-term Munc18-1 overexpression (Munch et al., 2016). Western blotting of cultured chromaffin cells confirmed overexpression of Munc18-1 (Figure 6K).
We conclude that overexpression of Munc18-1, the essential syntaxin-binding protein, which controls the entry into the exocytotic cascade, does not modulate Ca2+ currents in adrenal chromaffin cells. This underscores the correlation between syntaxin immunoavailability and calcium current depression.
Extraction of cholesterol from the membrane disperses syntaxin-1 clusters and inhibits VGCCs
The foregoing findings obtained using overexpression of syntaxin-1 or syntaxin-1–binding proteins for 6–8 h indicate that there is a strong negative correlation between syntaxin-1 immunoavailability and calcium current magnitude. Finally, we wanted to investigate whether acute dispersal of syntaxin-1 clusters might lead to the same effects without the intricacies of an expression system. Methyl-β-cyclodextrin (MBCD) readily extracts cholesterol from plasma membranes without affecting phospholipids (Ohtani et al., 1989), which leads to dispersal of syntaxin-1 (and SNAP-25) clusters in PC12 cells (Lang et al., 2001). We therefore treated chromaffin cells with 15 mM MBCD for 30 min and subjected cells to either staining for syntaxin-1 or electrophysiological analysis.
Syntaxin-1 staining increased in intensity on treatment with MBCD (Figure 6, L and M), which was observed previously and attributed to cluster dispersal (Lang et al., 2001), consistent with our interpretation. Moreover, calcium currents were reduced in amplitude (Figure 6N).
These experiments show that rapid syntaxin-1 cluster dispersal by chemical means also achieves correlated increases in syntaxin-1 immunolabeling and reductions in Ca2+ current magnitude.
DISCUSSION
We investigated the mechanism behind SNAP-25–induced suppression of Ca2+ currents in secretory cells. We find that BoNT/C expression blocks the ability of SNAP-25 to inhibit calcium currents in embryonic mouse adrenal chromaffin cells and that both overexpression of SNAP-25, and the endogenous SNAP-25 expression level cause correlated changes in syntaxin-1 immunoavailability and clustering. Dispersal of syntaxin-1 clusters results in increased immunoavailability (Lang et al., 2001), leading us to the suggestion that it is the dispersal of syntaxin-1 clusters that causes both increased intensity of syntaxin-1 staining and calcium current down-regulation. This was further shown by direct visualization of syntaxin-1 clusters using 3D-SIM. Our conclusion is supported by the observation that overexpression of two syntaxin-1–binding proteins, Doc2B and ubMunc13-2, also causes correlated reductions in Ca2+ current and increases in syntaxin-1 immunoavailability. However, only the endogenous expression level of SNAP-25 suffices to change calcium current amplitudes, whereas calcium influx/currents were unchanged in the Munc13-2 KO and Doc2B KO cells compared with WT littermates (Supplemental Figure S1; unpublished data). A similar effect was found upon treatment with MBCD, which expands syntaxin-1 clusters chemically within several minutes (Lang et al., 2001) and inhibits secretion (Lang et al., 2001; Churchward et al., 2005). Cholesterol is required for several processes involved in exocytosis and also plays a direct role due to its curvature (Churchward et al., 2005). Moreover, MBCD might have cholesterol-independent effects (Ormerod et al., 2012). Therefore MBCD application is a crude manipulation, but with this reservation in mind, the correlated increases in syntaxin-1 labeling and decreases in calcium current induced by MBCD overall support the notion that syntaxin-1 cluster expansion leads to VGCC inhibition. An alternative explanation of our data could be based on the observation that calcium influx can induce syntaxin-1 expression (Sutton et al., 1999). Therefore, if deletion of SNAP-25 induces changes in calcium channel gating, it might change the overall expression of syntaxin-1; however, the expression level of syntaxin-1 was normal in SNAP-25 KO cells (Figure 5C), ruling this out. It is possible, however, that this effect could cause the reduction in syntaxin-1 expression upon SNAP-25 overexpression (Figure 4E), which reduces calcium currents (Figure 1).
Our findings argue against the suggestion that free SNAP-25 inhibits VGCC, whereas the formation of a ternary SNARE complex with syntaxin-1 and synaptotagmin relieves the inhibition (Zhong et al., 1999): first, we found that syntaxin-1 inhibits VGCCs independently of SNAP-25, whereas SNAP-25 requires syntaxin-1 to inhibit channels. Second, we demonstrated that three separate manipulations (SNAP-25b overexpression, Munc18-1 overexpression, ubMunc13-2 overexpression), which all increase the number of primed vesicles having a partially assembled SNARE complex (Sørensen et al., 2003; Gulyas-Kovacs et al., 2007), do not increase Ca2+ currents. In fact, two of those manipulations (SNAP-25b and ubMunc13-2 overexpression) decreased Ca2+ currents while causing massive vesicle priming (Supplemental Figure S3; Sørensen et al., 2003). The Ca2+ currents in the Munc18-1 KO cells, which do not have any primed vesicles (Voets et al., 2001), were unchanged compared with the Munc18-1 overexpression, which rescues the primed pool (Gulyas-Kovacs et al., 2007). Thus, in a native secretory cell containing secretory vesicles, the proposed direct regulation of VGCCs by SNAP-25 and relief by ternary SNARE-complex formation do not dominate the macroscopic Ca2+ current, but it should be kept in mind that the previous experiments were conducted after expression of P/Q-type VGCCs, which constitute a minority population of the VGCCs in embryonic mouse chromaffin cells (Figure 3). Therefore a type-specific effect on P/Q channels, which might be highly relevant in synapses, might not have been picked up in our experiments.
We propose that SNAP-25 binds to syntaxin-1 and forms a 1:1 complex (or possibly a complex with two syntaxins; Xiao et al., 2001; Fasshauer and Margittai, 2004), which results in a net recruitment of syntaxin-1 out of clusters on the plasma membrane The recruited syntaxin-1 leads to inhibition of VGCCs (Supplemental Figure S4). Indeed, syntaxin:SNAP-25 dimers in different configurations can be readily detected in the plasma membrane of secretory cells (Halemani et al., 2010; Rickman et al., 2010), and it has been suggested that the first SNARE motif (Qb) in SNAP-25 can extract syntaxin-1 from clusters (Halemani et al., 2010). This fits well with our finding that the SNAP-25 expression level regulates syntaxin-1 clusters, but syntaxin-1 does not regulate SNAP-25 clusters. However, the function of these spontaneously forming SNAP-25:syntaxin dimers in exocytosis are unclear because reconstitution experiments indicate that productive SNARE complexes form from syntaxin-1 bound to Munc18-1 (Ma et al., 2013). Because syntaxin-1 and SNAP-25 exist in a large excess over Munc18-1 (Schutz et al., 2005; Wilhelm et al., 2014), the majority of syntaxin-1:SNAP-25 dimers would not be involved in fusing vesicles, but a dynamic balance between clustered, free, and SNAP-25 bound syntaxin-1 (Bar-On et al., 2012) might be used by the cell to regulate calcium influx (Supplemental Figure S4). According to this idea, the SNAP-25 effect on VGCC might depend on the clustering of syntaxin-1 and therefore be different in different cell types; in cell types with a low degree of syntaxin-1 clustering, SNAP-25 expression might relieve the syntaxin-1–dependent inhibition of VGCC (Jarvis and Zamponi, 2001) by driving free syntaxin-1 into syntaxin-1:SNAP-25 dimers, whereas in cells (presumably secretory cells) in which syntaxin-1 is strongly clustered, SNAP-25 might recruit syntaxin-1 out of clusters to cause channel inhibition. Thus it is hard to predict the outcome of this mechanism without knowing the distribution of syntaxin-1 (and SNAP-25) inside and outside of clusters. Therefore it remains an open question whether the mechanism we propose here is at work in neurons.
A few other observations point toward interaction with syntaxin-1 as part of the mechanism for SNAP-25–mediated down-regulation of VGCC activity and calcium currents: both in our study and in previous work, the SNAP-25b isoform was more effective than the SNAP-25a isoform in down-regulating Ca2+ channels (Figure 1; Pozzi et al., 2008). SNAP-25b interacts slightly stronger with syntaxin-1 (Nagy et al., 2005). Furthermore, the S187E (phosphomimetic) mutation in SNAP-25 also is more effective than the S187A mutant in down-regulating channels (Pozzi et al., 2008), and it also interacts more strongly with syntaxin-1 (Yang et al., 2007). Thus the ability of SNAP-25 variants to interact with syntaxin-1 seems to correlate with their efficacy in down-regulating Ca2+ currents.
The mechanism of syntaxin-1–induced inhibition of N-type channels has been extensively studied. Syntaxin-1A potentiates the inhibition of N-type channels by the Gβγ-subunit (Stanley and Mirotznik, 1997), leading to both a shift in the inactivation curve and tonic inhibition (Jarvis et al., 2000). Further experiments showed that syntaxin-1B affects channel gating but not G-protein modulation, whereas syntaxin-1A affects both (Jarvis and Zamponi, 2001; Lu et al., 2001); both syntaxin-1 isoforms are expressed in adrenal chromaffin cells (Baltazar et al., 2003). The SNARE domain of syntaxin-1A binds to Gβ, whereas the N-terminal domain binds to the synprint motif of the calcium channel (Jarvis et al., 2002). Syntaxin-1A might therefore increase the modification of the channel by increasing the availability of Gβ. In our study, the inactivation curves in the presence or absence of SNAP-25 or after syntaxin-1A overexpression were only slightly changed, if at all. This is consistent with the finding that the shift in inactivation curve is susceptible to coexpression of other syntaxin-1–interacting proteins, including nsec-1 (Munc18; Jarvis and Zamponi, 2001). Because we used native secretory cells expressing the full battery of secretory proteins (including Munc18-1), this might be the reason for the lack of a clear effect on inactivation potential. However, a shift in inactivation potential was found after BoNT/C treatment of chicken ciliary ganglion calyx synapses, which almost exclusively express N-type channels (Stanley, 2003). Therefore another reason for the small effect might be the broad expression of channel types in embryonic mouse chromaffin cells (Figure 3). A final reason can be found in the observation made for chicken N-type channels, in which low (endogenous) levels of Gβγ—as we presumably have here—resulted in voltage-independent inhibition, whereas voltage dependence was only seen after overexpression of Gβγ (Lu et al., 2001). Overall the voltage-dependent effect does not seem to account for the inhibition of Ca2+ currents upon SNAP-25 expression. A similar conclusion was reached upon examination of weakly inactivating N-type Ca2+ channels in bovine chromaffin cells inhibited by syntaxin-1 (Hurley et al., 2004).
Overall SNAP-25 is expected to have both positive and negative effects on exocytosis due to its obligatory involvement in exocytosis (Sørensen et al., 2003) and its negative regulation of Ca2+ influx. The reduction of calcium currents reduces the triggering of catecholamine release but also the pacemaker current via VGCCs (especially L-type channels Cav1.2 and Cav1.3), which in turn drives the opening of highly expressed BK and SK channels, resulting in action potential bursting (Marcantoni et al., 2010; Mahapatra et al., 2012; Vandael et al., 2015a). Owing to the very high input resistance of adrenal chromaffin cells, a few picoamperes of current is sufficient to make a difference for burst firing, which effectively increases catecholamine secretion (Vandael et al., 2015a, b). The consequences of shifting from SNAP-25a to SNAP-25b expression will be a compromise between the larger primed vesicle pool supported by the b isoform (Sørensen et al., 2003) and the stronger resulting inhibition of calcium currents. It is interesting that the postnatal shift from SNAP-25a to SNAP-25b expression (Bark et al., 1995; Bark et al., 2004) coincides with maturation of synapses, which involves a tighter colocalization of vesicles with VGCCs (Tachenberger and von Gersdorff, 2000). SNAP-25b might potentially contribute to this process by increasing vesicle priming while keeping VGCCs under tighter control.
The ability of syntaxin-1 or SNAP-25 to down-regulate VGCCs has been seen as part of the exocytotic cascade, and mechanisms have been proposed to incorporate the ability of SNAREs to inhibit Ca2+ influx into the pathway leading to vesicle fusion. Based on our data, the regulation of VGCCs by SNAP-25 and syntaxin-1 might take place in a reaction parallel to the exocytotic cascade and be executed by t-SNARE dimers or monomers, which do not at the same time participate in ternary SNARE-complex formation (Ma et al., 2013). Thus the bulk of the SNAP-25 and syntaxin-1, which are among the most highly expressed presynaptic proteins (Wilhelm et al., 2014), might act as gatekeepers to regulate Ca2+ influx, whereas the essential role in exocytosis is played by a minority of the protein population forming the small number of SNARE complexes needed for membrane fusion (Mohrmann et al., 2010; Sinha et al., 2011).
MATERIALS AND METHODS
Cell culture, expression constructs, and transfection
SNAP-25 KO and Munc18-1 KO embryonic mice and Doc2B KO postnatal (P2) mice of either sex were obtained by crossing of heterozygotes. Mice were killed by cervical dislocation or (for embryos) by decapitation. Embryonic mice were recovered by cesarean section at embryonic day 18 (E18). WT littermates were used as controls. All genotypes were identified by PCR genotyping. NMRI P2 mice were used for Western blotting. Adrenal chromaffin cells were enzymatically isolated as described previously (Sørensen et al., 2003). After isolation, adrenal chromaffin cells were seeded out on glass slides (25 mm), acute expression of protein of interest was induced with SFV, and the cells were used for electrophysiological experiments at days in vitro (DIV) 2–4. For immunohistochemistry, cells were cultured on poly-d-lysine (Sigma-Aldrich)–coated coverslips (25 mm). For each Western blot of cultured chromaffin cells expressing different construct, cells from six embryos were pooled, grown in Petri dishes, and lysed at DIV3. Generation of SFV particles followed standard protocols. SNAP-25A, SNAP-25B, Doc2B, Munc18-1, syntaxin-1A (syx), syntaxin-1A(L165A,E166A) (LE mutant), ubMunc13-2, and BoNT/C1 were expressed from SFV1 plasmids containing a poliovirus internal ribosomal entry site (PV-IRES) and EGFP. A plasmid harboring EGFP alone was used as a control. All constructs were verified by DNA sequencing, and 6–8 h was allowed for expressing the proteins of interest after infection with SFV.
Immunocytochemistry
Cells stained against syntaxin-1 (1:500; 110011/110302; Synaptic Systems), SNAP-25 (1:1000; 111011; Synaptic Systems), or EGFP (1:1500; ab13970; Abcam) were isolated and prepared as described. Cells were fixed in 4% paraformaldehyde (PFA) for 15 min, washed, permeabilized in 0.1% Triton X-100 (T8787; Sigma-Aldrich), and blocked in phosphate-buffered saline (PBS) containing 3% bovine serum albumin (BSA; A4503; Sigma-Aldrich). The cells were incubated with primary antibodies overnight at 4°C, washed, and incubated with secondary antibodies (Alexa Fluor 488–conjugated goat anti-chicken [A11039; Molecular Probes], Alexa Fluor 546–conjugated goat anti-rabbit [A11810; Invitrogen], Alexa Fluor 647–conjugated goat anti-mouse [A21235; Molecular Probes], and Alexa Fluor 647–conjugated goat anti-rabbit [A21245; Invitrogen]) for 2 h at room temperature, washed, and mounted on a microscope slide. Finally, samples were mounted with Fluorsave (Dako). Micrographs were recorded at room temperature.
Confocal microscopy
All transfected cells were identified by EGFP fluorescence. Micrographs were recorded using a Zeiss LSM710 point laser (Argon Lasos RMC781272) scanning confocal microscope with a Zeiss Plan-Apochromat 63×/numerical aperture (NA) 1.4 oil objective (Carl Zeiss, Oberkochen, Germany). All micrographs were sampled in a linear frame scan mode, with a frame size of 512 × 512, an image size of 33.7 × 33.7 μm, and a bit depth of 16. We collected z-stacks of ∼8–12 confocal sections (0.7–1 μm). For analysis, maximum projections from whole z-stacks were created using ImageJ 1.47q (National Institutes of Health, Bethesda, MD). Fluorescence levels were quantified as the integrated density of a circular region of each individual image containing a cell, with the intensity of a background region of the same size subtracted.
Syntaxin 1 cluster size analysis: sample preparation, 3D-SIM microscopy, and image analysis
For 3D-SIM analysis of E18 SNAP-25 KO and WT chromaffin cells (Figure 5), cells were fixed at DIV3. To avoid shrinking of cells and folding of plasma membrane, which is apparent on 3D-SIM micrographs and interferes with cluster analysis, cells were first prefixed for 5 min in isosmotic fixative containing 0.9% PFA in 0.05 M 1,4-piperazinediethanesulfonic acid (PIPES)/NaOH (pH 7.4, 310 mOsm). This was followed by 8 min of incubation in 3,7% PFA in 0.05 M PIPES/NaOH (pH 7.4). Afterward, the cells were washed three times with PBS (pH 7.4) and permeabilized for 10 min in PBS containing 0.1% Triton X-100 (Sigma-Aldrich), followed by 30 min of blocking in PBS containing 3% BSA. The blocking solution was used for diluting antibodies. The chromaffin cells were incubated overnight with anti-syntaxin1 antibodies (110011; Synaptic Systems) at room temperature, washed three times with PBS, and incubated with secondary goat anti-mouse Alexa 546 antibodies (Life Technologies) overnight at 4°C. After extensive washings with PBS, cells were rinsed with water and mounted on microscopic slides with Prolong Gold (Invitrogen). For this series of experiments, confocal images (Figure 5A) were acquired with a HCX PL APO CS lens (NA 1.4; Leica Microsystems A/S) with frame size of 512 × 512 and a zoom factor 10 on a LEICA SP5-X Confocal microscope (Leica Microsystems). 3D-SIM was performed using the Elyra PS.1 platform (Carl Zeiss) equipped with a 63× Plan-Apochromat lens (NA 1.46; Carl Zeiss). The 3D-SIM–specific background was subtracted by setting the intensity threshold above the intensity of a nucleus. For each cell, a region of interest (ROI) was defined as the inner edge of the plasma membrane. The content of the ROI was removed to isolate the intensities from the plasma membrane. The images were analyzed using Volocity software (Volocity Software), and clusters were identified as objects above a threshold of 700, which corresponds to ∼1% of the 16-bit dynamic range. Touching-object separation routine was applied to isolate single clusters, and objects <0.0038 μm2 (corresponding to four 31 nm × 31 nm pixels on a 3D-SIM image) were excluded from further analysis. A mean area of one syntaxin-1 cluster was determined for each cell. Results from four independent experiments were included in the final analysis.
Electrophysiology
Controls and cells expressing target proteins were measured from the same preparations in order to cancel out between-preparation variability. Transfected cells were identified on their expression of EGFP. The patch-pipette solution included (in mM) 112.5 CsGlut, 36 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES), 9 NaCl, 3 MgATP, 0.45 Na2GTP, and 10 EGTA (300 mOsm, pH 7.2). The standard extracellular medium consisted of (in mM) 135 NaCl, 10 HEPES, 2.8 KCl, 10 CaCl2, 1 MgCl2, 11 d-glucose, and 1 μM tetrodotoxin (ab120055; Abcam; 310 mOsm, pH 7.2). Cells were whole-cell voltage clamped at −70 mV with an EPC-10 USB amplifier (HEKA Elektronik, Lambrecht/Pfalz, Germany) under control of PatchMaster software (HEKA Elektronik). Currents were filtered at 8.3 kHz and sampled at 33.3 kHz. The series resistance was compensated 70%. Only cells with series resistances <17 MΩ were used for recordings and following analysis. Pipette resistance ranged from 3 to 5 MΩ. All recordings were done at room temperature. To analyze the standing currents and activation properties of the channels, cells were depolarized for 5 ms from holding at −70 to 0 mV. The amplitudes of the total standing currents and the subsequent tail currents were analyzed. An I/V protocol was executed with the test potential being applied as increments of 10 mV from −70 to +100 mV. Before execution of the inactivation protocol, cells were preconditioned with pulses of variable amplitudes of fixed duration (4 s) as P/-4 leak correction was used. Membrane capacitance measurements, ratiometric intracellular calcium [Ca2+]i measurements, and amperometry were performed using a depolarization train as previously described (Sørensen et al., 2006). The release of catecholamines after cleavage of syntaxin-1 by BoNT/C was triggered by ultraviolet flash photolysis (JML-C2; Rapp Optoelektronik) of a caged-calcium compound, nitrophenyl-EGTA (Synaptic Systems), which was infused into the cell via the patch pipette. The depolarizations in the Doc2B cells were introduced using a 6+4 protocol (Voets et al., 1999). For isolation of Ca2+ channels subtypes, subtype-specific blockers were sequentially applied to the cells via a local application system. The following concentrations of blockers were used: 3 μM nifedipine (L type; ab120135; Abcam), 1 μM ω-conotoxin (N type; ab120215; Abcam), 1 μM ω-agatoxin (P/Q type; 120210; Abcam), 300 nM SNX482 (R type; ab120259; Abcam), and 40 μM NiCl2 (T type).
Electrophoresis and immunoblotting
Adrenal glands were collected from P0-2 NMRI or E18 SNAP-25 mice and lysed in RIPA buffer (Invitrogen) supplemented with Protease Inhibitor Cocktail (Invitrogen). The protein concentrations were determined by use of a BCA Protein Assay Kit (23227; Pierce). A 30-μg (from adrenal gland samples) or 10-μg (SNAP-25 overexpression experiments) amount of protein was resolved by SDS–PAGE, transferred to nitrocellulose, and blotted using anti–SNAP-25 (mouse, dilution 1:1000; 111011, Synaptic Systems), syntaxin-1 (mouse, 1:250; 3265, Abcam), Cavα2δ4 (rabbit, 1:200; ACC-104, Alomone Labs), or actin horseradish peroxidase–conjugated (as loading control; mouse, 1:10,000; 3854, Sigma-Aldrich) antibodies. The blots were developed by chemiluminescence using the ECL Plus Western blotting substrate system (Pierce). Immunoreactive bands were detected using the FluorChemE image acquisition system (ProteinSimple) and quantified with ImageJ 1.47q.
Statistical analysis
GraphPad Prism 5.0 was used for all statistical analysis. Means ± SEM are displayed. For statistical analysis of two groups, a two-tailed Student’s t test was used. For analysis of several groups, one-way analysis of variance (ANOVA) with a Tukey’s post hoc test was used.
Supplementary Material
Acknowledgments
We thank Anne Marie Nordvig Petersen for excellent technical assistance, the Verhage lab for the viral construct expressing BoNT/C, and the Wojcik/Brose lab for viral particles expressing ubMunc13-2. This work was supported by grants from the Lundbeck Foundation (J.B.S.), the Novo Nordisk Foundation (J.B.S.), and the Danish Medical Research Council (J.B.S. and S.H.).
Abbreviations used:
- 3D-SIM
three-dimensional structured illumination microscopy
- ANOVA
analysis of variance
- BoNT/C
botulinum neurotoxin serotype C
- EGFP
enhanced green fluorescent protein
- EGTA
ethylene glycol tetraacetic acid
- HEPES
4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid
- KO
knockout/null
- MBCD
methyl-β-cyclodextrin
- PFA
paraformaldehyde
- PIPES
1,4-piperazinediethanesulfonic acid
- SFV
Semliki Forest virus
- SNAP-25
synaptosomal-associated protein of 25 kDa
- VGCC
voltage-gated calcium channel
- WT
wild type.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E16-03-0184) on September 7, 2016.
REFERENCES
- Albillos A, Neher E, Moser T. R-Type Ca2+ channels are coupled to the rapid component of secretion in mouse adrenal slice chromaffin cells. J Neurosci. 2000;20:8323–8330. doi: 10.1523/JNEUROSCI.20-22-08323.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldea M, Jun K, Shin HS, Andres-Mateos E, Solis-Garrido LM, Montiel C, Garcia AG, Albillos A. A perforated patch-clamp study of calcium currents and exocytosis in chromaffin cells of wild-type and alpha(1A) knockout mice. J Neurochem. 2002;81:911–921. doi: 10.1046/j.1471-4159.2002.00845.x. [DOI] [PubMed] [Google Scholar]
- Antonucci F, Corradini I, Morini R, Fossati G, Menna E, Pozzi D, Pacioni S, Verderio C, Bacci A, Matteoli M. Reduced SNAP-25 alters short-term plasticity at developing glutamatergic synapses. EMBO Rep. 2013;14:645–651. doi: 10.1038/embor.2013.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baltazar G, Carvalho AP, Duarte EP. Differential expression of syntaxin 1A and 1B by noradrenergic and adrenergic chromaffin cells. Neurochem Res. 2003;28:1453–1457. doi: 10.1023/a:1025658005937. [DOI] [PubMed] [Google Scholar]
- Barg S, Knowles MK, Chen X, Midorikawa M, Almers W. Syntaxin clusters assemble reversibly at sites of secretory granules in live cells. Proc Natl Acad Sci USA. 2010;107:20804–20809. doi: 10.1073/pnas.1014823107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bark C, Bellinger FP, Kaushal A, Mathews JR, Partridge LD, Wilson MC. Developmentally regulated switch in alternatively spliced SNAP-25 isoforms alters facilitation of synaptic transmission. J Neurosci. 2004;24:8796–8805. doi: 10.1523/JNEUROSCI.1940-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bark IC, Hahn KM, Ryabinin AE, Wilson MC. Differential expression of SNAP-25 protein isoforms during divergent vesicle fusion events of neural development. Proc Natl Acad Sci USA. 1995;92:1510–1514. doi: 10.1073/pnas.92.5.1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-On D, Wolter S, van de Linde S, Heilemann M, Nudelman G, Nachliel E, Gutman M, Sauer M, Ashery U. Super-resolution imaging reveals the internal architecture of nano-sized syntaxin clusters. J Biol Chem. 2012;287:27158–27167. doi: 10.1074/jbc.M112.353250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu J, Shen N, Dulubova I, Lu J, Guan R, Guryev O, Grishin NV, Rosenmund C, Rizo J. A minimal domain responsible for Munc13 activity. Nat Struct Mol Biol. 2005;12:1017–1018. doi: 10.1038/nsmb1001. [DOI] [PubMed] [Google Scholar]
- Bergsman JB, Tsien RW. Syntaxin modulation of calcium channels in cortical synaptosomes as revealed by botulinum toxin C1. J Neurosci. 2000;20:4368–4378. doi: 10.1523/JNEUROSCI.20-12-04368.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezprozvanny I, Tsien RW. Voltage-dependent blockade of diverse types of voltage-gated Ca2 +channels expressed in Xenopus oocytes by the Ca2+ channel antagonist mibefradil (Ro 40-5967) Mol Pharmacol. 1995;48:540–549. [PubMed] [Google Scholar]
- Bezprozvanny I, Zhong P, Scheller RH, Tsien RW. Molecular determinants of the functional interaction between syntaxin and N-type Ca2+ channel gating. Proc Natl Acad Sci USA. 2000;97:13943–13948. doi: 10.1073/pnas.220389697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasi J, Chapman ER, Yamasaki S, Binz T, Niemann H, Jahn R. Botulinum neurotoxin C1 blocks neurotransmitter release by means of cleaving HPC-1/syntaxin. EMBO J. 1993;12:4821–4828. doi: 10.1002/j.1460-2075.1993.tb06171.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronk P, Deak F, Wilson MC, Liu X, Sudhof TC, Kavalali ET. Differential effects of SNAP-25 deletion on Ca2+ -dependent and Ca2+ -independent neurotransmission. J Neurophysiol. 2007;98:794–806. doi: 10.1152/jn.00226.2007. [DOI] [PubMed] [Google Scholar]
- Carbone E, Calorio C, Vandael DH. T-type channel-mediated neurotransmitter release. Pflugers Arch. 2014;466:677–687. doi: 10.1007/s00424-014-1489-z. [DOI] [PubMed] [Google Scholar]
- Catterall WA. Interactions of presynaptic Ca2+ channels and snare proteins in neurotransmitter release. Ann NY Acad Sci. 1999;868:144–159. doi: 10.1111/j.1749-6632.1999.tb11284.x. [DOI] [PubMed] [Google Scholar]
- Churchward MA, Rogasevskaia T, Hofgen J, Bau J, Coorssen JR. Cholesterol facilitates the native mechanism of Ca2+-triggered membrane fusion. J Cell Sci. 2005;118:4833–4848. doi: 10.1242/jcs.02601. [DOI] [PubMed] [Google Scholar]
- Condliffe SB, Corradini I, Pozzi D, Verderio C, Matteoli M. Endogenous SNAP-25 regulates native voltage-gated calcium channels in glutamatergic neurons. J Biol Chem. 2010;285:24968–24976. doi: 10.1074/jbc.M110.145813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condliffe SB, Matteoli M. Inactivation kinetics of voltage-gated calcium channels in glutamatergic neurons are influenced by SNAP-25. Channels. 2011;5:304–307. doi: 10.4161/chan.5.4.16228. [DOI] [PubMed] [Google Scholar]
- Corradini I, Verderio C, Sala M, Wilson MC, Matteoli M. SNAP-25 in neuropsychiatric disorders. Ann NY Acad Sci. 2009;1152:93–99. doi: 10.1111/j.1749-6632.2008.03995.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies JN, Jarvis SE, Zamponi GW. Bipartite syntaxin 1A interactions mediate CaV2.2 calcium channel regulation. Biochem Biophys Res Commun. 2011;411:562–568. doi: 10.1016/j.bbrc.2011.06.185. [DOI] [PubMed] [Google Scholar]
- Degtiar VE, Scheller RH, Tsien RW. Syntaxin modulation of slow inactivation of N-type calcium channels. J Neurosci. 2000;20:4355–4367. doi: 10.1523/JNEUROSCI.20-12-04355.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado-Martinez I, Nehring RB, Sorensen JB. Differential abilities of SNAP-25 homologs to support neuronal function. J Neurosci. 2007;27:9380–9391. doi: 10.1523/JNEUROSCI.5092-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulubova I, Sugita S, Hill S, Hosaka M, Fernandez I, Sudhof TC, Rizo J. A conformational switch in syntaxin during exocytosis: role of munc18. EMBO J. 1999;18:4372–4382. doi: 10.1093/emboj/18.16.4372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan RR, Don-Wauchope AC, Tapechum S, Shipston MJ, Chow RH, Estibeiro P. High-efficiency Semliki Forest virus-mediated transduction in bovine adrenal chromaffin cells. Biochem J. 1999;342:497–501. [PMC free article] [PubMed] [Google Scholar]
- Fasshauer D, Margittai M. A transient N-terminal interaction of SNAP-25 and syntaxin nucleates SNARE assembly. J Biol Chem. 2004;279:7613–7621. doi: 10.1074/jbc.M312064200. [DOI] [PubMed] [Google Scholar]
- Foran P, Lawrence GW, Shone CC, Foster KA, Dolly JO. Botulinum neurotoxin C1 cleaves both syntaxin and SNAP-25 in intact and permeabilized chromaffin cells: correlation with its blockade of catecholamine release. Biochemistry. 1996;35:2630–2636. doi: 10.1021/bi9519009. [DOI] [PubMed] [Google Scholar]
- Frassoni C, Inverardi F, Coco S, Ortino B, Grumelli C, Pozzi D, Verderio C, Matteoli M. Analysis of SNAP-25 immunoreactivity in hippocampal inhibitory neurons during development in culture and in situ. Neuroscience. 2005;131:813–823. doi: 10.1016/j.neuroscience.2004.11.042. [DOI] [PubMed] [Google Scholar]
- Friedrich R, Groffen AJ, Connell E, van Weering JR, Gutman O, Henis YI, Davletov B, Ashery U. DOC2B acts as a calcium switch and enhances vesicle fusion. J Neurosci. 2008;28:6794–6806. doi: 10.1523/JNEUROSCI.0538-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber SH, Rah JC, Min SW, Liu X, de Wit H, Dulubova I, Meyer AC, Rizo J, Arancillo M, Hammer RE, et al. Conformational switch of syntaxin-1 controls synaptic vesicle fusion. Science. 2008;321:1507–1510. doi: 10.1126/science.1163174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant NJ, Hepp R, Krause W, Aunis D, Oehme P, Langley K. Differential expression of SNAP-25 isoforms and SNAP-23 in the adrenal gland. J Neurochem. 1999;72:363–372. doi: 10.1046/j.1471-4159.1999.0720363.x. [DOI] [PubMed] [Google Scholar]
- Groffen AJ, Martens S, Arazola RD, Cornelisse LN, Lozovaya N, de Jong APH, Goriounova NA, Habets RLP, Takai Y, Borst JG, et al. Doc2b is a high-affinity Ca2+ sensor for spontaneous neurotransmitter release. Science. 2010;327:1614–1618. doi: 10.1126/science.1183765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulyas-Kovacs A, de Wit H, Milosevic I, Kochubey O, Toonen R, Klingauf J, Verhage M, Sorensen JB. Munc18-1: sequential interactions with the fusion machinery stimulate vesicle docking and priming. J Neurosci. 2007;27:8676–8686. doi: 10.1523/JNEUROSCI.0658-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halemani ND, Bethani I, Rizzoli SO, Lang T. Structure and dynamics of a two-helix SNARE complex in live cells. Traffic. 2010;11:394–404. doi: 10.1111/j.1600-0854.2009.01020.x. [DOI] [PubMed] [Google Scholar]
- Harkins AB, Cahill AL, Powers JF, Tischler AS, Fox AP. Deletion of the synaptic protein interaction site of the N-type (CaV2.2) calcium channel inhibits secretion in mouse pheochromocytoma cells. Proc Natl Acad Sci USA. 2004;101:15219–15224. doi: 10.1073/pnas.0401001101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugo S, Dembla E, Halimani M, Matti U, Rettig J, Becherer U. Deciphering dead-end docking of large dense core vesicles in bovine chromaffin cells. J Neurosci. 2013;33:17123–17137. doi: 10.1523/JNEUROSCI.1589-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurley JH, Cahill AL, Wang M, Fox AP. Syntaxin 1A regulation of weakly inactivating N-type Ca2+channels. J Physiol. 2004;560:351–363. doi: 10.1113/jphysiol.2004.068817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahn R, Fasshauer D. Molecular machines governing exocytosis of synaptic vesicles. Nature. 2012;490:201–207. doi: 10.1038/nature11320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvis SE, Barr W, Feng ZP, Hamid J, Zamponi GW. Molecular determinants of syntaxin 1 modulation of N-type calcium channels. J Biol Chem. 2002;277:44399–44407. doi: 10.1074/jbc.M206902200. [DOI] [PubMed] [Google Scholar]
- Jarvis SE, Magga JM, Beedle AM, Braun JE, Zamponi GW. G protein modulation of N-type calcium channels is facilitated by physical interactions between syntaxin 1A and Gbetagamma. J Biol Chem. 2000;275:6388–6394. doi: 10.1074/jbc.275.9.6388. [DOI] [PubMed] [Google Scholar]
- Jarvis SE, Zamponi GW. Distinct molecular determinants govern syntaxin 1A-mediated inactivation and G-protein inhibition of N-type calcium channels. J Neurosci. 2001;21:2939–2948. doi: 10.1523/JNEUROSCI.21-09-02939.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keith RK, Poage RE, Yokoyama CT, Catterall WA, Meriney SD. Bidirectional modulation of transmitter release by calcium channel/syntaxin interactions in vivo. J Neurosci. 2007;27:265–269. doi: 10.1523/JNEUROSCI.4213-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DK, Catterall WA. Ca2+-dependent and -independent interactions of the isoforms of the alpha1A subunit of brain Ca2+ channels with presynaptic SNARE proteins. Proc Natl Acad Sci USA. 1997;94:14782–14786. doi: 10.1073/pnas.94.26.14782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klingauf J, Neher E. Modeling buffered Ca2+ diffusion near the membrane: implications for secretion in neuroendocrine cells. Biophys J. 1997;72:674–690. doi: 10.1016/s0006-3495(97)78704-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles MK, Barg S, Wan L, Midorikawa M, Chen X, Almers W. Single secretory granules of live cells recruit syntaxin-1 and synaptosomal associated protein 25 (SNAP-25) in large copy numbers. Proc Natl Acad Sci USA. 2010;107:20810–20815. doi: 10.1073/pnas.1014840107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochubey O, Lou X, Schneggenburger R. Regulation of transmitter release by Ca(2+) and synaptotagmin: insights from a large CNS synapse. Trends Neurosci. 2011;34:237–246. doi: 10.1016/j.tins.2011.02.006. [DOI] [PubMed] [Google Scholar]
- Lang T, Bruns D, Wenzel D, Riedel D, Holroyd P, Thiele C, Jahn R. SNAREs are concentrated in cholesterol-dependent clusters that define docking and fusion sites for exocytosis. EMBO J. 2001;20:2202–2213. doi: 10.1093/emboj/20.9.2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Schirra C, Edelmann L, Matti U, Rhee J, Hof D, Bruns D, Brose N, Rieger H, Stevens DR, Rettig J. Two distinct secretory vesicle-priming steps in adrenal chromaffin cells. J Cell Biol. 2010;190:1067–1077. doi: 10.1083/jcb.201001164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Font I, Torregrosa-Hetland CJ, Villanueva J, Gutierrez LM. t-SNARE cluster organization and dynamics in chromaffin cells. J Neurochem. 2010;114:1550–1556. doi: 10.1111/j.1471-4159.2010.06872.x. [DOI] [PubMed] [Google Scholar]
- Lu Q, AtKisson MS, Jarvis SE, Feng ZP, Zamponi GW, Dunlap K. Syntaxin 1A supports voltage-dependent inhibition of alpha1B Ca2+ channels by Gbetagamma in chick sensory neurons. J Neurosci. 2001;21:2949–2957. doi: 10.1523/JNEUROSCI.21-09-02949.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C, Su L, Seven AB, Xu Y, Rizo J. Reconstitution of the vital functions of Munc18 and Munc13 in neurotransmitter release. Science. 2013;339:421–425. doi: 10.1126/science.1230473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahapatra S, Calorio C, Vandael DH, Marcantoni A, Carabelli V, Carbone E. Calcium channel types contributing to chromaffin cell excitability, exocytosis and endocytosis. Cell Calcium. 2012;51:321–330. doi: 10.1016/j.ceca.2012.01.005. [DOI] [PubMed] [Google Scholar]
- Man KM, Imig C, Walter AM, Pinheiro PS, Stevens DR, Rettig J, Sorensen JB, Cooper BH, Brose N, Wojcik SM. Identification of a Munc13-sensitive step in chromaffin cell large dense-core vesicle exocytosis. Elife. 2015;4 doi: 10.7554/eLife.10635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcantoni A, Vandael DH, Mahapatra S, Carabelli V, Sinnegger-Brauns MJ, Striessnig J, Carbone E. Loss of Cav1.3 channels reveals the critical role of L-type and BK channel coupling in pacemaking mouse adrenal chromaffin cells. J Neurosci. 2010;30:491–504. doi: 10.1523/JNEUROSCI.4961-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochida S, Sheng ZH, Baker C, Kobayashi H, Catterall WA. Inhibition of neurotransmission by peptides containing the synaptic protein interaction site of N-type Ca2+ channels. Neuron. 1996;17:781–788. doi: 10.1016/s0896-6273(00)80209-3. [DOI] [PubMed] [Google Scholar]
- Mochida S, Westenbroek RE, Yokoyama CT, Zhong H, Myers SJ, Scheuer T, Itoh K, Catterall WA. Requirement for the synaptic protein interaction site for reconstitution of synaptic transmission by P/Q-type calcium channels. Proc Natl Acad Sci USA. 2003;100:2819–2824. doi: 10.1073/pnas.262787699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohrmann R, de Wit H, Verhage M, Neher E, Sorensen JB. Fast vesicle fusion in living cells requires at least three SNARE complexes. Science. 2010;330:502–505. doi: 10.1126/science.1193134. [DOI] [PubMed] [Google Scholar]
- Munch AS, Kedar GH, van Weering JR, Vazquez-Sanchez S, He E, Andre T, Braun T, Sollner TH, Verhage M, Sorensen JB. Extension of helix 12 in Munc18-1 induces vesicle priming. J Neurosci. 2016;36:6881–6891. doi: 10.1523/JNEUROSCI.0007-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy G, Milosevic I, Fasshauer D, Muller EM, de Groot BL, Lang T, Wilson MC, Sorensen JB. Alternative splicing of SNAP-25 regulates secretion through nonconservative substitutions in the SNARE domain. Mol Biol Cell. 2005;16:5675–5685. doi: 10.1091/mbc.E05-07-0595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher E. Merits and limitations of vesicle pool models in view of heterogeneous populations of synaptic vesicles. Neuron. 2015;87:1131–1142. doi: 10.1016/j.neuron.2015.08.038. [DOI] [PubMed] [Google Scholar]
- Ohtani Y, Irie T, Uekama K, Fukunaga K, Pitha J. Differential effects of alpha-, beta- and gamma-cyclodextrins on human erythrocytes. Eur J Biochem. 1989;186:17–22. doi: 10.1111/j.1432-1033.1989.tb15171.x. [DOI] [PubMed] [Google Scholar]
- Ormerod KG, Rogasevskaia TP, Coorssen JR, Mercier AJ. Cholesterol-independent effects of methyl-beta-cyclodextrin on chemical synapses. PLoS One. 2012;7:e36395. doi: 10.1371/journal.pone.0036395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padin JF, Fernandez-Morales JC, de Diego AM, Garcia AG. Calcium channel subtypes and exocytosis in chromaffin cells at early life. Cur Mol Pharmacol. 2015;8:81–86. doi: 10.2174/1874467208666150507094537. [DOI] [PubMed] [Google Scholar]
- Pinheiro PS, de Wit H, Walter AM, Groffen AJ, Verhage M, Sorensen JB. Doc2b synchronizes secretion from chromaffin cells by stimulating fast and inhibiting sustained release. J Neurosci. 2013;33:16459–16470. doi: 10.1523/JNEUROSCI.2656-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozzi D, Condliffe S, Bozzi Y, Chikhladze M, Grumelli C, Proux-Gillardeaux V, Takahashi M, Franceschetti S, Verderio C, Matteoli M. Activity-dependent phosphorylation of Ser187 is required for SNAP-25-negative modulation of neuronal voltage-gated calcium channels. Proc Natl Acad Sci USA. 2008;105:323–328. doi: 10.1073/pnas.0706211105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rettig J, Heinemann C, Ashery U, Sheng ZH, Yokoyama CT, Catterall WA, Neher E. Alteration of Ca2 +dependence of neurotransmitter release by disruption of Ca2+ channel/syntaxin interaction. J Neurosci. 1997;17:6647–6656. doi: 10.1523/JNEUROSCI.17-17-06647.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rettig J, Sheng ZH, Kim DK, Hodson CD, Snutch TP, Catterall WA. Isoform-specific interaction of the alpha1A subunits of brain Ca2+ channels with the presynaptic proteins syntaxin and SNAP-25. Proc Natl Acad Sci USA. 1996;93:7363–7368. doi: 10.1073/pnas.93.14.7363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickman C, Medine CN, Dun AR, Moulton DJ, Mandula O, Halemani ND, Rizzoli SO, Chamberlain LH, Duncan RR. t-SNARE protein conformations patterned by the lipid microenvironment. J Biol Chem. 2010;285:13535–13541. doi: 10.1074/jbc.M109.091058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizo J, Sudhof TC. The membrane fusion enigma: SNAREs, Sec1/Munc18 proteins, and their accomplices–guilty as charged? Annu Rev Cell Dev Biol. 2012;28:279–308. doi: 10.1146/annurev-cellbio-101011-155818. [DOI] [PubMed] [Google Scholar]
- Sato M, Mori Y, Matsui T, Aoki R, Oya M, Yanagihara Y, Fukuda M, Tsuboi T. Role of the polybasic sequence in the Doc2alpha C2B domain in dense-core vesicle exocytosis in PC12 cells. J Neurochem. 2010;114:171–181. doi: 10.1111/j.1471-4159.2010.06739.x. [DOI] [PubMed] [Google Scholar]
- Schutz D, Zilly F, Lang T, Jahn R, Bruns D. A dual function for Munc-18 in exocytosis of PC12 cells. Eur J Neurosci. 2005;21:2419–2432. doi: 10.1111/j.1460-9568.2005.04095.x. [DOI] [PubMed] [Google Scholar]
- Scullin CS, Tafoya LC, Wilson MC, Partridge LD. Presynaptic residual calcium and synaptic facilitation at hippocampal synapses of mice with altered expression of SNAP-25. Brain Res. 2012;1431:1–12. doi: 10.1016/j.brainres.2011.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieber JJ, Willig KI, Kutzner C, Gerding-Reimers C, Harke B, Donnert G, Rammner B, Eggeling C, Hell SW, Grubmuller H, Lang T. Anatomy and dynamics of a supramolecular membrane protein cluster. Science. 2007;317:1072–1076. doi: 10.1126/science.1141727. [DOI] [PubMed] [Google Scholar]
- Sinha R, Ahmed S, Jahn R, Klingauf J. Two synaptobrevin molecules are sufficient for vesicle fusion in central nervous system synapses. Proc Natl Acad Sci USA. 2011;108:14318–14323. doi: 10.1073/pnas.1101818108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sørensen JB, Nagy G, Varoqueaux F, Nehring RB, Brose N, Wilson MC, Neher E. Differential control of the releasable vesicle pools by SNAP-25 splice variants and SNAP-23. Cell. 2003;114:75–86. doi: 10.1016/s0092-8674(03)00477-x. [DOI] [PubMed] [Google Scholar]
- Sørensen JB, Wiederhold K, Muller EM, Milosevic I, Nagy G, de Groot BL, Grubmuller H, Fasshauer D. Sequential N- to C-terminal SNARE complex assembly drives priming and fusion of secretory vesicles. EMBO J. 2006;25:955–966. doi: 10.1038/sj.emboj.7601003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spafford JD, Munno DW, Van Nierop P, Feng ZP, Jarvis SE, Gallin WJ, Smit AB, Zamponi GW, Syed NI. Calcium channel structural determinants of synaptic transmission between identified invertebrate neurons. J Biol Chem. 2003;278:4258–4267. doi: 10.1074/jbc.M211076200. [DOI] [PubMed] [Google Scholar]
- Stanley EF. Syntaxin I modulation of presynaptic calcium channel inactivation revealed by botulinum toxin C1. Eur J Neurosci. 2003;17:1303–1305. doi: 10.1046/j.1460-9568.2003.02536.x. [DOI] [PubMed] [Google Scholar]
- Stanley EF, Mirotznik RR. Cleavage of syntaxin prevents G-protein regulation of presynaptic calcium channels. Nature. 1997;385:340–343. doi: 10.1038/385340a0. [DOI] [PubMed] [Google Scholar]
- Stevens DR, Wu ZX, Matti U, Junge HJ, Schirra C, Becherer U, Wojcik SM, Brose N, Rettig J. Identification of the minimal protein domain required for priming activity of Munc13-1. Curr Biol. 2005;15:2243–2248. doi: 10.1016/j.cub.2005.10.055. [DOI] [PubMed] [Google Scholar]
- Sudhof TC. A molecular machine for neurotransmitter release: synaptotagmin and beyond. Nat Med. 2013;19:1227–1231. doi: 10.1038/nm.3338. [DOI] [PubMed] [Google Scholar]
- Sutton KG, McRory JE, Guthrie H, Murphy TH, Snutch TP. P/Q-type calcium channels mediate the activity-dependent feedback of syntaxin-1A. Nature. 1999;401:800–804. doi: 10.1038/44586. [DOI] [PubMed] [Google Scholar]
- Tafoya LC, Mameli M, Miyashita T, Guzowski JF, Valenzuela CF, Wilson MC. Expression and function of SNAP-25 as a universal SNARE component in GABAergic neurons. J Neurosci. 2006;26:7826–7838. doi: 10.1523/JNEUROSCI.1866-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tafoya LC, Shuttleworth CW, Yanagawa Y, Obata K, Wilson MC. The role of the t-SNARE SNAP-25 in action potential-dependent calcium signaling and expression in GABAergic and glutamatergic neurons. BMC Neurosci. 2008;9:105. doi: 10.1186/1471-2202-9-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taschenberger H, von Gersdorff H. Fine-tuning an auditory synapse for speed and fidelity: developmental changes in presynaptic waveform, EPSC kinetics, and synaptic plasticity. J Neurosci. 2000;20:9162–9173. doi: 10.1523/JNEUROSCI.20-24-09162.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaidyanathan VV, Yoshino K, Jahnz M, Dorries C, Bade S, Nauenburg S, Niemann H, Binz T. Proteolysis of SNAP-25 isoforms by botulinum neurotoxin types A, C, and E: domains and amino acid residues controlling the formation of enzyme-substrate complexes and cleavage. J Neurochem. 1999;72:327–337. doi: 10.1046/j.1471-4159.1999.0720327.x. [DOI] [PubMed] [Google Scholar]
- Vandael DH, Marcantoni A, Carbone E. Cav1.3 channels as key regulators of neuron-like firings and catecholamine release in chromaffin cells. Curr Mol Pharmacol. 2015a;8:149–161. doi: 10.2174/1874467208666150507105443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandael DH, Ottaviani MM, Legros C, Lefort C, Guerineau NC, Allio A, Carabelli V, Carbone E. Reduced availability of voltage-gated sodium channels by depolarization or blockade by tetrodotoxin boosts burst firing and catecholamine release in mouse chromaffin cells. J Physiol. 2015b;593:905–927. doi: 10.1113/jphysiol.2014.283374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verderio C, Pozzi D, Pravettoni E, Inverardi F, Schenk U, Coco S, Proux-Gillardeaux V, Galli T, Rossetto O, Frassoni C, Matteoli M. SNAP-25 modulation of calcium dynamics underlies differences in GABAergic and glutamatergic responsiveness to depolarization. Neuron. 2004;41:599–610. doi: 10.1016/s0896-6273(04)00077-7. [DOI] [PubMed] [Google Scholar]
- Voets T, Neher E, Moser T. Mechanisms underlying phasic and sustained secretion in chromaffin cells from mouse adrenal slices. Neuron. 1999;23:607–615. doi: 10.1016/s0896-6273(00)80812-0. [DOI] [PubMed] [Google Scholar]
- Voets T, Toonen RF, Brian EC, de Wit H, Moser T, Rettig J, Sudhof TC, Neher E, Verhage M. Munc18–1 promotes large dense-core vesicle docking. Neuron. 2001;31:581–591. doi: 10.1016/s0896-6273(01)00391-9. [DOI] [PubMed] [Google Scholar]
- Weiss N, Hameed S, Fernandez-Fernandez JM, Fablet K, Karmazinova M, Poillot C, Proft J, Chen L, Bidaud I, Monteil A, et al. A Ca(v)3.2/syntaxin-1A signaling complex controls T-type channel activity and low-threshold exocytosis. J Biol Chem. 2012;287:2810–2818. doi: 10.1074/jbc.M111.290882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelm BG, Mandad S, Truckenbrodt S, Krohnert K, Schafer C, Rammner B, Koo SJ, Classen GA, Krauss M, Haucke V, et al. Composition of isolated synaptic boutons reveals the amounts of vesicle trafficking proteins. Science. 2014;344:1023–1028. doi: 10.1126/science.1252884. [DOI] [PubMed] [Google Scholar]
- Wiser O, Bennett MK, Atlas D. Functional interaction of syntaxin and SNAP-25 with voltage-sensitive L- and N-type Ca2+ channels. EMBO J. 1996;15:4100–4110. [PMC free article] [PubMed] [Google Scholar]
- Wiser O, Trus M, Hernandez A, Renstrom E, Barg S, Rorsman P, Atlas D. The voltage sensitive Lc-type Ca2+ channel is functionally coupled to the exocytotic machinery. Proc Natl Acad Sci USA. 1999;96:248–253. doi: 10.1073/pnas.96.1.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao W, Poirier MA, Bennett MK, Shin YK. The neuronal t-SNARE complex is a parallel four-helix bundle. Nat Struct Biol. 2001;8:308–311. doi: 10.1038/86174. [DOI] [PubMed] [Google Scholar]
- Yang Y, Craig TJ, Chen X, Ciufo LF, Takahashi M, Morgan A, Gillis KD. Phosphomimetic mutation of Ser-187 of SNAP-25 increases both syntaxin binding and highly Ca2+-sensitive exocytosis. J Gen Physiol. 2007;129:233–244. doi: 10.1085/jgp.200609685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama CT, Myers SJ, Fu J, Mockus SM, Scheuer T, Catterall WA. Mechanism of SNARE protein binding and regulation of Cav2 channels by phosphorylation of the synaptic protein interaction site. Mol Cell Neurosci. 2005;28:1–17. doi: 10.1016/j.mcn.2004.08.019. [DOI] [PubMed] [Google Scholar]
- Yokoyama CT, Sheng ZH, Catterall WA. Phosphorylation of the synaptic protein interaction site on N-type calcium channels inhibits interactions with SNARE proteins. J Neurosci. 1997;17:6929–6938. doi: 10.1523/JNEUROSCI.17-18-06929.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong H, Yokoyama CT, Scheuer T, Catterall WA. Reciprocal regulation of P/Q-type Ca2+ channels by SNAP-25, syntaxin and synaptotagmin. Nat Neurosci. 1999;2:939–941. doi: 10.1038/14721. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.